

Abstract
Use of false cell lines remains a major problem in biological research. Short tandem repeat (STR) profiling represents the gold standard technique for cell line authentication. However, mismatch repair (MMR) deficient cell lines are characterized by microsatellite instability, which could force allelic drifts in combination with a selective outgrowth of otherwise persisting side lines, and thus, are likely to be misclassified by STR-profiling.
Based on the high-throughput Luminex platform, we developed a 24-plex SNP-profiling assay, called Multiplex Cell Authentication (MCA), for determining authentication of human cell lines. MCA was evaluated by analysing a collection of 436 human cell lines from the DSMZ, previously characterised by eight loci STR profiling. Both assays showed a very high degree of concordance and similar average matching probabilities (~1 × 10−8 for STR-profiling and ~1 × 10−9 for MCA). MCA enabled the detection of less than 3% contaminating human cells. Analysing MMR deficient cell lines, evidence was obtained for a higher robustness of the MCA compared to STR profiling.
In conclusion, MCA could complement routine cell line authentication and replace the standard authentication STR technique in case of MSI cell lines.
Keywords: Multiplex cell authentication (MCA), SNP, STR profiling, Luminex, cell line, cross-contamination, MMR deficiency
Introduction
Cell culture contaminations are a serious problem in biological research. In principle, cell lines can be contaminated by a wide range of agents including i) bacteria, ii) viruses and iii) other cell lines. While frequent bacterial, viral and inter-species contamination can be rapidly detected by, for example, a recently described Multiplex cell Contamination Test (McCT) 1, contaminations of, for instance, one human cell type by another human cell (intra-species contamination) are more difficult to be determined. If the cell lines are similar in morphology, this cross-contamination or a final replacement of the slower growing cell line may remain unnoticed. When occurring during or early after establishment of a new cell line, this may result in a world-wide spread of a falsely assigned cell line 2–6. Additionally, mislabelling of cells during routine culture and cryopreservation has added to this problem. The result is that 15–20% of cell lines in current use are estimated to not be what they are thought to be 7, 8.
Short tandem repeat (STR) profiling is widely used at Biological Resource Centers including DSMZ (The German Resource Centre for Biological Material), as well as the ATCC (The American Type Culture Collection), JCRB (Japanese Collection of Research Bioresources), and RIKEN (The Institute of Physical and Chemical Research) to verify the authentication of human cell lines. Standardized STR profiling of human cell lines is based on analysis of at least eight multi-locus tetranucleotide repeats by multiplex PCR and subsequent size separation of the products by capillary electrophoresis 9. The alleles are scored and compared to a database. STR typing functions also for the authentication of recently established cell lines, if concordant STR typing can be obtained from paired donor and derived cell line. However, widely used cell lines, for instance Jurkat, carrying various defects in the mismatch repair system leading to a loss of MutS or MutL function, have been shown to be able to change their STR profile upon long-term culturing leading to false authentication results 10. This indicated the need for additional methods for cell line authentication, such as SNP typing 11.
Based on the high-throughput medium-density array Luminex platform, we developed a 24-plex single nucleotide polymorphisms (SNP) profiling assay, called Multiplex Cell Authentication (MCA), for determining authentication of human cell lines. MCA was evaluated by analysing a collection of 436 human cell lines from the DSMZ, previously characterised by STR-profiling.
Methods
Cell lines
A total of 436 available human cell lines were obtained by the DSMZ (Braunschweig, Germany). All cell lines were tested for Mycoplasma and characterised by STR-profiling as indicated by the DSMZ online catalogue 12.
Cell line DNA extraction
Genomic DNA from 3–5 × 106 cells was purified using the High Pure PCR Template Preparation Kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions. One aliquot of each cell line DNA (15–30ng/μL) was transferred to the DKFZ for the MCA test. Cell line DNA was tested blindly by MCA.
STR-profiling
At the DSMZ, STR Profiling of the 8 STR loci was done as recently described 13. In brief, fluorescent PCR in combination with capillary electrophoresis enable a robust method designed to obtain a STR DNA profile. Using different colors, the PowerPlexR 1.2 system was modified in order to run a two-color DNA profiling allowing the simultaneous single-tube amplification of eight polymorphic STR loci and Amelogenin for gender determination. STR loci of CSF1PO, TPOX, TH01, vWA and Amelogenin were amplified by primers labelled with the Beckman/Coulter dye D3 (green), while the STR loci D16S539, D7S820, D13S317 and D5S818 were amplified using primers labelled with D2 (black). All of the loci except the Amelogenin gene in this set are true tetranucleotide repeats. To facilitate analysis of the data, the CEQ 8000 software enables an automatic assignment of genotypes and automatic export of resulting numeric allele codes into the reference DNA database of DSMZ. Cell lines are assigned as mislabelled if the obtained STR-profile changes exceed the 20% cutoff proposed by Masters et al. 14. The matching criteria of 80 % similarity is based on the algorithm as follows: ((generated peaks of cell line A) × 2) / (all peaks of cell line A and reference B) 14. The questioned STR profile should be always a generated profile of the suspicious cell line A, while the reference profile B of the parental counterparts are hosted by the major global cell banks ATCC (USA), DSMZ (Germany), JCRB (Japan), KCLB (Korea) and RIKEN (Japan).
MCA-SNP typing
MCA was performed by multiplex PCR and subsequent Luminex hybridization using similar conditions as described for other assays developed in our laboratory 1, 15–17. The genes, marker ID and minor allele frequency of the SNP included in MCA have been described recently 15. The 24 SNP were located on chromosomes 1, 2, 3, 4, 6, 7, 10, 11, 16, 17 and X (Table S1, supplementary Appendix). Briefly, a total of 15–30ng of cell line DNA or 1μL of crude cell lysates were used per PCR reaction. As one of the primers in a primer pair was 5′-biotinylated, amplimers with a size of 100 to 200bp were detected by hybridization to SNP-specific probes coupled to fluorescence-labelled polystyrene beads (Luminex Corp., Austin, TX) followed by labelling the complexes with Strep-PE reporter conjugate, washing and analysis in the Luminex 100. Results were expressed as the median fluorescence intensity (MFI) of at least 100 beads analyzed per set and per reaction. The assay was calibrated with a reference panel of 90 DNA samples from the European population cohort of the HAPMAP Project 18 (Figure 1).
Figure 1. Assignment of genotypes using the HAPMAP reference panel.
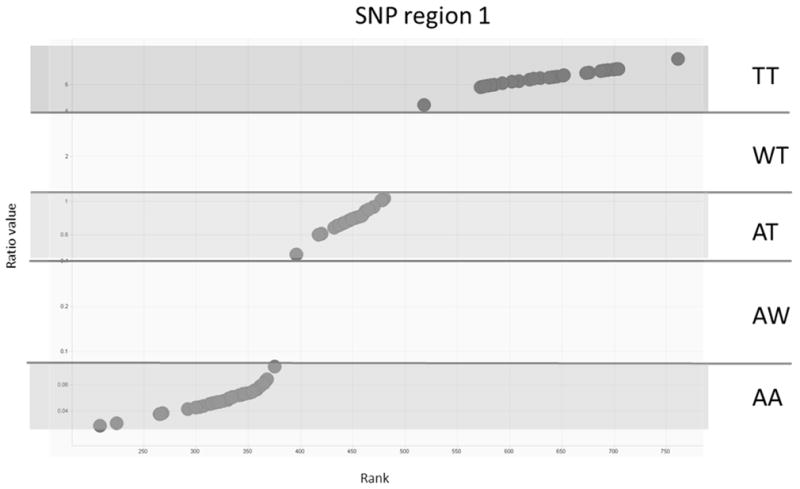
The ranked ratios of two allelic probes of one SNP region are shown for 90 HAPMAP samples. The three clusters indicate the genotypes: homozygous TT, heterozygous AT and homozygous AA.
Quality controls were used to monitor the assay performance. Negative PCR controls were run on all 96-well plates to confirm the absence of PCR contaminations. Positive controls (at least two cell line DNAs with known fingerprints and good DNA quality) were used on all runs to control both the amplification and hybridisation performance. For each positive control, the median MFI value of all SNP-specific probes was calculated. The criteria for repeating a complete run was a value of less than 100 median MFI for any of the positive controls. In addition, the same median MFI value was calculated for test samples to identify cell lines with inadequate DNA quality. Test samples with a value of less than 100 median MFI or more than two no-call positions were excluded from the analysis and repeated.
Statistics
For each SNP locus, genotypes were assigned by calculating the ratio of both MFI values of the allele-specific probes for each tested sample (see Table S2, supplementary Appendix) and comparing the ratio to ratios obtained by the HAPMAP reference panel (Figure 1). The genotyping data (genotyping code) of cell lines for all 24 SNP tested was generated by summing up all genotypes of the individual SNP. The alleles of each SNP were coded by A and T, so that genotypes of all SNPs were either AA, AT or TT. W was used, when the genotype could not be discriminated between one homozygous and the heterozygous group (ratio of the two signals for a SNP located between the two groups). Thus, the genotype is given as WT or AW, where W indicates the indeterminate allele (Figure 1). NN was used for no-call positions when amplification and detection of the respective SNP failed.
The genotyping code of a sample was defined by a 48-letter fingerprint sequence analysable by DNA sequence analysis programs. Based on pairwise comparison of all 436 cell lines we used a threshold (Percentage (%) genotype homology or number of matches and mismatches) to identify related or identical cell line pairs and groups. SNP genotyping and analyses for genotyping homology were performed blinded. Subsequent evaluations of the discriminatory ability of the assay and selection of unique cells (all different cells, excluding replicates or subclones) were performed unblinded by assigning the identification code of each specific cell provided by DSMZ.
In order to obtain the distribution of all pair-wise comparisons of the genotype codes of each unique cell line, we used ClustalW2 that generates a percentage score alignment for each of the comparisons 19. The match random probability (RMP) that two different cell lines will have the same multi-locus genotype was calculated as described 20 using our cell line panel as a reference population (assuming that our set of cells may represent a single population).
Results
Analysis of DSMZ cell line collection by MCA
Using MCA, all 24 loci tested could be typed simultaneously. The call rate in the 436 DSMZ cell lines was 99.3%. In summary, 77 out of 10,464 genotypes were not typed (NN). In 26 cases the same genotyping code was obtained for a pair of DSMZ cell lines. These pairs were correctly scored as identical because they were derived from the same donor or have been derivatives of a parental cell line. In addition to the cell line pairs described above, the same genotyping code was also found for 7 of 7 cell lines being HeLa or HeLa subclones, 5 of 5 cell lines being Namalwa or derivatives thereof, and 3 of 3 cell lines being U-937 or derivatives thereof.
The collection comprised additionally 5 monoclonal human choriocarcinoma-trophoblast hybrid cell lines (AC-1M88, AC-1M59, AC-1M81, AC-1M32 and AC-1M46) together with the original cell population ACH1P. These 6 cell lines showed distinct STR profiles. Using MCA, all lines clustered together with a genotype homology of at least 98% (only a single mismatch). However, they contained high numbers of Ws in 5 SNP loci (discrimination between one homozygous and the heterozygous allele status not possible) indicating a quantitative allele imbalance.
The other 393 cell lines could be discriminated from each other with a genotype homology ≤94%.
Probability of discrimination and pair-wise analysis
The average random match probability (RMP) using 24 SNPs was 2.6 × 10−09 (between 1.5 × 10−07 and 4.9 ×10−15), corresponding to a probability of discrimination of >99.9999997 %.
We performed pair-wise comparisons using 393 cell lines where known subclones were excluded by selecting parental profiles only. The average alignment score of the pair-wise comparisons between two different cell lines was 64.4% with a minimum of 34% and a maximum of 94% of alignment (Figure 2). These data suggested that a homology score of at least 96%, corresponding to 0–2 mismatches, is indicative for identical cell lines.
Figure 2. Frequency distribution of pairwise identity alignment scores from 24 SNPs genotyped on 393 unique paternal cell lines.
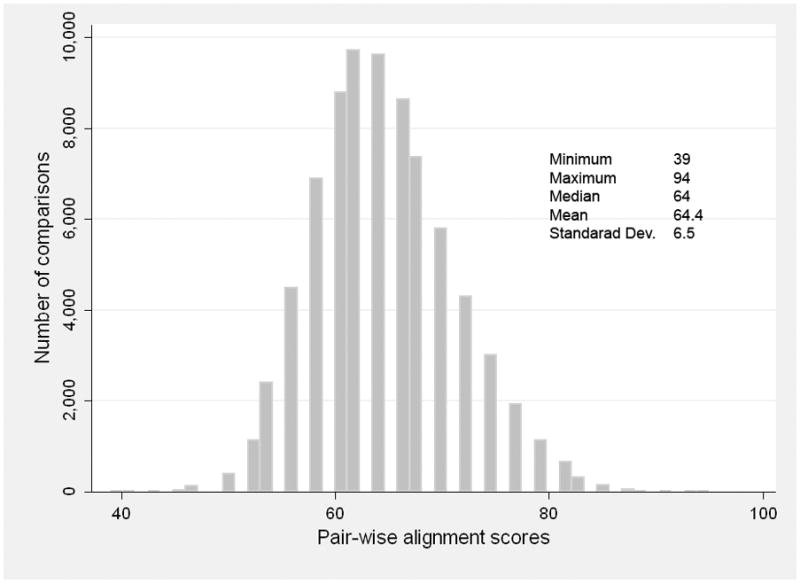
Total number of comparisons was 77028. Multiple alignment was performed using ClustalW2.
Reproducibility of MCA
Over a period of 4 weeks, a total number of 42 cell line DNA obtained from the DSMZ, were subjected twice to MCA. All replicates were genotyped with 100% homology (data not shown).
Detection of intra-human cross-contaminations
To mimic intra-human cell culture contaminations, we two-fold diluted purified DNA of distinct human cell lines (KYSE-520 and WSU-DLCL2, respectively) in a background of other human cell lines (CAPAN-1 and S-117, respectively) (Table 1). Both cell line pairs were randomly selected. Cell equivalents were computed based on the molecular weight of a diploid genome (6.6 pg per cell). The total number of contaminating and background cells (n~4000) was in accordance to the expected amount of cell equivalents per PCR reaction when following the DNA purification protocol. When both cell lines were present in equimolar quantities, a heterozygous allele status was observed for SNPs for which the original cell lines were homozygous AA and TT. In this study, we observed that cross-contaminations greater 3% contaminating cells showed at least 5 Ws or if less, multiple mismatches to the original cell line, representing a possibly useful cutoff for future studies.
Table 1.
Detection limit of intra-species cell line cross-contamination
| Cell line a
|
dilution factor | MCA fingerprint of 24 SNP regions | # of Ws | # of mismatches to background line | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| background (cell equivalents/PCR) | contaminating (cell equivalents/PCR) | ||||||||||||||||||||||||||||
| KYSE-520 | 0 | 4000 | TT | AA | TT | TT | AA | AA | AA | TT | AA | AA | AA | TT | AA | AA | AT | AA | AA | AA | AA | TT | AA | AA | AT | AA | |||
| CAPAN-1 | 4000 | 0 | TT | AA | TT | TT | TT | AA | AA | TT | TT | TT | AA | TT | TT | TT | AA | AA | TT | AT | AA | TT | AA | AA | TT | TT | |||
| Mix1 D1 | 2000 | 2000 | 0.500 | TT | AA | TT | TT | AT | AA | AA | TT | AT | AT | AA | TT | AT | AT | AW | AA | AT | AW | AA | TT | AA | AA | WT | WT | 4 | 6 |
| Mix1 D2 | 3000 | 1000 | 0.333 | TT | AA | WT | TT | W T | AA | AA | TT | WT | WT | AA | TT | WT | AT | AW | AA | WT | AW | AA | TT | AA | AA | WT | WT | 10 | 1 |
| Mix1 D3 | 3500 | 500 | 0.143 | TT | AA | TT | TT | W T | AA | AA | TT | WT | TT | AA | TT | TT | AT | AW | AA | WT | AT | AA | TT | AA | AA | WT | TT | 5 | 1 |
| Mix1 D4 | 3750 | 250 | 0.067 | TT | AA | TT | TT | W T | AA | AA | TT | WT | WT | AA | TT | TT | WT | AW | AA | WT | AT | AA | TT | AA | AA | TT | WT | 7 | 0 |
| Mix1 D5 | 3875 | 125 | 0.032 | TT | AA | TT | TT | TT | AA | AA | TT | WT | WT | AA | TT | TT | WT | AA | AA | WT | AT | AA | TT | AA | AA | TT | WT | 5 | 0 |
| Mix1 D6 | 3938 | 63 | 0.016 | TT | AA | TT | TT | TT | AA | AA | TT | WT | TT | AA | TT | TT | WT | AA | AA | TT | AT | AA | TT | AA | AA | TT | TT | 2 | 0 |
| Mix1 D7 | 3969 | 31 | 0.008 | TT | AA | TT | TT | TT | AA | AA | TT | TT | TT | AA | TT | TT | WT | AA | AA | TT | AT | AA | TT | AA | AA | TT | TT | 1 | 0 |
| Mix1 D8 | 3984 | 16 | 0.004 | TT | AA | TT | TT | TT | AA | AA | TT | TT | TT | AA | TT | TT | TT | AA | AA | TT | AT | AA | TT | AA | AA | TT | TT | 0 | 0 |
|
| |||||||||||||||||||||||||||||
| WSU-DLCL2 | 0 | 4000 | AT | AA | AT | TT | AT | AT | AA | AA | AA | AA | TT | AA | AT | AA | AA | AT | TT | AA | AA | AT | AA | AA | AA | AA | |||
| S-117 | 4000 | 0 | TT | AA | TT | TT | TT | AA | TT | TT | TT | TT | TT | AT | TT | AA | AA | AA | TT | AA | AA | TT | AA | AA | TT | AA | |||
| Mix2 D1 | 2000 | 2000 | 0.500 | WT | AA | WT | TT | WT | AT | AT | AT | AT | AT | TT | AW | WT | AA | AA | AW | TT | AA | AA | WT | AA | AA | AT | AA | 7 | 6 |
| Mix2 D2 | 3000 | 1000 | 0.333 | WT | AA | WT | TT | WT | AW | WT | AT | AT | AT | TT | AT | TT | AA | AW | AW | TT | AA | AA | WT | AA | AA | WT | AA | 9 | 3 |
| Mix2 D3 | 3500 | 500 | 0.143 | WT | AA | WT | TT | WT | AW | WT | WT | WT | WT | TT | AT | TT | AW | AA | AW | TT | AA | AA | WT | AA | AA | WT | AA | 12 | 0 |
| Mix2 D4 | 3750 | 250 | 0.067 | WT | AA | WT | TT | WT | AA | WT | TT | WT | WT | TT | AT | TT | AA | AA | AW | TT | AA | AA | WT | AA | AA | TT | AA | 8 | 0 |
| Mix2 D5 | 3875 | 125 | 0.032 | TT | AA | TT | TT | TT | AA | WT | TT | WT | WT | TT | AT | TT | AA | AA | AW | TT | AA | AA | WT | AA | AA | TT | AA | 5 | 0 |
| Mix2 D6 | 3937.5 | 63 | 0.016 | TT | AA | WT | TT | TT | AA | WT | TT | WT | WT | TT | AT | TT | AA | AA | AA | TT | AA | AA | TT | AA | AA | TT | AA | 4 | 0 |
| Mix2 D7 | 3968.75 | 31 | 0.008 | TT | AA | TT | TT | TT | AA | TT | TT | TT | TT | TT | AT | TT | AA | AA | AA | WT | AA | AA | TT | AA | AA | TT | AA | 1 | 0 |
| Mix2 D8 | 3984.375 | 16 | 0.004 | TT | AA | TT | TT | TT | AA | TT | TT | TT | TT | TT | AT | TT | AA | AA | AA | TT | AA | AA | TT | AA | AA | TT | AA | 0 | 0 |
DNA was purified from cell lines and adjusted to ~25ng/μL corresponding to ~4000 cell equivalents per μL. Contaminating cell line DNA was serially diluted in a background cell line DNA.
Analysis of MMR deficient cell lines
Isolated subclones of the human HEK293T (n=13) with known epigenetically silenced PMS2, MLH3 and MLH1 expression causing a MMR failure were tested by STR-profiling and MCA. In addition, single cell-derived subclones were isolated from NALM-6 (n=13) and LOVO (n=20) with known deleterious mutations of MSH2 causing a deficiency of MSH2, MSH3 and MSH6 proteins, and MSH6-deficient DLD-1 (HCT-15) (n=13) were tested by both methods.
All of the 13 STR profiles of the HEK293T subclones differed in up to 5 of the 16 tested STR alleles (median = 3, range 1 to 5) from the parental culture (Table 2). Based on the 80% cutoff, one (8%) of the HEK293T passages was scored as not identical to HEK293T. All of the 13 STR profiles of the 13 NALM-6 subclones differed in up to 5 of the 16 tested STR alleles (median = 2, range 1 to 5) from the parental culture (data not shown). Based on the 80% cutoff, one (8%) of the NALM-6 subclones were scored as not identical to NALM-6. In 11 of the 20 STR profiles of the LOVO subclones up to 4 of the 16 tested STR alleles (median = 1, range 0 to 4) from the parental culture (data not shown). Based on the 80% cutoff, all subclones were scored as identical to LOVO. In one of the 13 STR profiles of the DLD-1 subclones one of the 16 tested STR alleles (median = 0, range 0 to 1) differed from the parental culture; all subclones were scored as identical to DLD-1 (data not shown).
Table 2.
Eight loci STR-profiling for HEK293T subclones
| Cell Line | D5 a | D5′ | D13 | D13′ | D7 | D7′ | D16 | D16′ | vWA | vWA′ | TH01 | TH01′ | TPOX | TPOX′ | CSF1 | CSF1′ | Amel | Amel′ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 293T pMC-G-PMS2 10 | 8 | 9 | 12 | 13, 14 | 11 | 11 | 9 | 13 | 16 | 19 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
| 293T pMC-G-MLH1 3 | 8 | 8 | 11 | 12, 14 | 11 | 11 | 9 | 13 | 16 | 18, 19 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
| 293T pMC-G-MLH1 2 | 8 | 9 | 11 | 12, 14 | 11 | 11 | 9 | 13 | 16 | 19, 20 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
| 293T MutLa (mCherry-MLH1 GFP-PMS2) | 8 | 9 | 12 | 12 | 11 | 11 | 9 | 13 | 16 | 19 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
| 293T MutLb 1 (mCherry-MLH1 GFP-PMS1) | 8 | 9 | 12 | 14 | 11 | 12 | 9 | 13 | 16 | 19, 20 | 7 | 9.3 | 11 | 11 | 11 | 12, 8 | X | X |
| 293T MutSy 8 (YFP-His-MSH4 CFP-Strep-MSH5) | 8 | 9 | 12 | 13, 14 | 9 | 11 | 9 | 14 | 16 | 19 | 7 | 9.3 | 11 | 11 | 11 | 11 | X | X |
| 293T MutLa8 (mCherry-MLH1 GFP-PMS2) | 8 | 9 | 12 | 14 | 11 | 11 | 9 | 13 | 16 | 19, 20 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
| 293T MutLb 1 (mCherry-MLH1 GFP-PMS1) | 8 | 9 | 12 | 14 | 11 | 12, 6 | 9 | 13 | 16 | 19, 20 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
| pMC-G-MLH3delta7 8 | 9 | 9 | 12 | 14 | 11 | 11 | 9 | 13 | 16 | 19, 20 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
| pMC-G-MLH3delta7 4 | 8 | 9 | 11 | 12, 14 | 11 | 11 | 9 | 10, 13 | 16 | 19 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
| 293T MutLa 1 (mCherry-MLH1 GFP-PMS2) | 8 | 9 | 12 | 14 | 11 | 11 | 5 | 9, 13 | 16 | 18, 19 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
| pMC-G-MLH3WT 8 | 8 | 9 | 12 | 14 | 11 | 11 | 9 | 13 | 16 | 19 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
| pMC-G-MLH3WT 12 | 8 | 9 | 12 | 14 | 11 | 11 | 8 | 9, 14 | 16 | 19 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
| Reference profile HEK293T | 8 | 9 | 12 | 14 | 11 | 12 | 9 | 13 | 16 | 19 | 7 | 9.3 | 11 | 11 | 11 | 12 | X | X |
For each STR locus, the detected numbers of tetranucleotide repeats is indicated for each allele. Differences found compared to the reference profile are highlighted.
In contrast, using MCA all HEK293T and LOVO subclones were genotyped with 100% homology. Twelve of 13 NALM-6 subclones were genotyped with 100% homology, while one subclone showed a homology of 96%. Twelve of 13 DLD-1 subclones were genotyped with 100% homology, while one subclone showed a homology of 98%. Based on the 96% cutoff, all HEK293T, NALM-6, LOVO and DLD-1 subclones were scored as identical to the reference lines (data not shown).
Discussion
Authentication of cell lines is considered a key criterion for the validity of biomedical research results and, consequently, is increasingly required before grants are awarded or manuscripts are accepted for publication by high ranking journals. To avoid the consequences of using cross-contaminated cell lines, scientist should (i) test regularly for common cell culture contaminations, such as Mycoplasma and inter-species contaminations (e.g. confirming that cells are human and not murine), and (ii) confirm the identity of the cell line of interest. (i) could be addressed by various singleplex assays or the recently described Multiplex cell Contamination Test (McCT) that identifies more than 30 most frequent cell culture contaminations in a single reaction 1.To fully confirm the identity of cell cultures (ii), STR profiling is regarded as the gold standard 21. However, as others have shown, STR profiling may come to its limits if long-term passages of cell lines with mutations of the MMR system are tested, e.g. JURKAT and CCRF-CEM 10. In these cell lines, STR profiling may lead to erroneous results when the numbers of tetranucleotides in the analysed STR loci are altered by a selective bottleneck, e. g. isolation of subclones or single clones upon transfection. To circumvent this limitation, the DSMZ performs additionally cytogenetic analyses and uses an alternative genotyping technique, which is not controlled by the MMR system22. The MCA technique, described in this report, may represent a promising alternative or complementary method for the authentication of cell lines. In contrast to STR-profiling that is affected by MMR defects, defective MMR did not negatively influence MCA results in the present study. Furthermore, hMutSα defects mostly compromise the single nucleotide mismatch repair system and, thus, could lead to altered SNP results. Surprisingly, all MutSα defective subclones derived from NALM-6, DLD-1 and LOVO were correctly identified by MCA highlighting the robustness of the assay.
MCA analyzed rapidly and efficiently the fingerprints of 24 SNPs in 436 authenticated human cell lines provided by the DSMZ in Braunschweig. The SNP fingerprints are incorporated into a database to allow an automated comparison of tested cell lines with all human cell lines present in the DSMZ collection. The MCA reference database is currently extended to other frequently used human cell lines. A confirmatory STR-profiling of the cell line material is a prerequisite for implementing new cell lines in the MCA database.
To simplify the data treatment, the following system was used, AA for one homozygous state, AT for the heterozygous and TT for the other homozygous state. AW and WT are used for intermediate results; NN is reserved for a missing SNP and to keep the “bar code” length for the alignment consistent. A simple database scan allows confirming the presence or absence of a cell line deposited already in the database. In addition, newly derived cell lines could be verified if a concordant result is obtained from paired donor and derived cell line samples.
Over the last years several SNP typing assays have been published for forensic applications 20, 23–26 or cell line authentication 11, 27. In addition, the Cancer Cell Line Project has published data on 63 SNP loci to more than 770 different cell lines28. Compared to the RMP of current forensic SNP typing techniques, which include larger numbers and strategically selected markers for all human populations 20, the RMP of MCA with 24 SNPs is weaker, however, is in the same range as the standard eight multi-locus STR profiling that exhibits a RMP of ~1 in 10−08. It is acknowledged by the ATCC Standards Development Organization (ATCC-SDO), that this RMP meets the requirements for cell line authentication 21, 29. In order to use MCA for cell line authentication, the ratio of the allele values obtained in a given SNP locus have to be compared to the ratio of reference alleles. Luminex-based SNP typing appears to be ideal for cell line authentication as it has the capacity to quantitatively analyse up to 50 SNP loci simultaneously.
For the detection of cross-contaminations by MCA, intermediate allele ratios located between the reference allele ratios must be identified. While DNA replication events may be the cause for a limited number of intermediate ratios in a clean cell line, accumulation of intermediate ratios was observed to be an indicator for cross-contaminated cell lines. MCA showed a good ability to resolve DNA mixtures which is a prerequisite for the detection of cross-contaminations in human cell lines. Using a newly proposed cutoff, 3% of contaminating cells could be detected which is well comparable to the sensitivity of STR profiling, where 1–10% of contaminating cells are detectable 9. Assuming that in a freshly cross-contaminated cell culture the growth rate of one cell lines exceeds the other; this would ultimately lead to either a replacement of the original cell line or the disposal of the contaminating cells. Accordingly, the sensitivity of MCA to detect cross-contaminations should be sufficient to detect ongoing cross-contaminations at an early state.
In conclusion, MCA appears to be a powerful tool in assessing cell line identity and intra-human cross-contaminations and would be valuable for specialized laboratories, such as core facilities at scientific institutions and cell depositories for routine authentication of cell lines. Given its robustness in genotyping even MMR deficient cell lines and its fully automated data management, MCA could replace or at least complement conventional cell line authentication techniques.
Supplementary Material
Novelty.
Short tandem repeat (STR) profiling represents the gold standard technique for cell line authentication. However, mismatch repair (MMR) deficient cell lines may be misclassified by STR-profiling. The novel 24-plex SNP-profiling assay, called Multiplex Cell Authentication, showed a higher robustness for analysing MMR deficient cell lines and could complement routine cell line authentication and replace the standard authentication STR technique in case of MSI cell lines.
Acknowledgments
We thank Saskia Ziegler, Wojciech Leszczynski, Julia Peukes, Katrin Gärtner and Heike Schley for experimental help. We thank Dr. Ruth Pfeiffer from The National Cancer Institute (NCI) of the United States for the statistical advice.
Footnotes
Author contributions
M.S. and M.P. initiated and coordinated the study. W.D. and S.F. provided cell line DNA specimens and STR-profiling data. M.S. and F.C. developed the MCA assay, performed experiments. M.S. wrote the first draft of manuscript. F.C. and M.S. performed data analyses. All authors contributed to the final manuscript.
References
- 1.Schmitt M, Pawlita M. High-throughput detection and multiplex identification of cell contaminations. Nucleic acids research. 2009;37:e119. doi: 10.1093/nar/gkp581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boonstra JJ, van der Velden AW, Beerens EC, van Marion R, Morita-Fujimura Y, Matsui Y, Nishihira T, Tselepis C, Hainaut P, Lowe AW, Beverloo BH, van Dekken H, et al. Mistaken identity of widely used esophageal adenocarcinoma cell line TE-7. Cancer Res. 2007;67:7996–8001. doi: 10.1158/0008-5472.CAN-07-2064. [DOI] [PubMed] [Google Scholar]
- 3.Boonstra JJ, van Marion R, Beer DG, Lin L, Chaves P, Ribeiro C, Pereira AD, Roque L, Darnton SJ, Altorki NK, Schrump DS, Klimstra DS, et al. Verification and Unmasking of Widely Used Human Esophageal Adenocarcinoma Cell Lines. Journal of the National Cancer Institute. doi: 10.1093/jnci/djp499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chatterjee R. Cell biology. Cases of mistaken identity Science. 2007;315:928–31. doi: 10.1126/science.315.5814.928. [DOI] [PubMed] [Google Scholar]
- 5.Drexler HG, Dirks WG, Matsuo Y, MacLeod RA. False leukemia-lymphoma cell lines: an update on over 500 cell lines. Leukemia. 2003;17:416–26. doi: 10.1038/sj.leu.2402799. [DOI] [PubMed] [Google Scholar]
- 6.van Helden PD, Wiid IJ, Albrecht CF, Theron E, Thornley AL, Hoal-van Helden EG. Cross-contamination of human esophageal squamous carcinoma cell lines detected byDNA fingerprint analysis. Cancer Res. 1988;48:5660–2. [PubMed] [Google Scholar]
- 7.Freshney RI. Database of misidentified cell lines. International journal of cancer. 126:302. doi: 10.1002/ijc.24998. [DOI] [PubMed] [Google Scholar]
- 8.Capes-Davis A, Theodosopoulos G, Atkin I, Drexler HG, Kohara A, MacLeod RA, Masters JR, Nakamura Y, Reid YA, Reddel RR, Freshney RI. Check your cultures! A list of cross-contaminated or misidentified cell lines. International journal of cancer. 2010;127:1–8. doi: 10.1002/ijc.25242. [DOI] [PubMed] [Google Scholar]
- 9.Lins AM, Micka KA, Sprecher CJ, Taylor JA, Bacher JW, Rabbach DR, Bever RA, Creacy SD, Schumm JW. Development and population study of an eight-locus short tandem repeat (STR) multiplex system. J Forensic Sci. 1998;43:1168–80. [PubMed] [Google Scholar]
- 10.Parson W, Kirchebner R, Muhlmann R, Renner K, Kofler A, Schmidt S, Kofler R. Cancer cell line identification by short tandem repeat profiling: power and limitations. Faseb J. 2005;19:434–6. doi: 10.1096/fj.04-3062fje. [DOI] [PubMed] [Google Scholar]
- 11.Demichelis F, Greulich H, Macoska JA, Beroukhim R, Sellers WR, Garraway L, Rubin MA. SNP panel identification assay (SPIA): a genetic-based assay for the identification of cell lines. Nucleic acids research. 2008;36:2446–56. doi: 10.1093/nar/gkn089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DSMZ. Catalogue of human and animal cell lines. 2012. [Google Scholar]
- 13.Dirks WG, Faehnrich S, Estella IA, Drexler HG. Short tandem repeat DNA typing provides an international reference standard for authentication of human cell lines. Altex. 2005;22:103–9. [PubMed] [Google Scholar]
- 14.Masters JR, Thomson JA, Daly-Burns B, Reid YA, Dirks WG, Packer P, Toji LH, Ohno T, Tanabe H, Arlett CF, Kelland LR, Harrison M, et al. Short tandem repeat profiling provides an international reference standard for human cell lines. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8012–7. doi: 10.1073/pnas.121616198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castro FA, Haimila K, Sareneva I, Schmitt M, Lorenzo J, Kunkel N, Kumar R, Forsti A, Kjellberg L, Hallmans G, Lehtinen M, Hemminki K, et al. Association of HLA-DRB1, interleukin-6 and cyclin D1 polymorphisms with cervical cancer in the Swedish population--a candidate gene approach. International journal of cancer. 2009;125:1851–8. doi: 10.1002/ijc.24529. [DOI] [PubMed] [Google Scholar]
- 16.Schmitt M, Bravo IG, Snijders PJ, Gissmann L, Pawlita M, Waterboer T. Bead-based multiplex genotyping of human papillomaviruses. Journal of clinical microbiology. 2006;44:504–12. doi: 10.1128/JCM.44.2.504-512.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmitt M, Dondog B, Waterboer T, Pawlita M. Homogeneous amplification of genital human alpha papillomaviruses by PCR using novel broad-spectrum GP5+ and GP6+ primers. Journal of clinical microbiology. 2008;46:1050–9. doi: 10.1128/JCM.02227-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The_International_HapMap_C. The International HapMap Project. Nature. 2003;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 19.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, et al. Clustal W and Clustal X version 2.0. Bioinformatics (Oxford, England) 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 20.Kidd KK, Pakstis AJ, Speed WC, Grigorenko EL, Kajuna SL, Karoma NJ, Kungulilo S, Kim JJ, Lu RB, Odunsi A, Okonofua F, Parnas J, et al. Developing a SNP panel for forensic identification of individuals. Forensic science international. 2006;164:20–32. doi: 10.1016/j.forsciint.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 21.Alston-Roberts C, Barallon R, Bauer SR, Butler J, Capes-Davis A, Dirks WG, Elmore E, Furtado M, Kerrigan L, Kline MC, Kohara A, Los GV, et al. Cell line misidentification: the beginning of the end. Nature reviews. 2010;10:441–8. doi: 10.1038/nrc2852. [DOI] [PubMed] [Google Scholar]
- 22.Dirks WG, Drexler HG. Authentication of scientific human cell lines: easy-to-use DNA fingerprinting. Methods in molecular biology (Clifton, NJ) 2005;290:35–50. doi: 10.1385/1-59259-838-2:035. [DOI] [PubMed] [Google Scholar]
- 23.Budowle B, van Daal A. Forensically relevant SNP classes. BioTechniques. 2008;44:603–8. 10. doi: 10.2144/000112806. [DOI] [PubMed] [Google Scholar]
- 24.Musgrave-Brown E, Ballard D, Balogh K, Bender K, Berger B, Bogus M, Borsting C, Brion M, Fondevila M, Harrison C, Oguzturun C, Parson W, et al. Forensic validation of the SNPforID 52-plex assay. Forensic science international Genetics. 2007;1:186–90. doi: 10.1016/j.fsigen.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Pakstis AJ, Speed WC, Fang R, Hyland FC, Furtado MR, Kidd JR, Kidd KK. SNPs for a universal individual identification panel. Human genetics. 2010;127:315–24. doi: 10.1007/s00439-009-0771-1. [DOI] [PubMed] [Google Scholar]
- 26.Phillips C, Fang R, Ballard D, Fondevila M, Harrison C, Hyland F, Musgrave-Brown E, Proff C, Ramos-Luis E, Sobrino B, Carracedo A, Furtado MR, et al. Evaluation of the Genplex SNP typing system and a 49plex forensic marker panel. Forensic science international Genetics. 2007;1:180–5. doi: 10.1016/j.fsigen.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Josephson R, Sykes G, Liu Y, Ording C, Xu W, Zeng X, Shin S, Loring J, Maitra A, Rao MS, Auerbach JM. A molecular scheme for improved characterization of human embryonic stem cell lines. BMC biology. 2006;4:28. doi: 10.1186/1741-7007-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trust W. Cancer Genome Project. 2012. [Google Scholar]
- 29.ANSI/ATCC. Authentication of Human Cell Lines: Standardization of STR Profiling. ASN-0002-2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.