

Abstract
Cancers and tissue stem cells (SCs) share similar molecular pathways for their self-renewal and differentiation. The race is on to identify unique pathways to specifically target the cancer, while sparing normal SCs. Here, we uncover the transcription factor Runx1/AML1, a known haematopoietic and leukaemia factor, albeit dispensable for normal adult SC homeostasis, as being important for some mouse and human epithelial cancers. We implicate Runx1 as a SC-intrinsic gene in mouse hair follicle and oral epithelia by genetic lineage tracing in adulthood. Runx1-expressing SCs, but not other cells that ectopically upregulate Runx1 by injury and inflammation, are at the skin tumour origin. Runx1 loss impairs tumour initiation and maintenance and the growth of oral, skin, and ovarian epithelial human cancer cells. Runx1 stimulates Stat3 signalling via direct transcriptional repression of SOCS3 and SOCS4 and this is essential for cancer cell growth. Thus, Runx1 is a broader epithelial SC and cancer factor than previously recognized, and qualifies as an attractive potential target for both prevention and therapy of several epithelial cancers.
Keywords: hair follicle, Jak/Stat pathway, Runx1/AML1, squamous cell carcinoma, stem cells
Introduction
Increasing evidence, including genetic lineage tracing, seems to support the theory that some cancers, such as skin basal and squamous cell carcinoma (SCC) (Youssef et al, 2010; Lapouge et al, 2011; White et al, 2011), intestinal cancer, adenocarcinoma, and leukaemia originate in tissue stem cells (SCs; Visvader, 2011). Alternatively, some cancers might arise through de-differentiation of cells that hijack the normal SC self-renewal pathways (Visvader, 2011). In either case, for cancer therapies to work we must identify factors that are essential for the survival and proliferation of cancer cells, yet dispensable for normal SCs. There could be selective pressure among cancer cells to preserve the genomic locus of factors essential for their own growth and survival, and thus these factors might not surface in conventional human genetic mutation screens. In fact, such factors could be difficult to identify without in depth knowledge about the genetic control of normal and cancer SCs, which are difficult to access directly in humans.
We recently identified Runx1 as a factor involved, but dispensable for mouse skin epithelial hair follicle SCs (HFSCs) proliferation in vivo (Osorio et al, 2008). In contrast, Runx1 was strictly required for mouse skin papilloma and SCC formation and for cultured keratinocyte proliferation (Hoi et al, 2010). This makes Runx1 an attractive potential target for cancer that would not affect normal tissue homeostasis. However, the significance of our mouse finding to human cancer biology remained unclear. Human skin SCC is the second most common cancer after skin basal cell carcinoma (BCC), with ∼0.8 million new cases per year in the United States alone, of which ∼8% are metastatic and cannot be treated with current methods (Alam and Ratner, 2001). Head and neck (oral) SCC are extremely invasive and currently uncurable, and have several regulatory pathways in common with skin SCC (Molinolo et al, 2009).
Runx1 is recognized as a human leukaemia and haematopoietic stem cell factor (Mangan and Speck, 2011), but its role in human SCC and potentially other epithelial cancers is poorly understood. Recently, Runx1 SNPs were associated with human colon and rectal cancer (Slattery et al, 2011) as well as with prostate cancer (Yeh et al, 2009; Huang et al, 2011), and Runx1 was found expressed in endometrial and ovarian cancer (Planagumà et al, 2006, 2011). Moreover, triple negative (−Her2, −ER, −PR) human breast tumour growth in culture depends partially on Runx1 expression (Wang et al, 2011).
This evidence began to suggest that the leukaemia factor Runx1 might be more broadly implicated in cancer than previously recognized. Here, we present a breadth of evidence that Runx1 is overexpressed in a significant fraction of human epithelial cancers, and is essential for the growth and survival of three of them: skin SCC, oral SCC, and in part for ovarian cancer. Our lineage tracing data in adult mice show for the first time by lineage tracing that Runx1 is expressed in adult HFSCs and in oral epithelium SCs. The oral epithelium SCs were previously poorly characterized. Runx1 appears essential for solid tumour initiation and maintenance by upstream stimulation of Stat3 signalling, a central cancer pathway implicated in several epithelial cancers and in leukaemia (Chan et al, 2004a, 2004b; Yu and Jove, 2004; Aziz et al, 2007; Chan et al, 2008; Kim et al, 2009; Li et al, 2011). Together with the notion that Runx1 is not critical for normal adult tissue function, our work places this gene as an important candidate for epithelial cancer targeting.
Results
Runx1 is overexpressed in various epithelial tumours
Previously, we found Runx1 expressed in the same pattern in normal mouse and human skin (Hoi et al, 2010). Runx1 has a dynamic expression in the hair follicle during its homeostatic cycle (hair cycle), which consists of the morphologically distinct stages of growth and proliferation (anagen), regression (catagen), and rest (telogen) (Supplementary Figure S1; Tumbar, 2012). During quiescence, Runx1 marks a subset of the hair follicle bulge and hair germ cells where the SCs reside (Osorio et al, 2008). Moreover, Runx1 protein is detected in the bulge during anagen when HFSCs self-renew (Osorio et al, 2008; Zhang et al, 2009). The embryonic precursors of HFSCs express Runx1 (Osorio et al, 2011). Lineage tracing to formally prove Runx1 expression in adult HFSCs has not yet been done, but loss of Runx1 at different stages of the hair cycle delays HFSC activation from quiescence (Osorio et al, 2008), and slows down bulge cell proliferation during anagen (Hoi et al, 2010). Finally, we found Runx1 expressed at high levels in mouse skin papilloma and SCC, and Runx1 deficiency impaired mouse skin tumorigenesis (Hoi et al, 2010).
To examine the expression pattern of Runx1 in human epithelial cancers, we first surveyed the Oncomine public database, which contains microarray data comparing primary tumours and normal tissue. We found Runx1 to be overexpressed in several types of cancers in 47 studies and underexpressed in only 5 out of 138 studies analysed. The data are summarized in Figure 1A and Supplementary Table S1. Notably, comparing tumour to normal tissue Runx1 was among the top 1% of all overexpressed genes in head and neck, and skin SCC, and was highly expressed in several other epithelial tumours such as oesophageal SCC, cervical carcinoma, and colon adenocarcinoma. Next, we verified Runx1 expression by western blot and immunofluorescence staining of a panel of human cancer cell lines from breast (MCF-7; MDA-MB-231), prostate (PC-3; DU-145), colon (HCT116; CCSC, Sikandar et al, 2010), ovary (OVCAR-3; SKOV3), head and neck (oral) SCC (SCC66, SCC74, SCC125; White et al, 2007), and skin SCC (SCC13; Rheinwald and Beckett, 1980, 1981), as well as a mouse skin SCC cell line (Pam212; Yuspa et al, 1980). All cancer cell lines tested expressed more Runx1 relative to normal mouse keratinocytes, while we detected close to no Runx1 expression in mouse fibroblast, which served as negative control (Figure 1B–D; Supplementary Figure S2A and B). Attesting to the specificity of our antibody, we detected no Runx1 signal by immunostaining of knockout (KO) mouse keratinocytes (not shown). The different cancer cell lines showed variable levels and some contained only a subfraction of Runx1-positive cells. Notably, all head and neck and skin SCC cell lines showed a consistently high level of Runx1 expression.
Figure 1.

Runx1 in human cancers. (A) Meta-analysis of microarray studies comparing Runx1 levels in tumour versus normal tissue. Increased Runx1 expression (fold-change) is marked red, whereas a decrease is blue. (B) Western blot of mouse keratinocytes and mouse and human SCC skin (13) and head and neck (125, 66, and 74) cells for Runx1 and actin. (C) Summary of Runx1 expression in cancer cell lines of various origins derived from western blot (B) and stainings (D). (D) Sample images of Runx1 staining in human tumour cell lines. (E) Sample panel of mouse and human skin tumours stained for Runx1 (red), Keratin 5, 8, 15, and Ki67 (green) and DNA (blue). Dashed line marks the interface of stroma (*) and tumour. A human HF at equal exposure is presented for comparison. Image with increased brightness of Runx1 illustrates that Runx1 is present in this HF.
Finally, we checked for Runx1 expression in human primary tumours of skin cylindroma, skin SCCs, and BCCs, as well as head and neck (oral) SCCs by immunofluorescence staining. We found Runx1 expressed in a subset of tumour cells including the proliferative edge, and these cells co-localized in part with Ki67 (a proliferation marker), K15 (an HFSC marker), or K8 (a malignancy marker; Casanova et al, 2004). The results are detailed in Table I, Figure 1E, and Supplementary Figure S2C.
Table 1. Runx1 expression in various human and mouse skin and oral cancer samples in vivo.
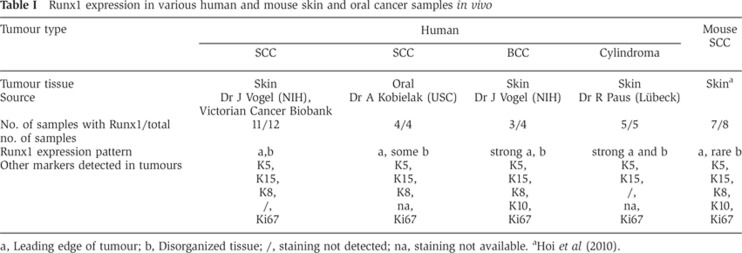
a, Leading edge of tumour; b, Disorganized tissue; /, staining not detected; na, staining not available.
The widespread expression of Runx1 in human epithelial cancers uncovered here, together with the strict requirement of Runx1 for the formation of mouse skin SCC (Hoi et al, 2010) warranted further examination of a potential role of Runx1 in human solid tumours and prompted us to delve deeper in the essentials of its mechanism. Specifically, we asked whether Runx1 expression is (a) required at the tumour initiation, promotion, or maintenance stage; (b) found in normal SCs and in cells of tumour origin; (c) ectopically raised by injury and inflammation in cells of tumour origin; (d) essential for human epithelial cancer cell growth and survival; (e) coupled with known signalling pathways of epithelial cancers.
Runx1 is required for tumour initiation but is dispensable for promotion
Previously, we showed that conditional deletion of Runx1 in mouse epidermis using a constitutively active K14-Cre allele drastically reduced papilloma and skin SCC formation (Hoi et al, 2010). If Runx1 is to be used as a cancer target, then it is important to determine the stage at which is acting in tumorigenesis (initiation, promotion, and maintenance). Thus, we deleted Runx1 by Tamoxifen (TM) injection in BL6 mice that carry the K14-CreERT2; Runx1fl/fl alleles at three different stages of tumorigenesis induced by a 2-step carcinogenesis protocol (Figures 2A and 5A). The KO targets the Runt domain in exon 4 rendering a null allele (Growney et al, 2005). Four days after TM injection, Runx1 KO was efficiently induced in over 90% of all HFs (Figure 2B). We applied 9,10-dimethyl-1,2-benzanthracene (DMBA) to induce point mutations in the DNA at first telogen (PD21) and 12-O-tetradecanoylphorbol-13-acetate (TPA) for the following 20 weeks to promote the mutation by robust proliferation of skin epithelial cells (Abel et al, 2009). As expected, after 20 weeks of TPA treatment, there were fewer tumours in the Runx1 KO mice TM treated at PD17 relative to both control groups (TM no CreER and oil only) (Figure 2C, top and middle; Figure 2D and G). We refer to this treatment as the initiation scheme (Figure 2A). Tumours that did form in the KO mice expressed Runx1 at high levels, likely from the rare cells that escaped Cre activity, developed later on and were ‘young’ in their development, as indicated by K10 expression (Figure 2C, middle; Supplementary Figure S2D; Santos et al, 1997). These data from inducible KO mice with a BL6 background were in line with our previous results obtained from mice carrying the K14-Cre allele, which were a mix of CD1 and BL6 genetic backgrounds, activated in embryogenesis (Hoi et al, 2010).
Figure 2.

Runx1 KO at tumour initiation or promotion impairs SCC formation. (A) Scheme of TM injection and DMBA/TPA treatments in mice to analyse Runx1 contribution to tumour initiation and promotion. Magnifying glass represents the end of monitoring mouse tumours. C, catagen, T, telogen, A, anagen. (B) PCR of genomic DNA from mice of indicated genotype to detect the Runx1 excision band (Δ4) and quantification as well as representative images of Runx1 staining in HFs from both schemes in (A). (C) Images of mice after 10, 15, and 20 weeks of TPA treatment in either scheme and staining of frozen section of corresponding tumours. Dashed line marks the interface of stroma (*) and tumour. (D) Tumour development of Runx1fl/fl; K14-CreER mice injected with TM (green) or oil (red) and of Runx1fl/fl injected with TM (orange) treated as in the initiation scheme in (A). P-values obtained by pairwise t-test (average±s.d.). (E) Same as (D) but for the promotion scheme in (A). A star represents a significant drop of tumour numbers after TPA cessation. (F) Tumour development in mixed FVB (average±s.d.). (G) Kaplan–Meier survival analysis for tumour onset in initiation (solid line, P-value=7.52 × 10−6) and promotion schemes (dashed lines, P-value=0.336) of BL6 mice. (H) Quantification of tumour numbers sorted by Runx1 expression (where ‘Present’ comprises ‘low’ and ‘high’ from I) in tumours derived from FVB/Bl6 mice. Ini, Initiation; Pro, Promotions; w, weeks. (I) Quantification of size, CD34, Ki67, and caspase 3 in tumours grouped by their Runx1 levels. (J) Staining of Runx1 and CD34 levels in FVB/BL6-derived promotion scheme tumours. Dashed line marks the interface of stroma (*) and tumour.
To differentiate between a role in tumour initiation and promotion, we injected TM 2.5–3 weeks after DMBA application when HF bulge cells divided at least once having passed through a full anagen (promotion scheme; Figure 2A and B). Surprisingly, Runx1 KO mice and control mice developed comparable numbers of tumours at a similar rate (Figure 2C bottom; Figure 2E and G), indicating that Runx1 is dispensable for tumour promotion or growth at least in this genetic background. However, 1 month after the final TPA treatment tumour counts dropped by 53% in the Runx1 KO mice as compared to 33% in WT mice (Figure 2E). Importantly, the only surviving tumours were those that still expressed Runx1 due to inefficient Cre activity (Figure 2C, bottom).
To check the robustness of our phenotypes in a uniform genetic background highly prone to skin cancer (Abel et al, 2009), we backcrossed our mice for five generations with Fvb background mice. As before, we found impaired tumorigenesis when Runx1 was deleted prior to tumour initiation (Figure 2F). This was true whether the Runx1 KO was induced 2 days before or 2 days after DMBA application either in telogen (PD18-20) or in anagen (PD21-23). When Runx1 was deleted 2.5 weeks after tumour initiation, 53% of tumours completely lacked Runx1 expression at week 7.5 of TPA treatment (Figure 2H and I). This was unlike our data from the initiation scheme, when no Runx1-negative tumours could be detected. This demonstrated that past the initiation stage tumours do not strictly require Runx1 for their growth under TPA treatment and was consistent with our data from the promotion scheme in the BL6 background. Suggestively, these Runx1-negative tumours were smaller in size (Figure 2I, bottom; Supplementary Figure S2E) and their number decreased steadily from 7.5 to 15 weeks of TPA treatment (Figure 2H), indicating that tumours are short lived in the absence of Runx1. Lower levels of Ki67 in these KO tumours support this loss, while apoptosis assayed by Caspase 3 staining does not seem to play a significant role (Figure 2I; Supplementary Figure S2F). Co-incidentally, the cancer SCs previously defined as a population of CD34-expressing papilloma cells (Malanchi et al, 2008), were greatly diminished in tumours that lacked Runx1 expression (Figure 2I and J; Supplementary Figure S2G). In general, cancer SCs are thought to maintain tumours and cause their re-occurrence for extended periods of time; CD34+ cancer SCs have been shown to initiate SCC formation upon serial transplantation for at least three generations (Malanchi et al, 2008). Our finding suggests that Runx1 expression is important for the maintenance of the CD34+ tumour SCs. Moreover, Runx1 is only essential for tumour growth in the absence of a proliferative agent such as TPA, which can at least temporarily overcome Runx1 loss.
Overall, our experiments clearly distinguish an irreplaceable function of Runx1 in tumour initiation from a somewhat more context-dependent function in tumour promotion.
Runx1-expressing cells are HFSCs
We previously reported that Runx1 loss impaired adult HF bulge-cell proliferation (Osorio et al, 2008; Hoi et al, 2010), but it remained unclear whether this effect is SC intrinsic or environmental, and if Runx1 is actually expressed in the adult long-term HFSCs, although we did find it expressed in the embryonic precursors of HFSC (Osorio et al, 2011). To address this question here, we used lineage tracing involving an inducible CreER recombinase previously knocked into the Runx1 genomic locus (Samokhvalov et al, 2007) and ROSA26R (Soriano, 1999) reporter mice. We injected TM at PD17 in these mice and sacrificed them at first telogen (PD21), first anagen (PD35), and third anagen (∼1-year old). In skin sections stained with X-Gal from the first telogen we observed labelling in ∼50% of HFs marking ∼5 cells per bulge (Figure 3A–C). Later on at the first and third anagen, we found prominent trails of X-Gal+ cells in the ORS (marked by K14) and the differentiated portion of the HF co-localizing with AE13 and AE15, as well as small clones of cells in the bulge (Figure 3B, top; Figure 3C–E). These X-Gal+ cells were completely absent in mice injected with oil only (Figure 3B, bottom). These data demonstrate for the first time by lineage tracing that original Runx1-expressing adult bulge cells are long-term self-renewing and differentiating HFSCs.
Figure 3.

Lineage tracing shows Runx1 expressed in HFSCs. (A) Schemes to trace the contribution of Runx1 bulge cells to HF homeostasis. C, catagen; T, telogen; A, anagen. (B) Representative images of the bulge lineage tracing (X-gal staining) 4 days after injection of oil or TM in Runx1-CreER xRosa26 mice sacrificed at Telogen and at first and third anagen. The inlay in third anagen TM image shows an additional high-magnification picture of an independent HF at that stage. (C) Quantification of bulge and germ (Telogen) and bulb/bulge (Anagen) labelling (average±s.d.). (D) Quantification of lineages labelled after long-term chase in Telogen and first and third anagen (average±s.d.). (E) Staining of traced HFs at third anagen for the differentiation markers AE13 and AE15, and the outer root sheath marker K14. Bottom right highlights the X-gal staining of multiple layers in a cross-sectioned HF.
Runx1-expressing HFSCs are at the origin of skin tumours
Since Runx1 is expressed in adult HFSCs, which can generate skin SCC, and Runx1 is important for tumour initiation it follows that it might be expressed in cells of tumour origin. However, epithelial skin tumours can originate in both HF and epidermis (Youssef et al, 2010; Lapouge et al, 2011; White et al, 2011). Our data so far suggest that tumour cells might hijack the self-renewal/proliferation pathways of normal HFSCs to promote their growth. Since Runx1 expression is confined to the HF in normal skin, it was intriguing to find that all skin tumours in our initiation scheme expressed high levels of Runx1 (Hoi et al, 2010). It is possible that some, but not all skin epithelial tumours generated in our protocol originated in HFSCs that normally expressed Runx1 and the remaining cells of tumour origin acquired ectopic Runx1 expression near the point of tumour initiation due to inflammation or injury induced by the treatment. We aimed to understand when Runx1 expression is aberrantly acquired in tumours and to what extent Runx1+ cells contribute to the cells of tumour origin. This would provide the context in which tumours are initiated and promoted.
The strategy was to induce genetic lineage tracing via TM injections in the Runx1-CreER; ROSA26R mice at different stages of tumorigenesis. We injected TM at PD17/18 in Runx1-CreER; ROSA26R mice and initiated the tumours with DMBA at PD21 (Figure 4A, Bulge scheme). This resulted in an average of 4.45 X-Gal+ cells/bulge in 80% of the HFs and 20% infundibular (Figure 4B and C, Bulge scheme) but no interfollicular epidermis labelling. After 20 weeks of TPA treatment, whole-mount X-Gal staining of the mouse back skin revealed 3/89 positive tumours in the TM-injected animals, as opposed to 0/71 in the oil controls (Supplementary Table S2). The positive tumours were completely blue suggesting monoclonal expansion, and expressed Runx1 (Figure 4D, Bulge scheme; Supplementary Figure S3A). Of important note only ∼2.3% of all bulge cells were X-Gal labelled upon TM induction due to low efficiency of the Runx1-CreER driven recombination. Using previously engineered Runx1-LacZ knock-in (KI) mice that report endogenous Runx1 promoter activity and expression (North et al, 1999) we estimate a conservative 50% of all bulge cells as normally being able to express Runx1 (Supplementary Figure S3B). These should have been labelled by the Runx1-CreER induction and we can thus infer that a significant fraction (∼70% if not more) of the skin tumours in this procedure may arise from Runx1+ bulge cells. The incidental labelling in 20% of infundibuli make these cells as potential alternative candidates for the origin of at least some of the tumours obtained in our treatment.
Figure 4.

Runx1 expressing HFSCs are the origin of SCCs. (A) Schemes of genetic lineage tracing in Runx1-CreER;Rosa26 mice to label and track bulge versus infundibular populations of Runx1+ cells contribution to tumorigenesis. Quantification of bulge (B) and infundibulum (C) labelling in mice treated as shown in each of the two schemes in (A) (average±s.d.; n>300HF). (D) Whole mount (left panels) and skin sections of X-Gal-positive and -negative tumours from the schemes in (A). Counterstain is haematoxylin. (E, F) Runx1-LacZ KI mice were subjected to a penetrating skin cut (A, red arrow) or TPA treatment (B, yellow arrows), sacrificed 4 days later, and skin sections analysed by X-Gal staining. Representative images show increased Runx1 expression in the HF infundibulum upon either physical or oncogenic assault. In E and H an asterisk (*) indicate a normal skin area, a hash (#) the wound area. (G, H) Runx1-CreER;Rosa26LacZ mice were subjected to TPA treatment (yellow arrows), followed by TM injection (syringe) to label the infundibulum. Bottom images show X-Gal staining of skin sections from mice after TM injection (G) or after TM injection and injury (H) at times indicated. Note lack of contribution from the infundibulum cells to either normal tissue homeostasis or injury repair.
Tissue injury and inflammation has long been recognized as an oncogenic factor. Intriguingly, we found that injury or short-term TPA treatment induced ectopic Runx1 expression in the hair infundibulum and epidermis just adjacent to the HF (Figure 4E and F; Supplementary Figure S13C). Micro-injury may be responsible for the 20% infundibular labelling present in our previous experiment. We asked whether the infundibular cells rendered to express Runx1 upon TPA induction or injury are SCs and may contribute to tissue homeostasis, injury repair, or tumour formation. Injection of TM in Runx1-CreER; ROSA26R mice at the onset of the second telogen (PD38-45) following ∼2.5 weeks of TPA treatment (Figure 4A, Infundibular scheme) resulted in the selective labelling of the majority of infundibular cells in each HF (Figure 4B, C, Infundibular scheme) but no labelling in the bulge or the interfollicular epidermis (n∼1000). Two weeks after TM injection, we sacrificed mice that had been injured by a penetrating cut and examined skin sections by X-gal staining. We determined that this population contributed neither to HF homeostasis (away from cut) nor to healing of the epidermal wound (Figure 4G and H). Therefore, we conclude that cells ectopically expressing Runx1 in the infundibulum do not acquire tissue SC properties.
We next asked if aberrant expression of Runx1 in these more differentiated HF cells had an oncogenic effect in the experimental conditions of our 2-stage skin carcinogenesis protocol. DMBA treatment 4 days after or 2.5–3 weeks before labelling by TM injection, followed by 20 weeks of TPA to generate tumours did not lead to any fully or partially X-Gal+ papilloma (Figure 4A and D, Infundibular schemes). These data together with the fact that we had achieved a much greater labelling efficiency in the infundibulum versus the bulge labelling schemes (Figure 4B and C), suggest that the ectopic Runx1 expression in populations of non-SCs do not render these cells capable of forming tumours. Additionally, the results indicate that Runx1 is essential for tumour formation only in HFSCs but not sufficient outside this niche, even if those more differentiated short-lived cells had been previously initiated with a mutagenic drug.
Absence of Runx1 leads to tumour regression
The data thus far demonstrated that Runx1 is in the cells of tumour origin, that these cells must be HFSC to generate tumours, and that Runx1 is essential in the initiation stage of tumour formation. Moreover, the loss of Runx1-negative tumours in the absence of a proliferative agent (Figure 2H) suggested a possible role for Runx1 in tumour maintenance.
To address this question, we attempted to remove Runx1 from tumours after they had formed and monitored their number and size over time. To remove Runx1, we used topical applications and monthly injections of 4-OHT in acetone directly into tumours (Figure 5A). One month after the first injection Runx1 was lost in 50% of 29 tumours analysed. We next measured the volume of 72 treated tumours bi-weekly for 26 weeks. We found that ∼75% of WT tumours (acetone only) increased 940% in size while 80% of 4-OHT injected tumours decreased to 8% of their original size, suggesting that Runx1 loss induced roughly 120-fold shrinkage of the tumour size (Figure 5B–D). This is likely an underestimation, as all remaining tumours in Runx1 KO mice at 26 weeks showed Runx1 expression due to incomplete KO (not shown). In conclusion, the removal of Runx1 from developed epithelial skin tumours drastically reduces the size of these tumours.
Figure 5.

Runx1 loss causes tumour regression. (A) Scheme of tumour regression study in Runx1 iKO mice. (B) Representative photos of Runx1 WT and KO mice at 0 and 26 weeks of 4OHT treatment. (C) q-q plot comparing the tumour distributions of WT (x axis) and KO (y axis) mice at 0 and 26 weeks. The black line indicates where the two genotypes would have equal distributions. A point to its right denotes more tumours of that size in WT mice and vice versa. (D) Quantification of growing or shrinking tumours in WT or KO mice including fractional tumour change (circle size).
Runx1 is important for oral tumour formation in LSL-KrasG12D mice and is expressed by oral epithelial SCs
Next, we aimed to expand our understanding of Runx1 role in SCs and tumours beyond that of skin SCC and HFSCs. Runx1 appeared highly expressed in oral epithelium human SCC lines and primary tumours (Figure 1). DMBA, which induces the initiating mutation in the two-stage carcinogenesis protocols utilized here, has been shown to target the Ras genes with high frequency (Abel et al, 2009) and a considerable fraction of human oral and skin SCCs show mutation in one of the Ras genes (Pierceall et al, 1991; Spencer et al, 1995). Expression of an activated form of K-Ras (KRasG12D) initiates squamous tumour formation in the oral and anal epithelium as well as in the skin (Vitale-Cross et al, 2004; Gilad et al, 2010; Lapouge et al, 2011). To connect our findings with genetic pathways of human SCC, confirming that tumours with activated Ras depend on Runx1 for their formation, and to analyse the role of Runx1 in the oral epithelium tumour formation, we utilized LSL-KRasG12D mice that can be activated by TM induced Cre activity (Jackson et al, 2001). We generated K14-CreERT2;LSL-KrasG12D;Runx1 KO, wild-type, and heterozygous mice and injected TM at PD21 to induce either the Runx1 KO, or K-Ras activation, or both. The KrasG12D mice rapidly developed sublingual and hard palate oral papillomas (Figure 6A–C) at low TM doses as previously reported (Gilad et al, 2010). Few K14-CreERT2-negative mice developed tumours as well, indicative of low frequency, leaky, non-Cre-dependent K-Ras activation. Oral tumour formation severely impaired the mice’s ability to take in food, ultimately leading to premature death of these mice, prior to skin tumour formation. Although we observed oral or anal tumours in all KrasG12D mice, tumour formation and subsequent death were significantly delayed in Runx1 KO mice. Moreover a dosage-dependent effect was apparent in mice with a single intact copy of Runx1 (Figure 6C). These data extend our results on the importance of Runx1 from skin to oral and anal epithelial tumorigenesis. Moreover, they underscore the function of Runx1 in a genetic context (KrasG12D) highly common in human skin and head and neck (oral) and skin SCC, complementing our 2-step skin carcinogenesis data.
Figure 6.

Runx1 marks oral stem cells and co-localizes with Lgr5 in the intestine. (A, B) Representative images and staining of sections from lip tumour tissue from KrasG12D mice 14 days after TM injection. (C) Kaplan–Meier survival analysis for K14-CreERT2;KrasG12D mice of all Runx1 genotypes (P-value=7.06 × 10−7) and Cre− controls. (D) Endogenous expression of Runx1 in oral tissue of Runx1LacZ-KI mice. Black arrows point to some epithelial LacZ+ cells. Inset shows a higher magnification of the marked tissue area. (E) Lineage tracing of oral (hard palate epithelium) Runx1 population using Runx1-CreER; tdTomato mice. (F) Same as (D) for colon and intestinal epithelium. (G) Runx1 and K8 staining of an intestinal villus and crypt in wild-type mice. (H) shows the close-up marked by the dashed line in (G). (I) Runx1 staining in crypts of Lgr5CreER-IRES-GFP mice where GFP expression highlights the intestinal stem cell population.
Given Runx1 function in oral tumorigenesis, we examined its expression in the oral epithelium using the Runx1-LacZ KI. At PD38 Runx1 expression is in the basal layer of the palate epithelium (Figure 6D). This area was previously shown to contain label-retaining cells, which were putative SCs (Bickenbach and Mackenzie, 1984).
Next, we traced the lineage of oral Runx1+ cells using mice containing the inducible Runx1-CreER and the tdTomato reporter (Madisen et al, 2010). Labelling was induced mostly in the basal layer of the hard palate 4 days after TM but not oil injection (Figure 6E). At 18.5 weeks after TM injection we detected rare clones consisting of basal layer (un-differentiated) and supra-basal (differentiated) cells (Figure 6E). This is the first study showing that indeed a subpopulation of oral basal layer cells differentiate and self-renew for extended periods of time. Oral epithelium SCs express Runx1, making this the third adult tissue SC type with lineage tracing evidence for Runx1 expression, after HFSCs (this work) and HSCs (Samokhvalov et al, 2007).
Runx1 expression in colon and intestine
Because we observed Runx1 expression in several human cancer cell lines of epithelial origin other than skin and oral, we decided to analyse Runx1 endogenous expression in a third tissue: the colon and intestine. We used the Runx1-LacZ KI mice at PD38 and found X-gal signal, and thus endogenous Runx1, in villus cells but more importantly, at the base of every crypt (Figure 6F) where intestinal SCs reside (Barker et al, 2008). We confirmed this by staining using a Runx1 antibody in wild-type intestinal tissue (Figure 6G and H) and in Lgr5CreER-IRES-GFP mice. Indeed, some Runx1 expressing crypt cells co-localize with GFP, suggesting they may be SCs (Figure 6I). Notably, staining of Runx1 and K8, an epithelial marker, in wild-type mice revealed an overall pattern of Runx1 expression that is similar to HFs: Runx1 is lightly expressed in a few SCs at the base of the crypt (analogous with the hair bulge) and strongly expressed in the activated cells or transit amplifying cells in the upper crypt (analogous with the hair germ). Extremely low labelling efficiency of Runx1-CreER in the intestine and colon with tdTomato, YFP or ROSA26R reporter prevented lineage tracing of these Runx1+ cells. It is possible that by studying the isoform structure of Runx1 in the intestine and different CreER KI strategy may overcome this limitation. These data suggest that Runx1 may be expressed in the intestine in a subfraction of the LGR5+ SCs.
Human cancer cells require Runx1 for growth
Thus far we presented evidence that Runx1 is a SC factor for at least two epithelial tissues, and is important for tumour initiation/formation in these tissues. Runx1 is also expressed in several kinds of human epithelial cancer cells (Figure 1E) and tumours, as shown by Oncomine data (Figure 1A). To test the significance of Runx1 in human epithelial cancers, we knocked down its expression in several cancer cell lines and monitored their cell growth and survival.
We used Luciferase shRNA (Luc) as a control and two Runx1 shRNAs. Runx1-1 and Luc expression, but not Runx1-2, can be assessed by detection of GFP (Materials and methods). Both Runx1 shRNAs (but not Luc) knocked down Runx1 levels (Runx1KD), albeit to a different degree, as determined by immuno-fluorescence staining with a Runx1-specific antibody (Figure 7A and B). Skin and head and neck (oral) SCC cell lines with Runx1KD failed to grow, confirming an essential role for Runx1 in tumour maintenance as seen in mouse papillomas (Figure 7C and D; Figure 5). Ovarian cell lines show a reduction in growth, whereas colon, breast, and prostate cancer cell lines were not affected (Figure 7D). Notably, cells react at different degrees to the two Runx1 shRNAs, likely due to variable knockdown efficiencies of the two target sequences. The effects of Runx1KD can be rescued by the overexpression of Runx1 (Figure 7C; Wang et al, 2011).
Figure 7.

Runx1 is important for cancer cell growth by modulating Stat signalling. (A) Images of Pam212 cancer cells transfected with control shRNA (Luc) and Runx1-1 shRNA reveal severe growth impairment in the latter (left and middle). Single colony detected with low levels of Runx1 (right) demonstrate the specificity of our antibody and the downregulation of Runx1. (B) Runx1 levels in MDA-231 cells with Runx1 shRNA 1 and 2. (C) Representative images of cell plates stained with Rhodanile Blue of Keratinocytes (Kera) and Pam212 cancer cells transfected with control or Runx1 shRNA. Note severe impairment in growth in the Runx1 KD cells. Cell growth was rescued with overexpression of Runx1, addition of OSM, and cStat3. (D) Summary of KD experiments in human cell lines and rescue with OSM where applicable. (E) Immunostaining of pStat3T705 levels in tumours with or without Runx1 expression. (F) Western blots of iKO keratinocytes treated either with ethanol (WT) or with 4-OHT for 4 or 5 days. (G) QRT–PCR of Runx1 RNA levels in the cells from (D) (average±s.d.). (H) Western blot of iKO (4 days) cells treated with OSM for 1 day. (I) Volcano plot of Jak/Stat pathway qPCR array. (J) Scheme for SOCS ChIP primers colour coded by the P-value determined for Runx1 binding (grey >0.07, yellow >0.05, green <0.05). (K) ChIP of WT and iKO cells (4 days) using Runx1 antibody for SOCS family members (average±s.d.). P-values for pair-wise comparisons are indicated. Significant P-values are highlighted in green. (L) Quantification of GFP+ cells in keratinocytes and Pam212 transfected with control or Runx1 shRNA and pLKO1-puro (control) and two shRNAs each for SOCS3 and SOCS4 (average±s.d.). Note lack of GFP+ cells when Runx1 is KD and their rescue by concomitant KD of Runx1 and either SOCS3 or SOCS4.
Since all cancer cell lines expressed Runx1 and their tissues of origin also displayed suggestive Runx1 expression we briefly looked into a possible mechanism for resistance to loss of Runx1. In a recent study in breast cancer Runx1 levels were inversely correlated with FOXO1 expression, compensating for Runx1 loss (Wang et al, 2011). However, we could not find a correlation between Runx1KD resistance and FOXO1 in any of the cell lines analysed here (Supplementary Figure S4A). Overall, these results show that loss of Runx1 is detrimental to growth of three human epithelial cancer cell types: head and neck (oral) SCC, skin SCC, and in part to ovarian cancer cells.
Runx1 acts upstream of Stat3 in cancer cell growth and survival
To understand the mechanism by which Runx1 affects cancer cell growth in mice and humans, we considered a possible interaction with Stat3, which appears to play a similar role as Runx1 in regulating the hair cycle, and in tumour formation (Sano et al, 1999, 2000; Chan et al, 2004a, 2004b; Osorio et al, 2008; Kim et al, 2009). The activated form of Stat3—pStat3 T705—is upregulated in skin upon TPA treatment in WT mice (Chan et al, 2004a). This upregulation appeared more variable and overall reduced in the Runx1 KO skin (Hoi et al, 2010), and is lost in tumours that form after loss of Runx1 during the tumour promotion stage (Figure 7E). Given the essential role of Stat3 in proliferation and survival of human and mouse SCC (Bito et al, 2011; Rho et al, 2011), this may explain why the Runx1-deficient tumours obtained here in the promotion scheme were short lived and could not be maintained in the absence of TPA (Figure 2H). Given the complex role of the microenvironment in regulating Stat3 activation, the manner in which Runx1 interacts with the Stat3 signalling pathway, and the functional relevance of this interaction to Runx1 mechanism of action are unclear. For example, Stat3 downregulation in the Runx1 KO skin treated with TPA could be an indirect effect to reduced inflammation potentially driven by a paracrine function downstream of Runx1, rather than a direct interaction of Runx1 with the Stat3 pathway itself.
To analyse whether Runx1 directly regulates Stat3 activation, we utilized cells removed from the complex skin and tumour environment and created inducible KO K14-CreER;Runx1fl/fl keratinocytes (iKO). After 4 days of 4-OHT treatment, Runx1 mRNA and protein level were significantly reduced as shown by western blot and by QRT-PCR (Figure 7F and G). Moreover, western blots for Stat3, pStat3 T705, and pStat3 S727 revealed a selective and striking de-phosphorylation of T705 upon Runx1 loss (Figure 7F). In this form, Stat3 is inactive because it cannot dimerize and translocate to the nucleus (Ihle, 1996).
To analyse if the disruption of the Jak/Stat pathway is responsible for the growth defect of normal and cancer cell lines with Runx1 deficiency, we attempted to restore Stat3 activity in Runx1KD cells. To this end, keratinocytes and Pam212 cells were either co-transfected with Runx1-1 shRNA and cStat3, the constitutively active form of Stat3 (Bromberg et al, 1999) or treated with Oncostatin M (OSM), a small signalling peptide that activates Stat3 signalling (Ishihara and Hirano, 2002). In both cases, the Runx1-deficient cell lines were then able to form large colonies (Figure 7D), and showed increased levels of T705 pStat3, but not Runx1, relative to untreated cells (Figure 7H).
OSM activates Jak and Tyk kinases through its receptors that in return activate the Stat3 and the MAP kinase pathway (Erk1/2) (Ishihara and Hirano, 2002). To see if the effect of OSM can be attributed to Stat3 or whether Erk phosphorylation plays a role in the rescue of growth defects in our cells, we examined P-Erk1/2 level by western blotting. We fail to see an increased phosphorylation of Erk1/2 in keratinocytes (Figure 7H), which suggests that it is the Stat3 activation that rescued the Runx1-deficient growth defect, further supporting the similar effect obtained with the cStat3 (Figure 7C).
Finally, we employed the human cancer cell lines that previously showed dependence on Runx1 levels, and found that OSM rescues their growth to different degrees (Figure 7D), with the most striking effects on skin and head and neck SCC lines and a milder effect on ovarian cancers. Hence, in normal keratinocytes as well as in three epithelial cancer cell types Runx1 loss appears to impair growth by directly preventing Stat3 activation.
Runx1 stimulates the Jak/Stat pathway by repression of SOCS3 and SOCS4
Next, we attempted to understand the mechanism by which Runx1 may stimulate Stat3 signalling. Since our western blots suggested stable Stat3 protein levels upon Runx1 loss, it seemed likely that some members of the Jak/Stat pathway involved in T705 phosphorylation might be Runx1 transcriptional targets. To find such putative targets, we surveyed the status of the Jak/Stat pathway using SA-Biosciences RT2 Profiler PCR array and compared mRNA levels in Runx1-CreER;Runx1fl/fl and Runx1fl/fl keratinocytes after 4 days of 4-OHT treatment. A volcano plot showing the fold changes between WT and KO versus the associate P-value for each tested gene revealed three genes significantly upregulated in the iKO cells: Crk, SOCS3, and SOCS4 (Figure 7I; raw data in Supplementary Table S3). Crk is a proto-oncogene thought to stimulate proliferation (Tanaka, 2009), and as such its upregulation cannot explain proliferation defects but is likely an attempt to compensate for Runx1 loss. On the other hand, SOCS3 and SOCS4 suppress Stat3 T705 phosphorylation via interference with Jak2 and EGFR kinase activity, respectively (Larsen and Ropke, 2002; Segatto et al, 2011).
To test if Runx1 directly binds to the SOCS3 and SOCS4 promoters to represses their transcription, we employed two independent K14-CreERT2;Runx1fl/fl keratinocyte lines and performed chromatin immunoprecipitation (ChIP) using Runx1-specific antibodies in KO and WT (Runx1fl/fl) cells. We followed with QRT-PCR using primers encompassing SOCS3 and SOCS4 genomic regions containing Runx1 binding sites (Figure 7J). Our data showed enrichment from one primer pair in each of the SOCS4 and SOCS3 gene loci (P=0.04 and 0.07, respectively). We used a region of the Gapdh promoter free of conserved Runx1-binding sites as a negative control (Figure 7K). Binding of Runx1 on the SOCS3 gene promoter, although here showed a P-value slightly lower than the conventional 0.05, was also reported by ChIP Seq data on a blood cell line (Wilson et al, 2010). Thus, we suggest that Runx1 represses SOCS3 and SOCS4 expression, likely via direct binding to their promoters, and ultimately upregulates Stat3 activity by enhancing phosphorylation at T705.
If Runx1 loss induced defects were mediated through SOCS3 and/or SOCS4, as indicated by the QRT-PCR and ChIP experiments described above, then simultaneous knockdown of SOCS3 or 4 and Runx1 should rescue the cell growth defect. This could be measured by the presence of GFP+ cells, which indicate Runx1 deficiency (Materials and methods). As expected, cells transfected with control Luc shRNA but not those transfected with Runx1-1 shRNA and the empty plasmid backbone (pLKO1) did not show a population of GFP+ keratinocytes or Pam212 cells after puromycin selection. In contrast, addition of either SOCS shRNA instead of the empty plasmid backbone rescued the GFP+ (Runx1-deficient) population, in which cells with high SOCS were able to grow without Runx1 (Figure 7L; Supplementary Figure S4B). Knockdown of SOCS3 or 4 alone did not affect cell growth as expected from low or absent SOC3/4 expression in WT cells and as shown in Supplementary Figure S4C and through equal amounts of GFP− cells in Luc+backbone or Runx1+SOCS shRNA-transfected cells. These data together suggested that Runx1 is a direct transcriptional repressor of SOCS4 and potentially SOCS3, which results in activation of Stat3 and thus allows growth of normal and cancer cells (Figure 8). In conclusion, Runx1 repression is accompanied by activation of SOCS genes, inhibition of Stat3 activity, and growth impairment of some epithelial cancer cells.
Figure 8.

Model. Model of Runx1 in tissue stem cells and its role in tumour formation regulation through Jak/Stat family members. Thick black bars highlight conclusions from this paper. Grey dashed line indicates a potential connection.
Discussion
Here we uncover Runx1, as a broader epithelial tissue SC and tumour factor than previously recognized. We not only confirmed its expression in adult mouse HFSCs by direct lineage tracing, but also we implicate Runx1 in previously uncharacterized oral epithelium SCs. We showed that mouse oral epithelium tumours, like their skin counterparts (Hoi et al, 2010, and this work), also appear dependent on Runx1 for their growth. We find that increased proliferation due to ectopic injury-induced expression of Runx1 in skin non-SCs does not render those cells tumorigenic, while Runx1-expressing SCs are at the tumour origin. We implicate Runx1 as a key molecule upstream of a central epithelial cancer pathway, Stat3 (Li et al, 2011), which Runx1 activates by direct transcriptional repression of SOC3/4. We documented for the first time the expression of Runx1 in primary skin and oral human epithelium SCC and suggest that several more human epithelial cancers may express Runx1. We demonstrate that three different human cancer cell types (skin SCC, oral SCC, and ovarian) depend upon Runx1 for their normal growth. We find Runx1 important for both skin tumour initiation and maintenance, while we previously showed that Runx1 was not essential for adult tissue maintenance (Osorio et al, 2008). This differential requirement for Runx1 places it as a promising target for skin, and potential other epithelial cancers.
Runx1 has been intensively studied for the past 4 decades as a cancer gene frequently mutated in acute myeloid leukaemia, which when studied in mice turned out to work as a master regulator of haematopoiesis (Mangan and Speck, 2011). Taking the opposite direction—from mouse to human—we first demonstrated that Runx1 is important for the timely emergence and maturation of HFSCs in embryogenesis (Osorio et al, 2011) and the activation and proliferation of adult HFSCs (Osorio et al, 2008). Moreover, Runx1 appears essential in mice for skin squamous epithelial tumour formation (Hoi et al, 2010). Importantly, with this work we now take Runx1 back to humans where we analysed the role of Runx1 in several epithelial cancers.
Runx1 surfaced among the top 10% highly expressed genes in 1/3 of all 138 applicable microarrays studies of human cancers found by us in the Oncomine database. These included the skin SCC and BCC, oral SCC, breast, oesophageal, lung, colon, and pancreatic cancer among others. We detected Runx1 expression by immunofluorescence staining of several kinds of skin human primary tumours as well as in oral epithelium SCCs. Moreover, we found Runx1 overexpressed in a whole panel of established human cancer cell lines. Importantly, Runx1 appears to be required for growth of skin and head and neck (oral) SCC, and to some extent ovary cancer cells, suggesting Runx1 as a novel putative target for treating these cancers. In contrast, although Runx1 was expressed at high levels it was dispensable for growth of breast, prostate, and colon cancers. In light of a recent study of breast cancer, these cells may have learnt to live without Runx1 by upregulating compensatory pathways (Wang et al, 2011). However, it appears that FOXO1, shown to compensate for Runx1 loss in MCF10A-5E breast cancer cells, is not upregulated in the cells we analysed here. This information will be important for future treatment of some cancer types in which a combination drug therapy against Runx1 and compensatory pathways may be required.
An attractive clue positioning Runx1 as a promising therapeutic target is our demonstration that loss of Runx1 shrinks tumours in mice and impairs human and mouse tumour cell growth in vitro. This potential adds an important angle to the role of Runx1 in tumour initiation, which could be exploited for tumour treatment and prevention, respectively. Runx1 loss is not essential for normal skin and oral epithelium homeostasis, and deletion of Runx1 in adult mice via a ubiquitous β-Actin-CreER allele or Mx1-Cre had mild effects on the mouse physiology (Appleford and Woollard, 2009; Hoi et al, 2010; and our unpublished data). The tolerance of the tissue to Runx1 loss in HFSCs is probably due to the unique protective niche environment, which is disrupted in cell culture and in tumours. Disrupting Runx1 binding to the DNA via specific drugs is already considered as a potentially beneficial approach to treating leukaemia (Mangan and Speck, 2011). A more targeted approach for local treatment of accessible skin or head and neck tumours would be even less invasive.
How is Runx1 working in regulating tumour formation and maintenance? One possibility is that Runx1 is expressed in the cell of tumour origin and remains expressed in some of the progeny cells subsequently generated. At least in the skin, HFSCs are known to be at the origin of some SCC (Youssef et al, 2010; Lapouge et al, 2011; White et al, 2011). Previously, we reported Runx1 expressed in a fraction of cells in bulge and hair germ cells, the regions of the HF where SCs are known to reside. However, it is unclear whether all cells in these regions are SCs. Even though loss of Runx1 impairs proliferation of HFSCs it was possible that Runx1 modulates the SC environment from neighbouring cells. For example, Runx1 is able to regulate production of several secreted Wnt molecules during embryogenesis (Osorio et al, 2011). Our lineage tracing work directly demonstrates here for the first time that adult Runx1-expressing cells are long-lived, self-renewing and differentiating HFSCs, which are at the tumour origin. Moreover, Runx1 also appears to be a marker for oral epithelium SCs. Our data suggest some overlap of Runx1 and LGR5 expression, and LGR5 is a well-recognized intestinal SC marker. Further substantiation by lineage tracing is needed for the intestine since the existing Runx1-CreER is extremely inefficient in this organ.
Using skin as a model system we demonstrate that Runx1-expressing HFSC is at the origin of tumours, and these tumours were likely monoclonal. It is recognized that epidermal cells may form skin SCC (Lapouge et al, 2011), and even though Runx1 is not expressed in the epidermis in normal conditions it appears that in our experimental conditions all mouse tumours expressed Runx1. We therefore hypothesized that a population of epidermal cells rendered by inflammation or injury caused by the carcinogenic treatment to ectopically express Runx1 might also generate tumours. However, we found this population located in the infundibulum of the HF to be short-lived progenitors that did not contribute to long-term homeostasis and injury repair. Moreover, this population was unable to generate tumours in our experimental conditions, even in the context of an initiating mutation induced by drug treatment. This underscores the crucial importance of the intrinsic epigenetic context or cellular make-up to initiation of carcinogenesis and adds to the theory that SCs, and not differentiated cells, are the most likely initiators of cancer (Magee et al, 2012).
Runx1 is known to regulate cell survival, proliferation, differentiation, and cell-cycle progression (Blyth et al, 2005; Mikhail et al, 2006; Friedman, 2009). Moreover, Runx1 directly binds and represses the promoter of cyclin-dependent kinase inhibitor p21 in skin epithelial cells (Lee J and Tumbar T, submitted; Hoi et al, 2010). Thus, it seemed surprising that Runx1 was essential for tumour initiation but dispensable for tumour promotion, in which over 50% of the tumours in Runx1 KO mice actually lacked Runx1. However, these tumours were lost after we ceased the TPA treatment, demonstrating that during the proliferative/maintenance stage other strongly proliferative agents can compensate for Runx1 loss. These short-lived Runx1-deficient tumours displayed downregulation of activated Stat3 and a striking absence of CD34+ cells, which were previously deemed cancer SCs, important for tumour growth and maintenance (Malanchi et al, 2008). These data together may suggest that Runx1 is expressed in the HFSCs that generate the tumours, and is continuously required in these tumours for the maintenance of their long-lived tumour SC population and for continuous tumour growth. Moreover, Runx1 role in this process is likely mediated via stimulation of Stat3 signalling.
Phosphorylation of Stat3 at T705 and S727 is necessary for full activity of Stat3 (Yokogami et al, 2000). However, phosphorylation at S727 alone prevents activation at T705 and thus locks Stat3 in its inactive state in the cytoplasm (Gartsbein et al, 2006). Upon Runx1 loss we observed a loss of T705 phosphorylation while S727 maintained its levels, indicating a double-safe mechanism to keep Stat3 inactive. Active Stat3 is associated with psoriasis in humans and mice (Sano et al, 2005) and its increased activity has been repeatedly associated with skin SCC formation (Chan et al, 2004a, 2004b, 2008; Aziz et al, 2007; Chan et al, 2008). Like Runx1, Stat3 is also considered essential for tumour initiation and maintenance by directly targeting the HFSCs in mouse models (Kim et al, 2009). Activated Stat3 is found in the vast majority of human oral cancers as well as in breast, lung, ovarian, pancreatic and prostate cancers and myelomas, leukaemias, and lymphomas (Yu and Jove, 2004). High levels of activated Stat3 have also been reported in oral SCC cell lines, and proliferation of these SCC cells could be further increased with OSM (Douglas et al, 2004), a potent activator of the Stat3 pathway. This connects well to our finding that addition of OSM rescues the proliferation defect of skin and oral SCC and ovarian cancer cells upon Runx1 loss, through activation of Stat3.
Here, we expose a novel regulatory pathway for Stat3 activity. We show that Runx1 regulates Stat3 activation by transcriptional repression of Stat3 inhibitors SOCS3 and SOCS4. Conversely, loss of Runx1 results in upregulation of SOCS3 and SOCS4 and Stat3 inactivation, which is detrimental to cancer cell growth. Importantly, we show that this mechanism is conserved in humans where it affects skin SCC, and head and neck (oral) SCC, and ovarian cancer. The connection between Runx1 and Stat3 is a putative axis in regulation of cell proliferation and tumorigenesis, suggesting a potential benefit for investigating Runx1 status in Stat3-dependent cancers. If Runx1 is upstream of Stat3 and potentially other major cancer pathway, such as Wnt signalling as we previously showed (Osorio et al, 2011), then it may be more beneficial to target the master regulator, Runx1, rather than its individual target signalling pathways.
Materials and methods
Samples
Human samples were obtained de-identified and stained as described in the supplement. K14-CreERT2, Runx1fl/fl, Runx1-CreER, LSL-KrasG12D, Lgr5CreER-IRES-GFP, ROSA26R, and tdTomato mice were crossed to obtain the various experimental mice. CreER activation by Tamoxifen (Sigma) and 2-step carcinogenesis DMBA/TPA protocol were previously described (Hoi et al, 2010). The Cornell University IACUC approved all the mouse work.
Rescue assays
All knockdown and rescue assays were duplicated or triplicated. Mouse keratinocytes or Pam212 was co-transfected with 1 μg DNA of each shRNA (Runx1-1 or Luc in pLKO1-pig) and cStat3 (Addgene plasmid 8722; Bromberg et al, 1999). At 90% confluency, we select for shRNA containing with puromycin. Oncostatin M (OSM) (Cell Signaling, human: #5367; mouse: #5371) was used at 10 ng/ml media at the time of puromycin selection. pLKO1-puro only or mouse-specific SOCS3 or 4 shRNA was co-transfected with pLKO1-pig Runx1-1 and Luc shRNA. All transfected cells will be puromycin resistant; however, only if growth of Runx1-1 (or Luc) KD cells is permissible, GFP+ cells are observed.
QRT–PCR
RNA from iKO cells treated with ethanol or 4-OHT (Sigma) for 4 days was extracted using Trizol and the RNeasy Mini kit (Qiagen) and converted to cDNA with the iScript kit (Bio-Rad). The mouse Jak/Stat RT2 Profiler PCR array (PAMM-039) was purchased from Qiagen, used as instructed, and the data were analysed using proprietary bioinformatics tools provided by Qiagen. Details for Runx1 QRT-PCR were described previously (Hoi et al, 2010).
ChIP
In all, 0.5–1 × 107 iKO or WT keratinocytes treated with 4-OHT for 4 days were used as starting material. SOCS3 and SOCS4 genomic regions were pulled down with 5 μg Runx1 antibody (Abcam ab23980). Control ChIP reactions were set up with 4 μg rabbit-IgG (Imgenex 20304) and 3 μg H3 (Abcam ab1791).
Statistics
Data was processed and analysed in Excel or R. The survival package in R was used for Kaplan–Meier analysis.
Supplementary Material
Acknowledgments
We thank the Victorian Cancer Biobank, Drs Jonathan Vogel, Agnes Kobielack and Ralf Paus for human tumour samples; Drs Igor Samokhvalov, Elaine Fuchs, Nancy Speck, Pierre Chambon, and Alexander Nikitin for transgenic mice. We thank Dr Stuart Yuspa for the mouse skin SCC cell line Pam212; Dr James Rheinwald for the human skin SCC cell line SCC13; Dr Susanne Gollin for the human oral SCC cell lines SCC66, 125, and 74; Dr Xiling Shen for colon CCSC and HCT116 and Dr Alexander Nikitin for all other human cancer cell lines. The Stem Cell and Transgenics Core at Cornell aided in the virus production. Funding for this work was from NYSTEM Grant C024354 and NIH Grant R01AR053201 to Tudorita Tumbar.
Author contributions: DJM performed and evaluated hair follicle stem cell homeostasis lineage tracing; TSL executed all skin tumor lineage tracing experiments and significantly contributed to monitoring the KrasG12D mice. CS designed the project, performed, analysed, and interpreted all other experiments and wrote the manuscript. TT designed the project, analysed, and interpreted data and wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Abel EL, Angel JM, Kiguchi K, DiGiovanni J (2009) Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc 4: 1350–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M, Ratner D (2001) Cutaneous squamous cell carcinoma. N Engl J Med 344: 975–983 [DOI] [PubMed] [Google Scholar]
- Appleford PJ, Woollard A (2009) RUNX genes find a niche in stem cell biology. J Cell Biochem 108: 14–21 [DOI] [PubMed] [Google Scholar]
- Aziz MH, Manoharan HT, Sand JM, Verma AK (2007) Protein kinase Cepsilon interacts with STAT3 and regulates its activation that is essential for the development of skin cancer. Mol Carcinog 653: 646–653 [DOI] [PubMed] [Google Scholar]
- Barker N, van de Wetering M, Clevers H (2008) The intestinal stem cell. Genes Dev 22: 1856–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickenbach JR, Mackenzie IC (1984) Identification and localization of label-retaining cells in hamster epithelia. J Invest Dermatol 82: 618–622 [DOI] [PubMed] [Google Scholar]
- Bito T, Sumita N, Ashida M, Budiyanto A, Ueda M, Ichihashi M, Tokura Y, Nishigori C (2011) Inhibition of epidermal growth factor receptor and PI3K/Akt signaling suppresses cell proliferation and survival through regulation of Stat3 activation in human cutaneous squamous cell carcinoma. J Skin Cancer 2011: 874571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyth K, Cameron ER, Neil JC (2005) The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer 5: 376–387 [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE (1999) Stat3 as an oncogene. Cell 98: 295–303 [DOI] [PubMed] [Google Scholar]
- Casanova ML, Bravo A, Martínez-Palacio J, Fernández-Aceñero MJ, Villanueva C, Larcher F, Conti CJ, Jorcano JL (2004) Epidermal abnormalities and increased malignancy of skin tumors in human epidermal keratin 8-expressing transgenic mice. FASEB J 18: 1556–1558 [DOI] [PubMed] [Google Scholar]
- Chan KS, Carbajal S, Kiguchi K, Clifford J, Sano S, DiGiovanni J (2004a) Epidermal growth factor receptor-mediated activation of Stat3 during multistage skin carcinogenesis. Cancer Res 64: 2382–2389 [DOI] [PubMed] [Google Scholar]
- Chan KS, Sano S, Kataoka K, Abel E, Carbajal S, Beltran L, Clifford J, Peavey M, Shen J, Digiovanni J (2008) Forced expression of a constitutively active form of Stat3 in mouse epidermis enhances malignant progression of skin tumors induced by two-stage carcinogenesis. Oncogene 27: 1087–1094 [DOI] [PubMed] [Google Scholar]
- Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N, Takeda J, Digiovanni J (2004b) Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest 114: 720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas WG, Tracy E, Tan D, Yu J, Hicks WL Jr, Rigual NR, Loree TR, Wang Y, Baumann H (2004) Development of head and neck squamous cell carcinoma is associated with altered cytokine responsiveness. Mol Cancer Res 2: 585–593 [PubMed] [Google Scholar]
- Friedman AD (2009) Cell cycle and developmental control of hematopoiesis by Runx1. J Cell Physiol 219: 520–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartsbein M, Alt A, Hashimoto K, Nakajima K, Kuroki T, Tennenbaum T (2006) The role of protein kinase C delta activation and STAT3 Ser727 phosphorylation in insulin-induced keratinocyte proliferation. J Cell Sci 119: 470–481 [DOI] [PubMed] [Google Scholar]
- Gilad O, Nabet BY, Ragland RL, Schoppy DW, Smith KD, Durham AC, Brown EJ (2010) Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res 70: 9693–9702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, Curley DP, Kutok JL, Akashi K, Williams IR, Speck NA, Gilliland DG (2005) Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 106: 494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoi C, Lee SE, Lu S-Y, McDermitt DJ, Osorio KM, Piskun C, Peters R, Paus R, Tumbar T (2010) Runx1 directly promotes proliferation of hair follicle stem cells and epithelial tumor formation in mouse skin. Mol Cell Biol 30: 2518–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S-P, Lan Y-H, Lu T-L, Pao J-B, Chang T-Y, Lee H-Z, Yang W-H, Hsieh C-J, Chen L-M, Huang L-C, Ting W-C, Bao B-Y (2011) Clinical significance of runt-related transcription factor 1 polymorphism in prostate cancer. BJU Int 107: 486–492 [DOI] [PubMed] [Google Scholar]
- Ihle JN (1996) STATs: signal transducers and activators of transcription. Cell 84: 331–334 [DOI] [PubMed] [Google Scholar]
- Ishihara K, Hirano T (2002) Molecular basis of the cell specificity of cytokine action. Biochim Biophys Acta 1592: 281–296 [DOI] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA (2001) Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15: 3243–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DJ, Kataoka K, Rao D, Kiguchi K, Cotsarelis G, Digiovanni J (2009) Targeted disruption of stat3 reveals a major role for follicular stem cells in skin tumor initiation. Cancer Res 69: 7587–7594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapouge G, Youssef KK, Vokaer B, Achouri Y, Michaux C, Sotiropoulou PA, Blanpain C (2011) Identifying the cellular origin of squamous skin tumors. Proc Natl Acad Sci USA 108: 7431–7436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen L, Ropke C (2002) Suppressors of cytokine signalling: SOCS. Review article. APMIS 110: 833–844 [DOI] [PubMed] [Google Scholar]
- Li N, Grivennikov SI, Karin M (2011) The unholy trinity: inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell 19: 429–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JA, Piskounova E, Morrison SJ (2012) Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 21: 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, Huber M, Hohl D, Cano A, Birchmeier W, Huelsken J (2008) Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature 452: 650–653 [DOI] [PubMed] [Google Scholar]
- Mangan JK, Speck NA (2011) RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making. Crit Rev Oncog 16: 77–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhail FM, Sinha KK, Saunthararajah Y, Nucifora G (2006) Normal and transforming functions of RUNX1: a perspective. J Cell Physiol 207: 582–593 [DOI] [PubMed] [Google Scholar]
- Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS (2009) Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol 45: 324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- North T, Gu T, Stacy T, Wang Q, Howard L, Binder M, Marin-Padilla M, Speck N (1999) Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development 126: 2563–2575 [DOI] [PubMed] [Google Scholar]
- Osorio KM, Lee SE, McDermitt DJ, Waghmare SK, Zhang YV, Woo HN, Tumbar T (2008) Runx1 modulates developmental, but not injury-driven, hair follicle stem cell activation. Development (Cambridge, England) 135: 1059–1068 [DOI] [PubMed] [Google Scholar]
- Osorio KM, Lilja KC, Tumbar T (2011) Runx1 modulates adult hair follicle stem cell emergence and maintenance from distinct embryonic skin compartments. J Cell Biol 193: 235–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierceall WE, Goldberg LH, Tainsky MA, Mukhopadhyay T, Ananthaswamy HN (1991) Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol Carcinog 4: 196–202 [DOI] [PubMed] [Google Scholar]
- Planagumà J, Gonzalez M, Doll A, Monge M, Gil-Moreno A, Baró T, García A, Xercavins J, Alameda F, Abal M, Reventós J (2006) The up-regulation profiles of p21WAF1/CIP1 and RUNX1/AML1 correlate with myometrial infiltration in endometrioid endometrial carcinoma. Hum Pathol 37: 1050–1057 [DOI] [PubMed] [Google Scholar]
- Planagumà J, Liljeström M, Alameda F, Bützow R, Virtanen I, Reventós J, Hukkanen M, Bützow R (2011) Matrix metalloproteinase-2 and matrix metalloproteinase-9 codistribute with transcription factors RUNX1/AML1 and ETV5/ERM at the invasive front of endometrial and ovarian carcinoma. Hum Pathol 42: 57–67 [DOI] [PubMed] [Google Scholar]
- Rheinwald JG, Beckett MA (1980) Defective terminal differentiation in culture as a consistent and selectable character of malignant human keratinocytes. Cell 22: 629–632 [DOI] [PubMed] [Google Scholar]
- Rheinwald JG, Beckett MA (1981) Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res 41: 1657. [PubMed] [Google Scholar]
- Rho O, Kim DJ, Kiguchi K, Digiovanni J (2011) Growth factor signaling pathways as targets for prevention of epithelial carcinogenesis. Mol Carcinog 50: 264–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samokhvalov IM, Samokhvalova NI, Nishikawa S-ichi (2007) Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 446: 1056–1061 [DOI] [PubMed] [Google Scholar]
- Sano S, Chan KS, Carbajal S, Clifford J, Peavey M, Kiguchi K, Itami S, Nickoloff BJ, DiGiovanni J (2005) Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med 11: 43–49 [DOI] [PubMed] [Google Scholar]
- Sano S, Itami S, Takeda K, Tarutani M, Yamaguchi Y, Miura H, Yoshikawa K, Akira S, Takeda J (1999) Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J 18: 4657–4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano S, Kira M, Takagi S, Yoshikawa K, Takeda J, Itami S (2000) Two distinct signaling pathways in hair cycle induction: Stat3-dependent and -independent pathways. Proc Natl Acad Sci USA 97: 13824–13829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos M, Ballestín C, Garcia-Martín R, Jorcano JL (1997) Delays in malignant tumor development in transgenic mice by forced epidermal keratin 10 expression in mouse skin carcinomas. Mol Carcinog 20: 3–9 [DOI] [PubMed] [Google Scholar]
- Segatto O, Anastasi S, Alemà S (2011) Regulation of epidermal growth factor receptor signalling by inducible feedback inhibitors. J Cell Sci 124: 1785–1793 [DOI] [PubMed] [Google Scholar]
- Sikandar SS, Pate KT, Anderson S, Dizon D, Edwards RA, Waterman ML, Lipkin SM (2010) NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res 70: 1469–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery ML, Lundgreen A, Herrick JS, Caan BJ, Potter JD, Wolff RK (2011) Associations between genetic variation in RUNX1, RUNX2, RUNX3, MAPK1 and eIF4E and risk of colon and rectal cancer: additional support for a TGF-β-signaling pathway. Carcinogenesis 32: 318–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21: 70–71 [DOI] [PubMed] [Google Scholar]
- Spencer JM, Kahn SM, Jiang W, DeLeo VA, Weinstein IB (1995) Activated ras genes occur in human actinic keratoses, premalignant precursors to squamous cell carcinomas. Arch Dermatol 131: 796–800 [PubMed] [Google Scholar]
- Tanaka S (2009) Biological roles of signaling adaptor protein CRK. Seikagaku 81: 361–376 [PubMed] [Google Scholar]
- Tumbar T (2012) Ontogeny and homeostasis of adult epithelial skin stem cells. Stem Cell Rev 8: 561–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE (2011) Cells of origin in cancer. Nature 469: 314–322 [DOI] [PubMed] [Google Scholar]
- Vitale-Cross L, Amornphimoltham P, Fisher G, Molinolo AA, Gutkind JS (2004) Conditional expression of K-ras in an epithelial compartment that includes the stem cells is sufficient to promote squamous cell carcinogenesis. Cancer Res 64: 8804–8807 [DOI] [PubMed] [Google Scholar]
- Wang GY, Wang J, Mancianti M-L, Epstein EH Jr (2011) Basal cell carcinomas arise from hair follicle stem cells in Ptch1+/− mice. Cancer Cell 19: 114–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AC, Tran K, Khuu J, Dang C, Cui Y, Binder SW, Lowry WE (2011) Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc Natl Acad Sci USA 108: 7425–7430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JS, Weissfeld JL, Ragin CCR, Rossie KM, Martin CL, Shuster M, Ishwad CS, Law JC, Myers EN, Johnson JT, Gollin SM (2007) The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines. Oral Oncol 43: 701–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NK, Foster SD, Wang X, Knezevic K, Schütte J, Kaimakis P, Chilarska PM, Kinston S, Ouwehand WH, Dzierzak E, Pimanda JE, de Bruijn MFTR, Göttgens B (2010) Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 7: 532–544 [DOI] [PubMed] [Google Scholar]
- Yeh H-Y, Cheng S-W, Lin Y-C, Yeh C-Y, Lin S-F, Soo V-W (2009) Identifying significant genetic regulatory networks in the prostate cancer from microarray data based on transcription factor analysis and conditional independency. BMC Med Genomics 2: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokogami K, Wakisaka S, Avruch J, Reeves SA (2000) Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol 10: 47–50 [DOI] [PubMed] [Google Scholar]
- Youssef KK, Van Keymeulen A, Lapouge G, Beck B, Michaux C, Achouri Y, Sotiropoulou PA, Blanpain C (2010) Identification of the cell lineage at the origin of basal cell carcinoma. Nat Cell Biol 12: 299–305 [DOI] [PubMed] [Google Scholar]
- Yu H, Jove R (2004) The STATs of cancer--new molecular targets come of age. Nat Rev Cancer 4: 97–105 [DOI] [PubMed] [Google Scholar]
- Yuspa SH, Hawley-Nelson P, Koehler B, Stanley JR (1980) A survey of transformation markers in differentiating epidermal cell lines in culture. Cancer Res 40: 4694–4703 [PubMed] [Google Scholar]
- Zhang YV, Cheong J, Ciapurin N, McDermitt DJ, Tumbar T (2009) Distinct self-renewal and differentiation phases in the niche of infrequently dividing hair follicle stem cells. Cell Stem Cell 5: 267–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.