

Abstract
Colorectal cancers are mostly sporadic; some cases of familial clustering and autosomal dominant conditions are also known to occur. Juvenile polyposis syndrome (JPS) is an autosomal dominant condition caused by the mutation of the SMAD4 or the BMPR1A genes. JPS is characterized by hamartomatous polyps developing in the upper and lower intestine. Contradicting previous studies, many of these polyps can go through malignant transformation.
This paper reports the case of a male patient who was continuously treated for juvenile polyposis. During the eighteen years of treatment, more than hundred polyps were endoscopically removed from his gastrointestinal tract. The patient’s care was interrupted for eight years due to insufficient compliance. He was subsequently referred to our Department of Gastroenterology in severe clinical condition caused by metastatic colorectal cancer. He died after a short palliative therapy at the age of 31. His first-degree accessible relatives were further examined for juvenile polyposis syndrome. Several gastrointestinal polyps of different histological origin were observed in the deceased patient’s brother, who subsequently had to undergo a left lateral hemicolectomy. Genetic analyses revealed mutations of the BMPR1A gene in the clinically affected brother, the brother’s daughter, and in the deceased proband’s daughter.
Indebt genetic analyses helped customize and deliver care to a very specific group of individuals. We were able to identify potential family members on whom preventive care and treatment could be focused and simultaneously prevented unnecessary clinical and invasive procedures on those who were healthy. Furthermore, these analyses helped prevent future unnecessary trauma or distress on the analyzed family.
Key Words: Juvenile polyposis syndrome (JPS), malignant transformation, BMPR1A gene mutation, mutation-carrier
Introduction
Colorectal cancer (CRC) is statistically known to be the fourth highest cause of cancer mortality in the world. Whereas most of these cancers are known to develop sporadically, there are some that are known to have genetic clustering. Genetic analyses of familial clustering with specific emphasis on the autosomal dominantly inherited colorectal cancers would facilitate care and treatment for CRC patients.
The Familial Adenomatous Polyposis Syndrome and its sub-types (Turcot-, Gardner-syndrome, AFAP, and lately MAP) (1) are the most illustrious kinds of polyposis syndromes in which malignancy develops from adenomatous polyps. Another well known syndrome is the Peutz-Jeghers syndrome which is characterized by multiple hamartomatous polyps scattered throughout the gastrointestinal tract. The hereditary nonpolyposis colorectal cancer (HNPCC) is another autosomal dominant familial syndrome that we have previously described in detail (2).
Juvenile polyposis syndrome (JPS) is a rare form of the hereditary polyposis syndrome. Recent studies on JPS have indicated a higher risk or an early malignant transformation by the age of 30-35 (3,4).
The first juvenile polyposis describing the histological analysis of a 30-month-old child’s rectal polyps was published in 1939 (5). The name ‘juvenile polyp’ was given by Horrilleno et al. in 1957 (6). The hamartomatous histological characteristic was suggested by Morson in 1962 as a differential marker to distinguish juvenile polyps from adenomatous polyps (7). In case of generalized JPS, the polyps were hamartomatous with adenomatous lesions reported in 20-30% of the cases and with dysplasia and malignancy in the more advanced stages. JPS had been considered as a benign disease until malignant transformation was reported in 1984 (8). Studies done in Britain have shown 17% gastrointestinal malignancy (3), and according to the American data, the cumulative risks of gastrointestinal and colorectal malignancy could be as high as 55% (4).
The presence of multiple juvenile polyps in the gastrointestinal tract was first reported in 1964 (9). Polyps are commonly known to occur in the large intestine and rectum, and may also appear in the stomach and small intestine (10).
Gastrointestinal bleeding, diarrhoea, protein losing enteropathy, and spontaneous autoamputation and elimination of polyps have been observed in JPS patients. In 20-50% of the cases, JPS is caused by germline mutations within the coding sequence of the TGFβ superfamily of genes, namely the SMAD4/DPC4 tumor suppressor gene on chromosome 18q21.1 (11,12) or the BMPR1A gene on chromosome 10q22-23 (13,14).
Typically, these mutations are present in 20% of JPS cases (14) and can be detected within epithelial cells but not in stromal cells. To diagnose JPS the following criteria are used: either the presence of 10 or more juvenile polyps in the colorectum or in other part of the gastrointestinal tract, or any number of juvenile polyps anywhere in the gastrointestinal tract accompanied with positive family anamnesis for JPS (15). Exceptions, however, may occur, like the example of a nine-year-old boy whose family anamnesis for JPS was negative. In spite of missing data, a role of a “de novo” germline mutation may be hypothesized in the boy’s case (16). Three subgroups of JPS have been differentiated on the basis of clinical features and pathogenesis: (I) childhood JP, (II) juvenile polyposis coli (the juvenile polyps are only present in the colon and the rectum), and (III) generalized JP (the polyps can be found anywhere between the stomach and rectum).
Case presentation
Pathogenesis of the proband
The proband was born in 1966. At the age of three, he was examined in a county hospital and had symptoms of diarrhoea, spontaneous polyp-elimination, and rectal bleeding. In the same hospital in 1970, he was diagnosed with ileus and treated with conservative, non-invasive methods that ended the symptoms, thus no further examinations followed. In 1971 he was again hospitalized due to ileus caused by intussusceptions; six hamartomatous polyps were removed from the colon by sigmoidoscopy. During the patient’s childhood, abdominal pain and spontaneous polyp-elimination were typical symptoms, nonetheless, serious bleeding and gravis anaemia were not observed and the patient’s general clinical status was good. During his teenage years, spontaneous polyp-amputations were observed together with rectal bleeding monthly or bimonthly; yet, the patient did not develop anaemia. During a continuous 18 years of care, 107 polyps were eliminated by endoscopy from the proband’s colon. Histological analyses showed hamartomatous lesions in each case without adenomatous elements (Figure 1). The accessible relatives were examined while researching the family anamnesis; however, no typical alteration was found. The inheritance of familial colorectal cancer could be excluded; the colonoscopy done on the proband’s elder, symptom-free brother was negative.
Figure 1.

Hamartomatous polyp from proband’s colon. A. Cystosus glands and regular colonic epithelial glands are visible (organotypic structure) (Hemtaoxilin & eosin (HE) staining). B. Higher magnification of figure 1A (HE staining; 80× magnificiation)
In 1985 several hamartomatous polyps with coloured cystic dilatation were removed from the proband’s colon by colonoscopy, whereas the results of gastroscopy and duodenoscopy were negative.
Although the proband was in a generally good condition, he was followed up yearly. In 1989, hamartomatouos polyps were found in the stomach and in the small intestine for the first time (Figure 2).
Figure 2.
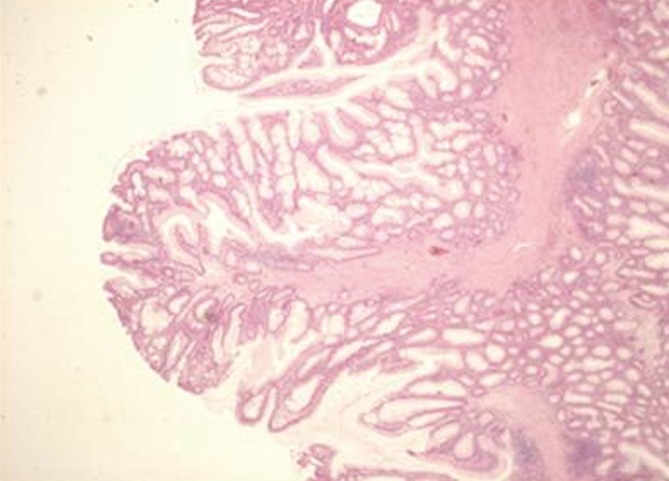
Juvenile polyp from proband’s small intestine. The submucosa is visible along the axis of the polyp. The epithelial glands are proliferating (HE staining)
The colonoscopy performed at that time revealed mucoid, glove shaped, and soft, mostly pedunculated polyps in the rectum and proximal sigmoid colon, whereas the other parts of the sigmoid and descending colon were polyp free. Furthermore, some polyps were also observed in the transversal colon. Two polypectomies were performed; one of them was a juvenile polyp and the other was characterized as adenomatous with second degree dysplasia (Figure 3). A year later, some polyps were detected in the rectosigmoid and even more in the stomach. Despite our yearly call, the proband did not come for controls between 1990 and 1997. The Hungarian political and economical changes as well as a significant growth in the proband’s and his family’s financials might have played a role in the insufficient compliance. The patient got married in 1993; his daughter was born in 1995. According to a follow up report, the proband did not show any signs or symptoms of the disease.
Figure 3.

A. Hyperplastic polyp with adenomatous transformation from proband’s colon. (framed region) (HE staining); B. Adenomatous glands, some of them next to a cystous gland from proband’s colon (HE staining; 80× magnificiation); C. Real neoplastic, tubular adenoma without evidence of malignancy from proband’s colon (HE staining; 80× magnificiation)
In April 1997 the proband checked into the local hospital with symptoms of grave anaemia and weight-loss. Gastroscopy revealed severe polyposis in the stomach, thus total gastrectomy was suggested by the local gastroenterologist. A month later, when the proband checked in our hospital, the total gastrectomy was rejected due to severely progressed polyposis which expanded to the duodenum. Four polypectomies were performed; histological analyses did not show malignancy. Colonoscopy was performed and revealed large polyps in the rectum which were suspected to be malignant. 15-20 polyps were removed; however, during the procedure arterial bleeding occurred that could not be controlled by coagulation. As a result, per rectum surgical intervention was needed; after the bleeding was under control, the colonoscopy was repeated and it revealed neoplastic tissue growth causing obstruction in the transversal colon. Staging tests, gastric ultrasonography and CT including the scan of the chest unveiled multiple hepatic metastases and a pulmonal metastasis in the right lobe. The patient was inoperable and was treated with palliative chemotherapy. In the same year, in 1997, he died at the age of 31 of a disease that was previously thought to be benign.
Pathogenesis of the proband’s brother
The proband’s elder brother (II.1., born in 1958) was first examined in 1971 when the proband was diagnosed with multiple polyposis. Endoscopy did not show any alteration in either the gastroduodenal or the colorectal tract. In January 1990 endoscopy detected several small polyps in the sigmoid colon as well as a larger (6 mm) polyp which was endoscopically removed; histological analysis confirmed it was hamartomatous. In the gastroduodenal tract, alterations were not detected. Despite our calls for regular, yearly controls, the brother did not check into our hospital until 1997 when he finally came in accompanying the seriously ill proband. The stomach ultrasonography and panendoscopy performed at that time showed no alterations in the elder brother, whereas during colonoscopy 6 small, villous polyps were removed from the rectosigmoid. Furthermore, the most part of an apple-sized, soft, villous polyp was removed from the descending colon in pieces. Due to the remaining tissue growth, repeated endoscopy and polypectomy were suggested. Histological analyses revealed hamartomatous lesions. Two months later a repeated colonoscopy was performed; due to the family anamnesis and the presence of an extremely large polyp in the border of the descending colon and the lienal flexure, subtotal colectomy was suggested (Figure 4).
Figure 4.

Adenoma from the colon of the proband’s brother. A. Neoplastic tubular adenoma with irregular, dysplastic glands (HE staining; 160× magnification) B. Adenoma with severe dysplasia. The structure of the glands corresponds to an early malignant state (HE staining; 160× magnification)
After the operation, regular endoscopic examinations were carried out in every two years. In 2001 a polyp was detected in the stomach and each time the patient was examined by endoscopy different numbers of polyps were detected in and removed from the remaining part of the colon. In March 2005, capsular endoscopy and colonoscopy were performed. Some adenomatous polyps were removed from the rectum. After treatment, the patient was free from lesions and symptoms. In 2006 the upper panendoscopy was negative, but six tiny polyps were eliminated from the rectum and five bigger polyps (the diameter of the largest one was 20 mm) were removed from the large intestine. Malignancy was not found based on histological data; at the same time, the amount of hamartomatous elements decreased whereas the adenomatous lesions and tubular architectures increased in the histological samples.
Family anamnesis
The family anamnesis of the two brothers is not typical. The patients’ father died at the age of 66, autopsy did not show any characteristics of JPS. The mother was thoroughly examined at the age of 42 years and was free of JPS; she is still alive and free of any symptoms. The proband’s paternal grandfather died of colon cancer at the age of 67 (his file is not accessible, autopsy was not performed). There are no data about the proband’s paternal grandmother. The proband’s daughter was first examined in 2006 after genetic analysis revealed positivity for JPS. She has been followed up every year since. Two children of the proband’s brother have also been examined in 2004. The proband’s niece was diagnosed with JPS and has been yearly followed up by endoscopy since 2006. Based on genetic analysis the nephew was negative for JPS.
Molecular diagnostic examination
The activity of the p53, c-MYC and H-ras genes was examined in blood samples derived from the proband, the proband’s daughter, his elder brother and one of his elder brother’s children. These results revealed increased activity of all three genes examined.
The increased expression of tumorsuppressor p53, c-MYC oncoprotein, and H-ras genes cannot be explained based on our current knowledge; even the latest publications can only hypothesize the possible causes of such aberrations (17,18). The activity of these genes was only elevated, however, in those people’s blood samples that carried a mutation in genes playing a role in the development of JPS (19,20).
James R. Howe and his colleagues examined the samples taken from the proband’s daughter, his brother, and the brother’s two children. The published genetic analyses revealed a mutation in the BMPR1A gene (21). Two substitutions were found in consecutive nucleotides of exon 7 (735-6 TG>AT) of the BMPR1A gene. Interestingly, each of these substitutions would change the corresponding amino acid into a stop codon. This genetic aberration has been diagnosed in the proband, in his daughter, in his elder brother, and in his brother’s daughter, but was not detected in the proband’s son (21).
Care
After receiving the genetic results, the risk-specific care of the proband’s family was planned. The results of the first surveillance are the following:
II.1. Proband’s brother (53 year-old man) - Multiple polyps in the colon. Subtotal colectomy cue to the presence of an extremely large polyp in the border of the descending colon and the lienal flexure.
II.2. Proband (49 year-old man) - The stomach is free of polyps. Two adenomatous polyps without dysplasia were removed during colonoscopy. Capsule endoscopy did not show alterations in the small intestine.
III.1. Proband’s niece (25 year-old single, childless woman) - The gastroscopy was negative; colonoscopy revealed two small, flat polyps which were hyperplastic based on the histologycal analysis.
III.2. Proband’s nephew (24 year-old single, childless man) - Oesophago-gastro-duodenoscopy and colonoscopy were performed, both with negative results.
III.3. Proband’s daughter (13 year-old girl) - Pathological alterations were not detected by endoscopy in the upper gastrointestinal tract. Five polyps were removed endoscopically and several pinhead-sized polyps were detected by total colonoscopy. The removed polyps were hamartomatous and typical for JPS.
The proband’s risk-specific family tree is shown in Figure 5.
Figure 5.
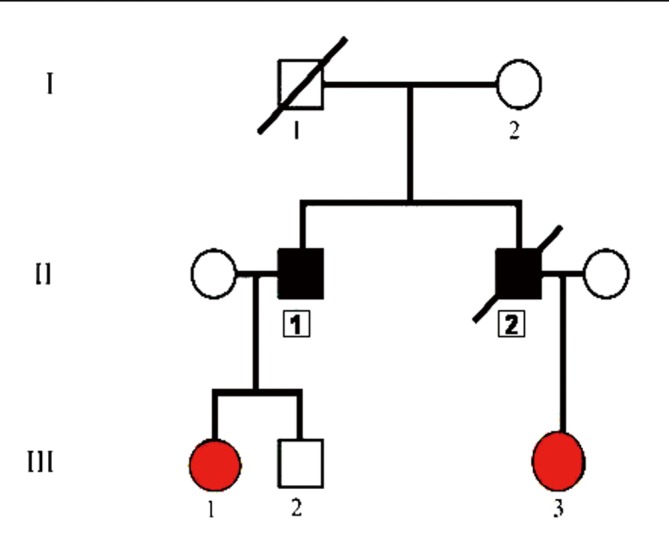
Proband’s family tree. Patient II/2 (proband) clearly has JPS. Mutation of the BMPRA1 gene was shown in patients III/1 (proband’s niece) and III/3 (proband’s daughter), therefore they need strict endoscopic surveillance. Mutation of the BMPRA1 gene was not detected in patient III/2, therefore his risk to develop carcinoma is the same as for average people
Discussion
In this study we have presented the case of a man who died of Juvenile Polyposis Syndrome. Several important clinical conclusions can be drawn from the case as well as many interesting questions have emerged.
Until recently, JPS was described as an illness caused by the inheritance of an autosomal dominant gene mutation. We have previously shown that two consecutive substitutions leaded to consecutive nonsense mutations in the BMPR1A gene in our proband’s family (21). The severity of disease in this family seemed more pronounced than in many others with germline BMPR1A mutations, as two affected individuals developed colon cancer, and one had gastric juvenile polyps, both of which are more common in JP patients with SMAD4 rather than BMPR1A mutations (22,23). Our case also contradicts previous studies in the sense that JPS is an autosomal dominant disorder, and inheritance of a single deleterious allele is sufficient to cause the JP phenotype (21).
Apart from their academic significance, genetic examinations are crucial in identifying children who are carriers but have no symptoms. In any case, a paradigm shift is indeed needed for autosomal dominantly inherited diseases such as JPS. Hamartomatous polyps are not benign lesions; thus, we need to provide more advanced carcinoma-prevention by endoscopy, invasive endoscopy, or surgical methods which may give these patients the opportunity for a better quality life with a longer life-span.
Acknowledgements
We thank Professor Istvan Ember (Medical University of Pecs, Hungary) for tumor marker analysis, Professor Istvan Racz (Petz Aladár Teaching Hospital of Győr, Hungary) for performing capsule endoscopy, and James R. Howe (University of Iowa Carver College of Medicine, Iowa City, IA, USA) for the genetic testing.
Disclosure: The authors declare no conflict of interest.
References
- 1.Sulová M, Zídková K, Kleibl Z, et al. Mutation analysis of the MYH gene in unrelated Czech APC mutation-negative polyposis patients. Eur J Cancer 2007;43:1617-21 [DOI] [PubMed] [Google Scholar]
- 2.Tam B SL. Polyposis syndromes and the herediter non-polyposis colorectal carcinoma, In: Tulassay Zs, eds. Prevention and treatment of colon cancer. Springer Tudományos Kiadó, 2004:87-93. [Google Scholar]
- 3.Coburn MC, Pricolo VE, DeLuca FG, et al. Malignant potential in intestinal juvenile polyposis syndromes. Ann Surg Oncol 1995;2:386-91 [DOI] [PubMed] [Google Scholar]
- 4.Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol 1998;5:751-6 [DOI] [PubMed] [Google Scholar]
- 5.Diamond M.Adenoma of the rectum in children: Report of a case in a thirty month old girl. Am J Dis Child 1939;57:360-7 [Google Scholar]
- 6.Horrilleno EG, Eckert C, Ackerman LV. Polyps of the rectum and colon in children. Cancer 1957;10:1210-20 [DOI] [PubMed] [Google Scholar]
- 7.Morson BC. Some prominent personalities in the history of St. Mark’s Hospital. Dis Colon Rectum 1962;5:173-83 [DOI] [PubMed] [Google Scholar]
- 8.Järvinen H, Franssila KO. Familial juvenile polyposis coli; increased risk of colorectal cancer. Gut 1984;25:792-800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mccoll I, Busxey HJ, Veale AM, et al. Juvenile polyposis coli. Proc R Soc Med 1964;57:896-7 [PubMed] [Google Scholar]
- 10.Howe J. Juvenile Polyposis Syndrome. In: Scriver CR, Sly WS, Valle D, et al. eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, 2001:805-818. [Google Scholar]
- 11.Howe JR, Ringold JC, Summers RW, et al. A gene for familial juvenile polyposis maps to chromosome 18q21.1. Am J Hum Genet 1998;62:1129-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998;280:1086-8 [DOI] [PubMed] [Google Scholar]
- 13.Howe JR, Bair JL, Sayed MG, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet 2001;28:184-7 [DOI] [PubMed] [Google Scholar]
- 14.Howe JR, Sayed MG, Ahmed AF, et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet 2004;41:484-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sachatello CR, Hahn IS, Carrington CB. Juvenile gastrointestinal polyposis in a female infant: report of a case and review of the literature of a recently recognized syndrome. Surgery 1974;75:107-14 [PubMed] [Google Scholar]
- 16.Teisseyre M, Plawski A, Podralska M, et al. Juvenile polyposis syndrome in a 9-year-old boy caused by new detected BMPR1A gene mutation. Gut 2007;56:A249 [Google Scholar]
- 17.Marsh DJ, Coulon V, Lunetta KL, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet 1998;7:507-15 [DOI] [PubMed] [Google Scholar]
- 18.Simon L. Risks of juvenile gastrointestinal polyposis. In Austro-Hungarian Gastroenterology Meeting. 1998: Vienna, Austria. [Google Scholar]
- 19.To MD, Perez-Losada J, Mao JH, et al. Crosstalk between Pten and Ras signaling pathways in tumor development. Cell Cycle 2005;4:1185-8. [DOI] [PubMed]
- 20.Dai MS, Jin Y, Gallegos JR, et al. Balance of Yin and Yang: ubiquitylation-mediated regulation of p53 and c-Myc. Neoplasia 2006;8:630-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howe JR, Chinnathambi S, Calva D, et al. A family with two consecutive nonsense mutations in BMPR1A causing juvenile polyposis. Cancer Genet Cytogenet 2008;181:52-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedl W, Uhlhaas S, Schulmann K, et al. Juvenile polyposis: massive gastric polyposis is more common in MADH4 mutation carriers than in BMPR1A mutation carriers. Hum Genet 2002;111:108-11 [DOI] [PubMed] [Google Scholar]
- 23.Sayed MG, Ahmed AF, Ringold JR, et al. Germline SMAD4 or BMPR1A mutations and phenotype of juvenile polyposis. Ann Surg Oncol 2002;9:901-6 [DOI] [PubMed] [Google Scholar]