

Abstract
Obesity/metabolic syndrome are common risk factors for overactive bladder. This study aimed to investigate the functional and molecular changes of detrusor smooth muscle (DSM) in high-fat insulin resistant obese mice, focusing on the role of protein kinase C (PKC) and Cav1.2 in causing bladder dysfunction. Male C57BL/6 mice were fed with high-fat diet for 10 weeks. In vitro functional responses and cystometry, as well as PKC and Cav1.2 expression in bladder were evaluated. Obese mice exhibited higher body weight, epididymal fat mass, fasting glucose and insulin resistance. Carbachol (0.001–100 µM), α,β-methylene ATP (1–10 µM), KCl (1–300 mM), extracellular Ca2+ (0.01–100 mM) and phorbol-12,13-dibutyrate (PDBu; 0.001–3 µM) all produced greater DSM contractions in obese mice, which were fully reversed by the Cav1.2 blocker amlodipine. Cystometry evidenced augmented frequency, non-void contractions and post-void pressure in obese mice that were also prevented by amlodipine. Metformin treatment improved the insulin sensitivity, and normalized the in vitro bladder hypercontractility and cystometric dysfunction in obese mice. The PKC inhibitor GF109203X (1 µM) also reduced the carbachol induced contractions. PKC protein expression was markedly higher in bladder tissues from obese mice, which was normalized by metformin treatment. The Cav1.2 channel protein expression was not modified in any experimental group. Our findings show that Cav1.2 blockade and improvement of insulin sensitization restores the enhanced PKC protein expression in bladder tissues and normalizes the overactive detrusor. It is likely that insulin resistance importantly contributes for the pathophysiology of this urological disorder in obese mice.
Introduction
Metabolic syndrome is a highly prevalent public health problem that is defined by the coexistence of central obesity, insulin resistance, glucose intolerance, dyslipidemia and arterial hypertension [1]. Metabolic syndrome increases the risk of developing type II diabetes and cardiovascular diseases [2]. Insulin resistance is the central feature of metabolic syndrome, whereby target tissues fail to respond efficiently to normal concentrations of insulin. Obesity is the main etiological factor that predisposes to insulin resistance and vascular diseases [3]–[5].
Along with the dramatic increase in the incidence of diabetes, obesity and metabolic syndrome, there has been a parallel increase in urological complications [6]. Obesity and metabolic syndrome have been established as common risk factors for lower urinary tract symptoms (LUTS) including overactive bladder and urinary incontinence [7]–[9]. Overactive bladder is a highly prevalent condition that affects millions of people worldwide with a profound effect on quality of life and considerable costs to health care systems. The third National Health and Nutritional Examination Survey (NHANES III) showed that an increase in body mass index (BMI) in people over 25 years of age is positively correlated with LUTS, and men with a larger waist circumference (>102 cm) are more likely to exhibit LUTS [10]. However, few studies have explored the mechanisms of bladder dysfunction in metabolic syndrome / diabetes. Male Wistar rats fed with 60% fructose-enriched chow for 6 weeks became overweight, hyperinsulinemic and hyperglycemic, and exhibited unstable premicturition bladder contractions that were suggestive of detrusor overactivity [11]. Female Wistar rats treated with the same high-fructose diet for prolonged periods (24 weeks) showed no increase in body weight, although insulin resistance was observed [12]. Bladders from these animals showed markedly lower bladder contractions to electrical-field stimulation (EFS), carbachol and KCl in vitro, along with higher contractions to ATP. A cystometric study carried out in conscious female obese Zucker rats revealed decreased voiding frequency and non-voiding contractions in both obese non-diabetic and obese diabetic animals, suggesting that chronic obesity itself reduces the activity of the bladder, with no significant roles in voiding outcomes due to diabetes [13]. Although human obesity is mostly derived from consumption of high-fat diets combined with low expenditure of energy [14], only a single experimental study has focused on bladder activity in high-fat diet-induced obesity. Male Sprague-Dawley rats fed with a hyperlipidemia diet for 24 weeks gained more weight and showed an increase in voiding and non-voiding bladder contractions as evaluated by cystometry, suggesting bladder overactivity [15]. Urinary bladder dysfunction during obesity has thus been poorly studied, with conflicting findings that may be due to differences in the strain and sex of test animals, and experimental paradigm.
Protein kinase C (PKC) isoforms have been established as important regulators of lipid-induced insulin resistance [16]. Defects in the diacylglycerol (DAG)-PKC pathway in skeletal muscle and adipocytes are implicated in the insulin-resistant states of obesity and type II diabetes, as observed in animals [17], [18] and humans [19]. Under physiological conditions, coupling of muscarinic receptors (mAChR) to Cav1.2 L-type Ca2+ channels in pig and mice bladder has recently been shown to involve an atypical signaling cascade involving associated PKC and Cav1.2 channels [20]. In the present study we have used a model of metabolic syndrome, in which mice on a high-fat diet develop obesity and insulin resistance, to further investigate the functional and molecular changes in the urinary bladder in this condition. Since elevated Ca2+ entry via Cav1.2 L-type Ca2+ channels plays a major role in the altered detrusor contractions observed under diabetic conditions [21], we have also focused on the role of PKC-mediated extracellular Ca2+ influx through Cav1.2 Ca2+ channels to bladder dysfunction and its relationship with insulin resistance in the high-fat fed obese mice.
Materials and Methods
Animals
All animal procedures and experimental protocols are in accordance with the Ethical Principles in Animal Research adopted by the Brazilian College for Animal Experimentation (COBEA) and were approved by the institutional Committee for Ethics in Animal Research/State University of Campinas (CEEA-UNICAMP, protocol number 2067-1). Four-week-old male C57BL6/J mice were provided by the Central Animal House Services of State University of Campinas (UNICAMP).
Diet-induced Obesity and Treatments
The animals were housed three per cage on a 12 h light–dark cycle, and fed for 10 weeks with either a standard chow diet (carbohydrate: 70%; protein: 20%; fat: 10%) or a high fat diet that induces obesity (carbohydrate: 29%; protein: 16%; fat: 55%), as previously described [22], [23].
Lean and obese mice were further divided into two additional groups, namely, animals treated chronically with either the L-type Ca2+ channel blocker amlodipine (25 mg/kg/day, given in the drinking water from the 7th to the 10th week) [24] or the insulin sensitizing agent metformin (300 mg/kg/day, given by gavage from the 8th to the 10th week) [25]. Tail-cuff pressure was evaluated in all groups, adapted to mice.
In vivo Insulin Sensitivity
After 6 h fasting, systemic insulin sensitivity was analyzed by the Insulin Tolerance Test (ITT) which was performed. Tail blood samples were collected before (0 min) and at 5, 10, 15, 20, 25 and 30 min after an intraperitoneal injection of 1.00 U/Kg of regular insulin (Novolin R, NovoNordisk, Bagsvaerd, Denmark). Glucose concentrations were measured using a glucometer (ACCUCHEK Performa; Roche Diagnostics, Indianapolis, IN, USA) and the values were used to calculate the constant rate for blood glucose disappearance (Kitt), based on the linear regression of the neperian logarithm of glucose concentrations obtained from 0 to 30 min of the test. Kitt was calculated using the formula 0.693/(t1/2)×−1×100.
In vitro Functional Assays
Mice were anesthetized with isoflurane, and urinary bladders removed and sectioned horizontally at the level of the ureters. Two longitudinal detrusor smooth muscle (DSM) strips with intact urothelium were obtained from each bladder. Strips of DSM were mounted in 10 ml organ baths containing Krebs-Henseleit solution with the following composition (mM): 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3 and 11 glucose, pH 7.4, at 37°C and bubbled with a gas mixture of 95% O2 and 5% CO2. Changes in isometric force were recorded using a Power Lab v.7.0 system (Colorado Springs, CO, USA). The resting tension was adjusted to 0.5 g at the beginning of the experiments. The equilibration period was 60 min and the bathing medium was changed every 15 min.
Nonlinear regression analysis to determine the pEC50 was carried out using GraphPad Prism (GraphPad Software, San Diego, CA, USA) with the constraint that Φ = 0. All concentration–response data were evaluated for a fit to a logistics function in the form:
E is the maximum response produced by agonists; c is the logarithm of the EC50, the concentration of drug that produces a half-maximal response; x is the logarithm of the concentration of the drug; the exponential term, n, is a curve-fitting parameter that defines the slope of the concentration–response line and Φ is the response observed in the absence of added drug. The values of pEC50 data represent the mean ± S.E. Maximal response (Emax) data were normalized to the wet weight of the respective urinary bladder strips, and the values of Emax were represented by mN per milligram wet weight.
Contractile Responses to Carbachol, α,β-methylene ATP, KCl, Extracellular CaCl2 and phorbol-12,13-dibutyrate (PDBu) in Mice DSM
Cumulative concentration-response curves to the mAChR agonist carbachol (0.001–100 µM), the PKC activator PDBu (0.001–3 µM) and KCl (1–300 mM) in DSM strips were performed. Contractile response to the P2X1 purinergic agonist α,β-methylene ATP were obtained through non-cumulative addition of three different drug concentrations (1, 3 and 10 µM).
Cumulative concentration-response curves to extracellular CaCl2 (0.01–30 mM) in depolarizing conditions were constructed. The strips were prepared and mounted in 10 ml organ baths containing Krebs-Henseleit Ca2+-free solution containing EGTA (1 mM) to sequester Ca2+ ions, and cyclopiazonic acid (CPA, 1 µM) to deplete sarcoplasmic reticulum Ca2+ stores. Next, bath solution was removed and replaced by Krebs-Henseleit Ca2+-free solution containing KCl (80 mM) and CPA (1 µM). After an equilibration period of 15 min, the cumulative curve to CaCl2 was constructed [26].
The concentration-response curves to carbachol, KCl, PDBu and extracellular CaCl2 in DSM strips obtained from both lean and obese mice were performed in the absence and in the presence of either the L-type Ca2+ channel blocker amlodipine (3 µM, 30 min) or antidiabetic agent metformin (1 µM, 30 min). Concentration-response curves to carbachol were also performed in the presence of the PKC inhibitor GF109203X (1 µM, 30 min). Contractile responses to PDBu was also evaluated in a Ca2+-free Krebs buffer medium. Contractile responses to these agents were also evaluated in DSM strips from lean and obese mice treated chronically with amlodipine (25 mg/kg/day for 21 days) or metformin (300 mg/kg/day, 14 days).
Electrical-field Stimulated-induced DSM Contractions
Frequency-response curves (1–32 Hz) were elicited by stimulating the tissues for 10 s with pulses of 1 ms width at 80 V, with 3 min interval between stimulations. Subsequently after 30 min incubation periods, frequency-response curves were repeated in the presence of the non-selective mAChR antagonist atropine (1 µM, 30 min) and the P2X receptor blocker PPADS (30 µM, 30 min), to confirm the mediation by mAChR and P2X receptor activation. Frequency-response curves were also performed in the presence of the voltage-gated sodium channel blocker tetrodotoxin (1 µM, 30 min) to confirm the neurogenic nature of the contractions.
Cystometry
Mice were anaesthetized with an intraperitoneal injection of urethane (1.8 g/kg). A 1 cm abdominal incision was made, the bladder was exposed and a butterfly cannula (25 G) was inserted into the bladder dome. The cannula was connected to a three-way tap, one port of which was connected to a pressure transducer and the other to the infusion pump through a catheter (PE50). Before starting the cystometry, the bladder was emptied via the third port. Continuous cystometry was carried out by infusing saline into the bladder at a rate of 0.6 ml/h. The following parameters were assessed: Threshold pressure (TP; the intravesical pressure immediately before micturition); Post-void pressure (PVP; the intravesical pressure immediately after micturition); Peak pressure (PP; the peak pressure reached during micturition); Capacity (CP; the volume of saline needed to induce the first micturition); Compliance (CO; the ratio of CP to TP); Frequency of voiding contractions (VC) and frequency of non-voiding contractions (NVCs). NVCs were defined as spontaneous bladder contractions greater than 4 mmHg from the baseline pressure that did not result in a void. Bladders from mice used in the cystometry were not used in the other experiments.
Western Blotting Detection of PKC and Cav1.2 Calcium Channels in Urinary Bladder
Bladder tissues were homogenized in a SDS lysis buffer with a Polytron PTA 20S generator (model PT 10/35; Brinkmann Instruments, Inc., Westbury, NY) operated at maximum speed for 30 sec and centrifuged (12,000×g, 4°C, 20 min) to remove insoluble material. Protein concentrations of the supernatants were determined by the Bradford assay, and equal amount of protein from each sample (50 µg) was treated with Laemmli buffer containing dithiothreitol 100 mM. Samples were heated in a boiling water bath for 15 min and resolved by SDS-PAGE. Electrotransfer of proteins to nitrocellulose membrane was performed for 60 min at 15 V (constant) in a semi-dry device (Bio-Rad, Hercules, CA, USA). Nonspecific protein binding to nitrocellulose was reduced by pre-incubating the membrane overnight at 4°C in blocking buffer (0.5% non-fat dried milk, 10 mM Tris, 100 mM NaCl, and 0.02% Tween 20). Detection using specific antibodies, HRP-conjugated secondary antibodies, and luminol solution was performed, as described previously [27]. Anti-PKC (ab59363), anti-DHPRα1 subunit (ab58552) antibodies were obtained from AbCam Technology (Cambridge, England, UK)and and anti GAPDH (SC25778) was from Santa Cruz Biotechnologie (Santa Cruz, CA, USA. Densitometry was performed using the Scion Image software (Scion Corporation, Frederick, MD) and results represented as the ratio of the density of the PKC / CaV1.2-α1 band to the density of the GAPDH band.
Drugs
Urethane, metformin, carbachol, pyridoxalphosphate-6-azophenyl-2’,4’-disulfonic acid (PPADS), α,β-methylene ATP, atropine, amlodipine, tetrodotoxin, phorbol-12,13-dibutyrate (PDBu) and cyclopiazonic acid were obtained from Sigma (St. Louis, MO, USA).
Statistical Analysis
Data are expressed as mean ± SEM of n experiments. In the cumulative concentration and frequency-response curves data were expressed as mean of the contraction in mN/mg of wet strip weight ± SEM of n experiments. The program Instat (GraphPad Software) was used for statistical analysis. One-way analysis of variances (ANOVA) followed by a Tukey test was used in all groups, and p<0.05 was accepted as significant.
Results
Body Weight, Epididymal Fat Mass and Insulin Sensitivity
Obese mice exhibited a significant increase in body weight and epididymal fat mass compared with lean mice (p<0.001; Table 1). Bladder weight was not significantly modified between lean and obese groups. Fasting glucose levels were increased by 70% in obese group (p<0.001; Table 1), whereas insulin sensitivity was markedly reduced when compared with lean mice (p<0.05), as evidenced by the curve representing glucose decay and Kitt values (Figure 1A and B). Tail-cuff pressure was not modified in obese compared with lean group (84±3.4 and 86±3.3 mmHg, respectively).
Table 1. Body weight, epididymal fat mass, bladder weight and glucose levels in lean and high-fat fed obese mice, treated or not with metformin (300 mg/kg/day, 14 days).
| Lean | Lean + Metformin | Obese | Obese + Metformin | |
| Body weight (g) | 28.5±0.3 (9) | 28.4±0.3 (10) | 45.7±1.1*** (9) | 43.7±0.9*** (10) |
| Epididymal fat mass (g) | 0.25±0.01 (9) | 0.39±0.3 (10) | 1.71±0.08*** (9) | 1.86±0.1*** (10) |
| Bladder weight (mg) | 24.9±4.1 (6) | 25.7±0.8 (7) | 27.9±6.9 (6) | 27.1±0.9 (7) |
| Glucose (mg/dl) | 139±6.6 (9) | 166±10 (5) | 238±12*** (10) | 162±5.2# (5) |
Data represent the means ± SEM for 5–10 mice.
p<0.001 compared with lean group;
p<0.001 compared with respective obese group.
Figure 1. Insulin tolerance test (ITT): (A) insulin sensitivity test after intraperitoneal injection of insulin (1.00 U/kg body wt) was performed on lean and obese groups.
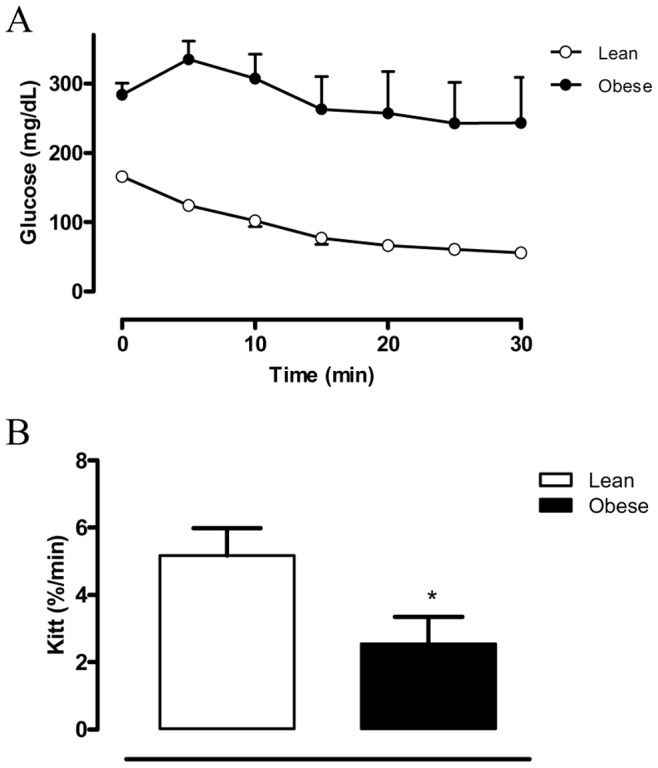
Blood samples were collected from the tail at indicated time points and analyzed for glucose concentration; (B) the constant rate for blood glucose disappearance (Kitt), based on the linear regression of the neperian logarithm of glucose concentrations, obtained from indicated time points. Results are expressed as mean ± SEM from 5–7 animals in each group.
In vitro Functional Assays
Figure 2A shows that the mAChR agonist carbachol (1 nM – 100 µM) produced concentration-dependent contractions in strips of isolated DSM with a maximal response (Emax) greater in the obese (p<0.01) than the lean group (5.02±0.89 and 1.83±0.36 mN/mg, respectively; n = 6–7). No significant differences for the pEC50 values for carbachol were found between the lean and obese groups (6.23±0.09 and 6.11±0.06, respectively).
Figure 2. Detrusor smooth muscle contraction in response to the muscarinic agonist carbachol (A), the purinergic P2X agonist α,β-methylene-ATP (B), electrical-field stimulation (C), KCl (D) and CaCl2 (E) in bladder strips from control and obese mice.

Data represent the mean ± SEM for 6 to 7 mice each group. * p<0.05, ** p<0.01 compared with control group.
Contractile responses to the P2X receptor agonist α,β-methylene ATP (1 – 10 µM) did not significantly differ between control and obese groups (n = 6–7; Figure 2B).
Electrical-field stimulation (EFS; 1–32 Hz) produced frequency-dependent DSM contractions in both groups, which were greater in obese mice at all frequencies employed (Figure 2C ; n = 6). Pre-treatment of DSM preparations with the mAChR antagonist atropine (1 µM) together with the purinergic receptor blocker PPADS (30 µM) markedly reduced (p<0.001) the EFS-induced contractions in both lean (Emax: 1.57±0.25 and 0.37±0.09 mN/mg for untreated and treated preparations, respectively; n = 6) and obese mice (Emax: 2.82±0.38 and 0.93±0.19 mN/mg for untreated and treated preparations, respectively; n = 6). Incubation with the voltage-gated sodium channel blocker tetrodotoxin (1 µM) almost abolished the EFS-elicited contractions at all frequencies tested (n = 4, data not shown).
Cumulative concentration-response curves to the receptor-independent agents KCl and CaCl2 were also obtained in DSM strips from obese and lean mice (Figure 2 D and E). Potassium chloride (KCl; 1 – 300 mM; n = 6–7) and CaCl2 (0.01 – 100 mM; n = 5–6) produced concentration-dependent DSM contractions with Emax significantly greater in obese mice for both agents (p<0.05) compared with the lean group (Figure 2D and E). No differences at the pEC50 levels for KCl and CaCl2 were found between lean (1.25±0.07 and 2.19±0.08, respectively) and obese groups (1.36±0.07 and 2.14±0.06, respectively).
Effect of Cav1.2 L-type Ca2+ Channels Blockade by Amlodipine
Since KCl and CaCl2-induced contractions were enhanced in strips taken from obese DSM, we hypothesized that an increase in Ca2+ entry through L-type voltage-operated Ca2+ channels is likely to play a key role in the overactive DSM. In vitro incubation of DSM with the dihydropyridine calcium channel blocker amlodipine (3 µM; n = 4–5) nearly normalized the enhanced contractile responses (Emax) to carbachol in obese mice, with a small but significant inhibition of contraction in the lean mice (Figure 3 A and B). Similarly to carbachol, the enhanced contractile responses to KCl (Figure 3 C and D; n = 5–7) and CaCl2 (Figure 3 E and F; n = 4–5) in obese mice were prevented by in vitro incubation with amlodipine.
Figure 3. Cumulative concentration-response curves to carbachol (A and B), KCl (C and D) and CaCl2 (E and F) in the presence of in vitro pre-incubated metformin (1 µM) or amlodipine (3 µM) in detrusor smooth muscle from lean and obese mice.

Data represent the mean ± SEM for 4 to 7 mice each group. * p<0.05, ** p<0.01 compared with untreated group.
In separate groups, obese and lean mice were treated orally with amlodipine (25 mg/kg/day, 21 days), and contractile responses of DSM strips to carbachol (n = 6–7), KCl (n = 5–10) and CaCl2 (n = 5–11) were obtained. Long-term amlodipine administration prevented the enhancement of contractions to carbachol (Figure 4 A and B), KCl (Figure 4 C and D) and CaCl2 (Figure 4 E and F) in obese mice. In lean group, oral amlodipine did not significantly alter the contractile responses to carbachol and KCl, but significantly reduced the CaCl2-induced DSM contractions.
Figure 4. Cumulative concentration-response curves to carbachol (A and B), KCl (C and D) and CaCl2 (E,F) in detrusor smooth muscle from lean and obese mice chronically treated with metformin (300 mg/kg/day, 14 days) or amlodipine (25 mg/kg/day, 21 days).

Data represent the mean ± SEM for 5 to 11 mice each group. * p<0.05, ** p<0.01 compared with untreated group.
Role for Insulin Resistance in the Overactive DSM in Obese Mice
Chronic treatment with the anti-hyperglycemic agent metformin (300 mg/kg/day, 14 days) did not significantly affect body weight and epididymal fat mass in obese or lean mice (Table 1, n = 10). However, metformin treatment normalized the lower insulin sensitivity seen in obese mice, restoring the Kitt to control values (5.17±0.81, 4.68±1.23, 2.54±0.80 and 5.76±0.38 % / min for lean untreated, lean treated, obese untreated and obese treated, respectively; n = 4 – 6), as well as the fasting blood glucose values (166±10 and 162±5.2 mg/dL for lean and obese treated, respectively; n = 5).
Chronic metformin treatment had no significant effect on DSM contractions in lean mice (Figure 4 A, C and E). However, the increase in DSM contractions to carbachol (Figure 4B), KCl (Figure 4D) and CaCl2 (Figure 4F) in obese mice was suppressed by chronic metformin treatment (n = 6 – 11). In contrast, in vitro incubation of DSM strips with metformin (1 µM, 30 min) had no effect on DSM contractions in lean or obese mice (Figure 3).
Cystometric Studies
During cystometry, lean mice showed regular micturition cycles with rare non-voiding contractions (Figure 5A). In contrast, obese mice exhibited an irregular micturition pattern (Figure 5B) and significant increases (p<0.01) in the frequency of voiding and non-voiding contractions (Figure 6 A and B; n = 5–7). The post-void pressure (PVP), which reflects the efficiency of bladder emptying, was markedly higher (p<0.01) in obese mice compared with the lean group (Figure 6C). The bladder capacity, threshold pressure, compliance and peak pressure did not significantly differ between lean and obese mice (Table 2).
Figure 5. Representative cystometric recordings from lean (A), obese (B), obese treated with amlodipine (C) and obese treated with metformin (D) mice.

Arrows in the cystometric trace indicate the micturition peaks.
Figure 6. Cystometric parameters in lean and obese mice treated or not with metformin (300 mg/kg/day, 14 days) or amlodipine (25 mg/kg/day, 21 days).
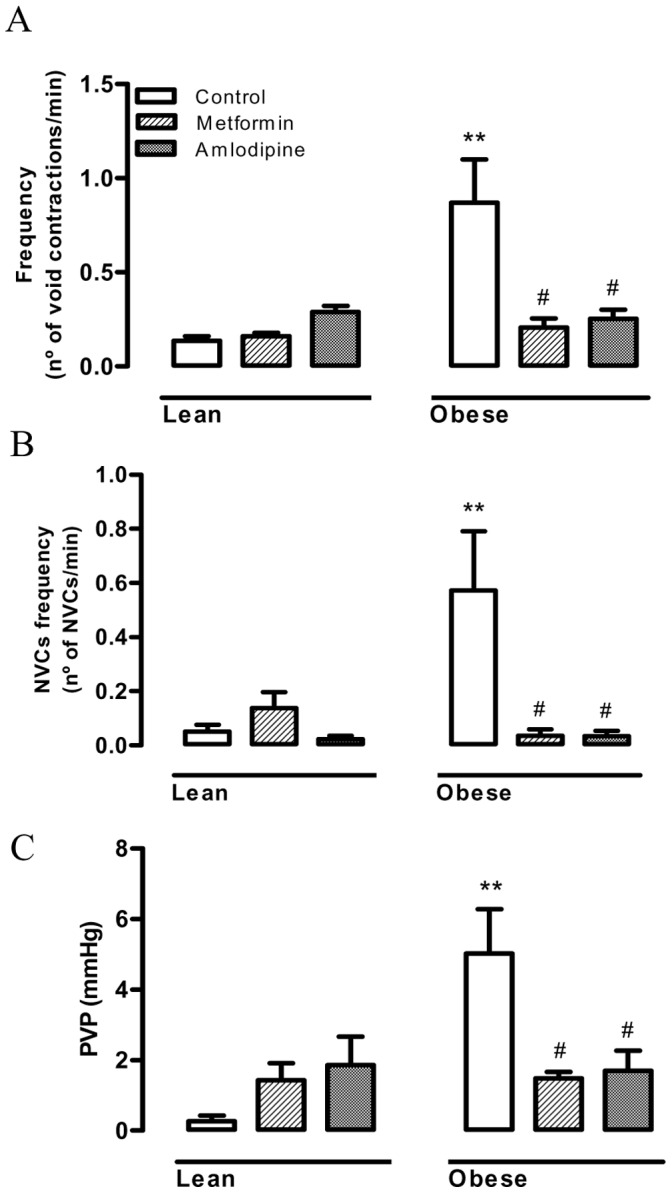
(A) Frequency of voiding contractions, (B) Frequency of non-voiding contractions and (C) Post-void pressure. Data represent the mean ± SEM for 5–7 mice each group. ** p<0.01 compared with control group, # p<0.01 compared with untreated obese mice.
Table 2. Values of cystometric parameters evaluated in lean and obese mice treated or not with amlodipine (25 mg/kg/day, 21 days) or metformin (300 mg/kg/day, 14 days).
| Lean mice | Obese mice | |||||
| Untreated | Amlodipine | Metformin | Untreated | Amlodipine | Metformin | |
| Capacity (ml) | 0.13±0.01 | 0.15±0.02 | 0.19±0.04 | 0.13±0.02 | 0.13±0.03 | 0.16±0.02 |
| Threshold pressure (mmHg) | 3.6±0.7 | 5.3±0.7 | 4.2±0.3 | 4.3±0.9 | 4.0±0.6 | 5.4±0.9 |
| Compliance (mmHg/mL) | 0.03±0.004 | 0.03±0.007 | 0.04±0.009 | 0.03±0.009 | 0.03±0.007 | 0.036±0.011 |
| Peak pressure (mmHg) | 12.2±0.82 | 9.3±1.02 | 13.9±1.44 | 12.2±2.3 | 9.9±0.88 | 16.2±1.9 |
Results are expressed as means ± SEM from 4–7 animals in each group.
Since amlodipine normalized the overactive DSM in obese mice, we further investigated its effects on the urodynamic changes. Long-term treatment with amlodipine had no significant effects on the cystometric parameters of lean mice (n = 5–7). However, this treatment prevented the increased frequencies of voiding and non-voiding contractions, and prevented the increased PVP seen in the obese group (Figures 5C and 6). The other cystometric parameters (capacity, threshold pressure, compliance, and peak pressure) remained unchanged in amlodipine-treated mice (Table 2).
As the improvement of insulin sensitivity by chronic treatment with metformin also normalized the overactive DSM in obese mice, we next investigated the effect of this anti-hyperglycemic agent on the cystometric alterations. The increased frequency of voiding and non-voiding contractions, as well as the increased PVP in obese mice was normalized by metformin treatment (Figures 5D and 6). Metformin treatment did not significantly affect these parameters in lean mice (Figure 6). The other cystometric parameters remained unchanged in metformin-treated mice (Table 2).
Role of Protein Kinase C (PKC) in the Enhanced Contractile-responses
As PKC regulates Cav1.2 calcium channel activity in various smooth muscle cell types, including DSM [20], we investigated the role of PKC in the enhanced DSM contractions in obese mice, by using functional and molecular approaches. The PKC inhibitor GF109203X (1 µM) normalized the enhanced carbachol-induced contractions in obese mice (Figure 7A). In addition, the PKC activator PDBu (0.001–3 µM) produced concentration-dependent contractions in strips of isolated DSM that were greater in the obese than the lean group (Figure 7B; n = 4–7; p<0.01). Prior incubation with amlodipine (3 µM) abolished the PDBu-induced DSM contractions in both obese and lean mice (Figure 7A). Similar data were obtained with chronic treatment with amlodipine (n = 4; not shown). No contractile responses to PDBu were observed in Ca2+-free medium (n = 3; not shown).
Figure 7. Contractile responses to carbachol (A) and phorbol-12,13-dibutyrate (PDBu; B) in detrusor smooth muscle from lean and obese mice.

Responses to carbachol were performed in the presence of the PKC inhibitor GF109203X (1 µM, 30 min), whereas responses to PDBu were performed in the presence of amlodipine (3 µM). Protein expression (Western blotting) for PKC and α1 subunit of Cav1.2 calcium channel in bladder tissues from lean and obese mice, treated or not with either amlodipine (3 µM, 30 min) or metformin (300 mg/kg/day, 14 days), is shown in panels C, D and E. Data represent the mean ± SEM for 4 to 7 mice each group. * p<0.05, ** p<0.01 compared with untreated lean group; *** p<0.001, † p<0.05 compared with respective amlodipine groups.
The PKC protein expression was significantly higher in bladder tissues from obese mice (p<0.05), and the increase in PKC expression was abrogated by amlodipine or metformin (Figure 7 C, D and E). Neither of these treatments affected the expression of PKC in the lean group. Expression of the α1 subunit of Cav1.2 was equivalent in all treatment groups (Figure 7C, D and E).
Discussion
The present study shows that high-fat fed obese mice display overactive bladder and enhanced PKC protein expression in bladder tissues that are normalized by blockade of Cav1.2 and improvement of insulin sensitivity. It is likely that insulin resistance in obese mice plays a major role in the pathophysiology of this urological disorder.
The term “metabolic syndrome” describes the combination of metabolic abnormalities or risk factors for type II diabetes and cardiovascular diseases, such as central obesity, dyslipidemia, hypertension, insulin resistance and glucose intolerance [2], [28]. Among these risk factors, central obesity is regarded to be the major determinant criteria for metabolic syndrome [4]. In the model of diet-induced obesity employed here, mice exhibited increased body weight, epididymal fat mass (corresponding to central obesity in human) and insulin resistance. Previous studies have also identified dyslipidemia and impaired glucose tolerance in high-fat fed mice [22], [23]. These changes suggest that this mouse model of metabolic syndrome closely mirrors the changes in humans with metabolic syndrome / type II diabetes. Obese mice did not show increased tail-cuff pressure, excluding arterial hypertension as a cause for the present functional bladder alterations.
Epidemiological studies support a strong causal link between obesity and the development of urinary incontinence [29]. Data from our urodynamic study revealed detrusor overactivity in obese mice, as evidenced by the higher frequencies of micturition and non-voiding contractions (NVC), as well as the increased post-voiding pressure (PVP). On the other hand, the bladder capacity, threshold pressure, compliance and peak pressure did not significantly change in the obese group. This contrasts with streptozotocin-induced type I diabetes in mice, where all of these parameters are increased in comparison with control animals [21], possibly as a consequence of the enhanced urine output (diuresis) and the resulting bladder remodeling [30]. Obesity / metabolic syndrome markedly increase the risk of benign prostatic hyperplasia (BPH) that is in turn a risk factor for detrusor overactivity resultant from bladder outlet obstruction [31]. High fat-fed insulin-resistant Sprague Dawley rats exhibit prostate enlargement that is prevented by treatment with the anti-hyperglycemic agent pioglitazone [32]. Therefore, in our study, whether the increased PVP and poor bladder emptying efficiency seen in obese mice reflect a failure to compensate for enhanced urethral resistance due to prostate enlargement requires further studies [32].
Urinary bladder function is regulated by a complex interaction of efferent and afferent fibers from the autonomic nervous system and somatic innervations [33]. The neurogenic contractions of the bladder mainly reflect the release of the excitatory transmitter acetylcholine (ACh) from parasympathetic fibers. The bladder contains all mAChR subtypes, but the M3 receptor is responsible for the urinary bladder contractions [34]. The excitatory transmitter ATP, through ionotropic P2X1 receptors, also mediates part of the atropine-resistant neurogenic bladder contractions under normal and pathophysiological conditions, although to varying extents across species from rodent to man [35]. There is also a muscarinic- and purinergic-resistant neurogenic component that has not yet been fully characterized [36], [37]. In our study, neurogenic- and carbachol-induced DSM contractions were greater in obese mice compared with lean mice, whereas no significant differences for the purinergic P2X1 agonist α,β-methylene ATP were found.
Muscarinic M3 receptors interact with Gq to elicit phosphoinositide hydrolysis and generation of the second messenger inositol-1,4,5-trisphosphate (IP3), which activates the IP3 receptor to release Ca2+ from internal stores [34]. Muscarinic agonist-induced contractions have also been shown to partly depend on Ca2+ entry through Cav1.2 channels, as evidenced in animal and human bladders under physiological conditions [38]–[42]. High levels of extracellular K+ depolarize the cell membrane and activate Cav1.2 channels, resulting in increased inward movement of Ca2+, which in turn activates contractile proteins [43]. In smooth muscle tissues, dihydropyridine Ca2+ channel blockers inhibit the increase in [Ca2+]i induced by high K+ or extracellular CaCl2. Our findings that DSM strips obtained from obese mice show greater contractile responses to both receptor–dependent (carbachol) and –independent agents (KCl and extracellular CaCl2) strongly suggest that enhanced extracellular Ca2+ entry via Cav1.2 channels plays a critical role in the overactive DSM in obese mice. This is reinforced by our data showing that pretreatment with the dihydropyridine Ca2+ channel blocker amlodipine fully reversed the enhanced contractile responses to carbachol, KCl and CaCl2 in obese mice. The urodynamic alterations in the obese group were also reversed by treatment of mice with amlodipine, which is consistent with the reversal of the enhanced DSM contractions in vitro to carbachol, KCl and extracellular CaCl2 by amlodipine.
The signaling cascade for M3 receptors in smooth muscle also involves generation of DAG leading to PKC activation and inhibition of myosin light-chain (MLC) phosphatase, thus enhancing the contractile response [43]. Dysfunctions of the DAG–PKC pathway have been associated with vascular abnormalities in diabetic and insulin resistant states [44]. In the present study, we have performed concentration-response curves to carbachol in the presence of the PKC inhibitor GF109203X, and found that enhanced DSM contractions in the strips taken from obese mice were nearly normalized by this inhibitor. Additionally, the PKC activator PDBu produced concentration-dependent DSM contractions that were markedly greater in the strips taken from obese mice, an effect nearly abolished by amlodipine. This is consistent with the higher PKC protein expression in the bladder tissues of obese mice. Our data that the α1 subunit of Cav1.2 channel expression remained unchanged between groups together with functional data suggest that overactive DSM in obese mice takes place through elevated Cav1.2 channel activity, increasing the extracellular Ca2+ influx. However, the lack of a direct measurement of Cav1.2 activity or extracellular Ca2+ influx by electrophysiological studies in the bladder limits our comprehension about the observed phenomenon. We cannot ascertain, for instance, if changes in each of the component of the M3/PKC/Cav1.2 axis act independently or as a signaling pathway to determine the overactive DSM. Cav1.2 channels have been suggested to serve as an anchoring target for PKC during translocation of this protein to the cell membrane after agonist stimulation [20]. The increased carbachol-induced bladder contractions observed under in vitro hyperglycemic conditions has also been associated to rho-kinase-mediated increased PKC activity [45]. Interestingly, in our study, amlodipine normalized the PKC protein levels in bladder tissue of obese mice. One may speculate that inactivation of Cav1.2 channels results in downregulation of PKC expression, which is consistent with a previous study showing that amlodipine decreases the phosphorylation and activity of PKC in cultured human endothelial cells stimulated with the PKC activator PMA [46].
Metformin is a first-line pharmacological treatment for patients with type II diabetes mellitus because of its favorable overall profile, including its glucose-lowering ability, weight-neutral effects, and low risk of hypoglycemia. In our study, oral treatment of obese mice with metformin normalized the insulin resistance, as well as the in vitro and the in vivo (cystometry) bladder dysfunction. Moreover, metformin reversed the enhanced PKC levels in the bladder tissues from obese mice. Accordingly, a previous study has shown that PKC-θ knockout mice are protected from fat-induced insulin resistance in skeletal muscle [47]. Our findings that in vitro incubation of DSM strips with metformin does not alter the overactive DSM indicate that normalization of bladder function resultant from the long-term treatment with this anti-hyperglycemic agent is secondary to the improvement of insulin sensitivity, rather than through a direct effect on the contractile machinery. Atypical and conventional PKC isoforms are classically related to mechanisms that contribute to insulin resistance in peripheral tissues responsible for glucose disposal and energy homeostasis [48]. Our present results evidence that PKC activation induced by high fat feeding plays a role in a very specific complication associated with insulin resistance. Thus, our data suggest that PKC activity plays an important pathological role that is beyond the impairment glucose homeostasis control. In agreement with our findings, endothelial insulin resistance and vascular hyperactivity in high fat diet fed mice was recently described to be ameliorated by PKC inhibition [44], [49], suggesting that the mechanism presently described is not specific for the bladder.
Obese mice treated with metformin remained overweight, suggesting that insulin resistance and subsequent hyperglycemia, rather than obesity, is the main cause of bladder overactivity. Although metformin reduces body weight and waist circumference in humans [50], no such effect has been reported in animals treated chronically with this agent [51]. Interestingly, in bladder dysfunction in female obese Zucker rats, it was the presence of chronic obesity rather than the onset of diabetes that led to impaired bladder function [13].
In summary, the present study shows that the contractile and urodynamic alterations as well as the enhanced PKC expression in bladder tissues seen in high-fat fed obese mice are normalized by blockade of Cav1.2 and improvement of insulin sensitivity. Up-regulation of PKC in obese mice is likely to simultaneously mediate the insulin resistance and hence overactive bladder.
Funding Statement
Funding was provided by the State of São Paulo Research Foundation, Grant Number: FAPESP 2010/01452-4. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Grundy SM, Brewer HB Jr, Cleeman JI, Smith SC Jr, Lenfant C (2004) American Heart Association; National Heart, Lung, and Blood Institute. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 109: 433–438. [DOI] [PubMed] [Google Scholar]
- 2. Opie LH (2007) Metabolic syndrome. Circulation 115: 32–35. [DOI] [PubMed] [Google Scholar]
- 3. Kahn BB, Flier JS (2000) Obesity and insulin resistance. J Clin Invest 106: 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carr DB, Utzschneider KM, Hull RL, Kodama K, Retzlaff BM, et al. (2004) Intra-abdominal fat is a major determinant of the national Cholesterol Education Program Adult Treatment Panel III Criteria for the Metabolic Syndrome. Diabetes 53: 2087–2094. [DOI] [PubMed] [Google Scholar]
- 5. Barton M, Baretella O, Meyer MR (2012) Obesity and Risk of Vascular Disease: Importance of Endothelium-Derived Vasoconstriction. Br J Pharmacol 165: 591–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moul S, McVary KT (2010) Lower urinary tract symptoms, obesity and the metabolic syndrome. Curr Opin Urol 20: 7–12. [DOI] [PubMed] [Google Scholar]
- 7. Kirby MG, Wagg A, Cardozo L, Chapple C, Castro-Diaz D, et al. (2010) Overactive bladder: Is there a link to the metabolic syndrome in men? Neurourol Urodyn 29: 1360–1364. [DOI] [PubMed] [Google Scholar]
- 8. Laven BA, Orsini N, Andersson SO, Johansson JE, Gerber GS, et al. (2008) Birth weight, abdominal obesity and the risk of lower urinary tract symptoms in a population based study of Swedish men. J Urol 179: 1891–1895. [DOI] [PubMed] [Google Scholar]
- 9. Richter HE, Kenton K, Huang L, Nygaard I, Kraus S, et al. (2010) The impact of obesity on urinary incontinence symptoms, severity, urodynamic characteristics and quality of life. J Urol 183: 622–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rohrmann S, Smit E, Giovannucci E, Platz EA (2005) Association between markers of the metabolic syndrome and lower urinary tract symptoms in the Third National Health and Nutritional Examination Survey (NHANES III). Int J Obes (Lond) 29: 310–316. [DOI] [PubMed] [Google Scholar]
- 11. Tong YC, Cheng JT (2007) Alterations of M2,3-muscarinic receptor protein and mRNA expression in the bladder of the fructose fed obese rat. J Urol 178: 1537–1542. [DOI] [PubMed] [Google Scholar]
- 12. Lee W, Chien C, Yu H, Lee S (2008) Bladder dysfunction in rats with metabolic syndrome induced by long-term fructose feeding. J Urol 179: 2470–2476. [DOI] [PubMed] [Google Scholar]
- 13. Gasbarro G, Lin DL, Vurbic D, Quisno A, Kinley B, et al. (2010) Voiding function in obese and type 2 diabetic female rats. Am J Physiol Renal Physiol 298: F72–77. [DOI] [PubMed] [Google Scholar]
- 14. Galgani J, Ravussin E (2008) Energy metabolism, fuel selection and body weight regulation. Int J Obes (Lond) 32: S109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rahman NU, Phonsombat S, Bochinski D, Carrion ED, Nunes L, et al. (2007) An animal model to study lower urinary tract and erectile dysfunction: the hyperlipidaemic rat. BJU Int 100: 658–663. [DOI] [PubMed] [Google Scholar]
- 16. Turban S, Hajduch E (2011) Protein kinase C isoforms: mediators of reactive lipid metabolites in the development of insulin resistance. FEBS Lett 585: 269–274. [DOI] [PubMed] [Google Scholar]
- 17. Yu CL, Chen Y, Cline GW, Zhang DY, Zong HH, et al. (2002) Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 277: 50230–50236. [DOI] [PubMed] [Google Scholar]
- 18. Chibalin AV, Leng Y, Vieira E, Krook A, Bjornholm M, et al. (2008) Downregulation of diacylglycerol kinase delta contributes to hyperglycemiainduced insulin resistance. Cell 132: 375–386. [DOI] [PubMed] [Google Scholar]
- 19. Itani SI, Ruderman NB, Schmieder F, Boden G (2002) Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and I kappa B-alpha. Diabetes 51: 2005–2011. [DOI] [PubMed] [Google Scholar]
- 20.Huster M, Frei E, Hofmann F, Wegener JW (2010) A complex of Ca(V)1.2/PKC is involved in muscarinic signaling in smooth muscle. FASEB J 24, 2651–2659. [DOI] [PubMed]
- 21. Leiria L, Mónica FZ, Carvalho F, Claudino MA, Franco-Penteado C, et al. (2011) Functional, morphological and molecular characterization of bladder dysfunction in streptozotocin-induced diabetic mice: Evidence of a role for L-type voltage-operated Ca2+ channels. Br J Pharmacol 163: 1276–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, et al. (2007) Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 56: 1986–1998. [DOI] [PubMed] [Google Scholar]
- 23. Calixto MC, Lintomen L, Schenka A, Saad MJ, Zanesco A, et al. (2010) Obesity enhances eosinophilic inflammation in a murine model of allergic asthma. Br J Pharmacol 159: 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Oliveira CF, Nathan LP, Metze K, Moreno H Jr, De Luca IM, et al. (1999) Effect of Ca2+ channel blockers on arterial hypertension and heart ischaemic lesions induced by chronic blockade of nitric oxide in the rat. Eur J Pharmacol 373: 195–200. [DOI] [PubMed] [Google Scholar]
- 25. Shore SA, Williams ES, Zhu M (2008) No effect of metformin on the innate airway hyperresponsiveness and increased responses to ozone observed in obese mice. J Appl Physiol 105: 1127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lagaud GJ, Randriamboavonjv V, Roul G, Stoclet JC, Andriantsitohaina R (1999) Mechanism of Ca2+ release and entry during contraction elicited by norepinephrine in rat resistance arteries. Am J Physiol 276: H300–308. [DOI] [PubMed] [Google Scholar]
- 27. Anhê GF, Hirabara SM, Turrer TC, Caperuto LC, Anhê FF, et al. (2007) Postpartum glycemic homeostasis in early lactating rats is accompanied by transient and specific increase of soleus insulin response through IRS2/AKT pathway. Am J Physiol Regul Integr Comp Physiol 292: R2225–2233. [DOI] [PubMed] [Google Scholar]
- 28. Hanley AJG, Wagenknecht LE, D’Agostino RB Jr, Zinman B, Haffner SM (2003) Identification of Subjects with Insulin Resistance and β-Cell Dysfunction Using Alternative Definitions of the Metabolic Syndrome. Diabetes 52: 2740–2747. [DOI] [PubMed] [Google Scholar]
- 29. Richter HE, Burgio KL, Brubaker L, Moalli PA, Markland AD, et al. (2005) Urinary Incontinence Treatment Network. Factors associated with incontinence frequency in a surgical cohort of stress incontinent women. Am J Obstet Gynecol 193: 2088–2093. [DOI] [PubMed] [Google Scholar]
- 30. Daneshgari F, Liu G, Birder L, Hanna-Mitchell AT, Chacko S (2009) Diabetic bladder dysfunction: current translational knowledge. J Urol 182: S18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parsons JK, Sarma AV, McVary K, Wei JT (2009) Obesity and benign prostatic hyperplasia: clinical connections, emerging etiological paradigms and future directions. J Urol 182: S27–S31. [DOI] [PubMed] [Google Scholar]
- 32. Vikram A, Jena G, Ramarao P (2010) Pioglitazone attenuates prostatic enlargement in diet-induced insulin-resistant rats by altering lipid distribution and hyperinsulinaemia. Br J Pharmacol 161: 1708–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Michel MC, Barendrecht MM (2008) Physiological and pathological regulation of the autonomic control of urinary bladder contractility. Pharmacol Ther 117: 297–312. [DOI] [PubMed] [Google Scholar]
- 34. Abrams P, Andersson KE, Buccafusco JJ, Chapple C, de Groat WC, et al. (2006) Muscarinic receptors: their distribution and function in body systems, and the implications for treating overactive bladder. Br J Pharmacol 148: 565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ford AP, Cockayne DA (2011) ATP and P2X purinoceptors in urinary tract disorders. Handb Exp Pharmacol 202: 485–526. [DOI] [PubMed] [Google Scholar]
- 36. Pinna C, Sanvito P, Puglisi L (2006) Altered neurogenic and mechanical responses to acetylcholine, ATP and substance P in detrusor from rat with outlet obstruction. Life Sci 79: 1301–1306. [DOI] [PubMed] [Google Scholar]
- 37. Kennedy C, Tasker PN, Gallacher G, Westfall TD (2007) Identification of atropine and P2X1 receptor antagonist-resistant, neurogenic contractions of the urinary bladder. J Neurosci 27: 845–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Uchida W, Masuda N, Shirai Y, Shibasaki K, Satoh N, et al. (1994) The role of extracellular Ca2+ in carbachol-induced tonic contraction of the pig detrusor smooth muscle. Naunyn Schmiedebergs Arch Pharmacol 350: 398–402. [DOI] [PubMed] [Google Scholar]
- 39. Masters JG, Neal DE, Gillespie JI (1999) The contribution of intracellular Ca2+ release to contraction in human bladder smooth muscle. Br J Pharmacol 127: 996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wegener JW, Schulla V, Lee TS, Koller A, Feil S, et al. (2004) An essential role of Cav1.2 L-type calcium channel for urinary bladder function. FASEB J 18: 1159–1161. [DOI] [PubMed] [Google Scholar]
- 41. Wuest M, Hiller N, Braeter M, Hakenberg OW, Wirth MP, et al. (2007) Contribution of Ca2+ influx to carbachol-induced detrusor contraction is different in human urinary bladder compared to pig and mouse. Eur J Pharmacol 565: 180–189. [DOI] [PubMed] [Google Scholar]
- 42. Frazier EP, Peters SLM, Braverman AS, Ruggieri MR, Michel MC (2008) Signal transduction underlying the control of urinary bladder smooth muscle tone by muscarinic receptors and β-adrenoceptors. Naunyn-Schmiedeberg’s Arch Pharmacol 377: 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Andersson KE, Arner A (2004) Urinary bladder contraction and relaxation: physiology and pathophysiology. Physiol Rev 84: 935–986. [DOI] [PubMed] [Google Scholar]
- 44. Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K, et al. (2006) Activation of vascular protein kinase C-β inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes 55: 691–698. [DOI] [PubMed] [Google Scholar]
- 45. Nobe K, Yamazaki T, Tsumita N, Hashimoto T, Honda K (2009) Glucose-dependent enhancement of diabetic bladder contraction is associated with a rho kinase-regulated protein kinase C pathway. J Pharmacol Exp Ther 328: 940–50. [DOI] [PubMed] [Google Scholar]
- 46. Lenasi H, Kohlstedt K, Fichtlscherer B, Mülsch A, Busse R, et al. (2003) Amlodipine activates the endothelial nitric oxide synthase by altering phosphorylation on Ser1177 and Thr495. Cardiovasc Res 59: 844–853. [DOI] [PubMed] [Google Scholar]
- 47. Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, et al. (2004) PKC-θ knockout mice are protected from fat-induced insulin resistance. J Clin Invest 114: 823–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stretton C, Evans A, Hundal HS (2010) Cellular depletion of atypical PKC{lambda} is associated with enhanced insulin sensitivity and glucose uptake in L6 rat skeletal muscle cells. Am J Physiol Endocrinol Metab. 299: E402–412. [DOI] [PubMed] [Google Scholar]
- 49. Lu X, Bean JS, Kassab GS, Rekhter MD (2011) Protein kinase C inhibition ameliorates functional endothelial insulin resistance and vascular smooth muscle cell hypersensitivity to insulin in diabetic hypertensive rats. Cardiovasc Diabetol 10: 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee M, Aronne LJ (2007) Weight management for type 2 diabetes mellitus: global cardiovascular risk reduction. Am J Cardiol 99: 68B–79B. [DOI] [PubMed] [Google Scholar]
- 51. Sena CM, Matafome P, Louro T, Nunes E, Fernandes R, et al. (2011) Metformin restores endothelial function in aorta of diabetic rats. Br J Pharmacol 163: 424–437. [DOI] [PMC free article] [PubMed] [Google Scholar]