

Abstract
Presynaptic terminals are specialized sites for information transmission where vesicles fuse with the plasma membrane and are locally recycled. Recent work has extended this classical view, with the observation that a subset of functional vesicles is dynamically shared between adjacent terminals by lateral axonal transport. Conceptually, such transport would be expected to disrupt vesicle retention around the active zone, yet terminals are characterized by a high-density vesicle cluster, suggesting that counteracting stabilizing mechanisms must operate against this tendency. The synapsins are a family of proteins that associate with synaptic vesicles and determine vesicle numbers at the terminal, but their specific function remains controversial. Here, using multiple quantitative fluorescence-based approaches and electron microscopy, we show that synapsin is instrumental for resisting vesicle dispersion and serves as a regulatory element for controlling lateral vesicle sharing between synapses. Deleting synapsin disrupts the organization of presynaptic vesicle clusters, making their boundaries hard to define. Concurrently, the fraction of vesicles amenable to transport is increased, and more vesicles are translocated to the axon. Importantly, in neurons from synapsin knock-out mice the resting and recycling pools are equally mobile. Synapsin, when present, specifically restricts the mobility of resting pool vesicles without affecting the division of vesicles between these pools. Specific expression of synapsin IIa, the sole isoform affecting synaptic depression, rescues the knock-out phenotype. Together, our results show that synapsin is pivotal for maintaining synaptic vesicle cluster integrity and that it contributes to the regulated sharing of vesicles between terminals.
Introduction
A prominent feature of most presynaptic terminals is the presence of large synaptic vesicle (SV) clusters next to the active zone (De Robertis and Bennett, 1955; Palay, 1956; Siksou et al., 2007). The availability of SVs for release is a key determinant of presynaptic function and serves as an important locus for regulating synaptic strength (Pan and Zucker, 2009). Recently it was shown that recycling SVs traffic along the axon between synapses and incorporate into new host terminals (Darcy et al., 2006a; Staras et al., 2010), extending the conventional view beyond the perception that transported vesicles serve mostly as building blocks for new synaptic contacts (Ahmari et al., 2000). Consequently, the synapse-spanning vesicle superpool was introduced alongside the readily releasable pool, the recycling pool, and the resting pool (Denker and Rizzoli, 2010). It is still poorly understood how these pools interchange vesicles. Currently, the synaptic terminal is viewed as a structure sculpted bidirectionally by dispersive and conservative processes (Tsuriel et al., 2006, 2009). Therefore, understanding vesicle mobility is of considerable interest. To clarify, we distinguish between intrasynaptic and intersynaptic SV mobility. The former has been probed by fluorescence-fluctuation-analysis (Jordan et al., 2005), fluorescence recovery after photobleaching (FRAP) (Gaffield et al., 2006), and most recently by stimulated-emission-depletion-microscopy (Kamin et al., 2010). The latter, which is the subject of the present work, was examined by whole-terminal FRAP in hippocampal neurons (Darcy et al., 2006a; Staras et al., 2010).
The synapsins are presynaptic proteins proposed to cluster SVs based on their dual interaction with SVs and cytoskeletal elements (Greengard et al., 1993). Synapsin triple knock-out (TKO) neurons contain markedly fewer vesicles at sites close to the synaptic active zone (Gitler et al., 2004a; Siksou et al., 2007), which may explain the accelerated synaptic depression observed in TKO excitatory neurons (Gitler et al., 2004a). Acute perturbation of the synapsins by antibodies (Pieribone et al., 1995) or interfering peptides (Hilfiker et al., 1998, 2005) also produces vesicle loss and synaptic transmission defects. This textbook view has been contested (Gaffield and Betz, 2007), and alternative functions, including stabilization of SV structure (Rosahl et al., 1995), or a role in endocytosis (Bloom et al., 2003) were suggested. Here we evaluate the role of synapsins in regulating SV distribution using light and electron microscopy, and their effect on intrasynaptic SV mobility using FRAP. We show that in the absence of synapsins, SV clusters at terminals have loose cohesion, and SVs are overrepresented in adjoining axonal segments. Furthermore, live imaging shows that deletion of the synapsins substantially increases the intersynaptic mobility of SVs. We demonstrate that it is specifically the resting vesicles that the synapsins immobilize. Surprisingly, and contrary to the original perception, these proteins do not determine the division of vesicles between the resting and recycling pools. We suggest that the synapsins contribute to the regulation of the total SV population by immobilizing resting vesicles, but that the size of the recycling pool is proportionally maintained by a separate unidentified mechanism.
Materials and Methods
Solutions, materials, and antibodies
Solutions.
Normal saline (in mm): NaCl 150, KCl 3, HEPES 10, CaCl2 2, MgCl2 2, and glucose 20 at pH 7.35. Hyperkalemic saline (in mm): like normal saline except NaCl 63, KCl 90. NH4Cl saline (in mm): like normal saline except NaCl 100, NH4Cl 50.
Materials.
2-Amino-5-phosphonopentanoic acid (APV) and 6,7-dinitroquinoxaline-2,3(1H,4H)-dione (DNQX; Sigma-Aldrich), FM 1–43/SynaptoGreen C4 (Biotium), ADVASEP-7 (CyDex), bafilomycin A1 (Enzo Life Sciences), and roscovitine (EMD Chemicals).
Antibodies.
Rabbit polyclonal anti-synaptobrevin 2 (Syb2; 1:2000) and mouse monoclonal anti-Bassoon (1:1000) (Synaptic Systems) and donkey anti-mouse IgG and donkey anti-Rabbit IgG, labeled with Northern Lights 637 or 557, respectively (1:1000) (R&D Systems).
Mice and Neuronal Cultures
Synapsin TKO mice and wild-type (WT) controls (Gitler et al., 2004b), both on a C57BL6/129 background, were grown at the Ben-Gurion University mouse facility. Animals were treated in accordance to the guidelines of the Ben-Gurion University Institutional Committee for Ethical Care and Use of Animals in Research. Hippocampal cultures from P0–P2 pups of either sex were performed essentially as described previously (Gitler et al., 2004b). Cells were grown for 11–18 d before imaging.
Molecular constructs, transfections, and infections
Viral particles were prepared as described previously (Groh et al., 2008) in HEK cells using both the pD1 and pD2 helper plasmids. cDNAs of interest—EYFP-synapsin Ia, EGFP-synapsin IIa, synaptophysin I-EGFP (SypI-EGFP), TagBFP-Synapsin IIa—were subcloned into a plasmid containing adeno-associated virus 2 inverted terminal repeats flanking a cassette consisting of the 1.1 kb cytomegalovirus enhancer/chicken β-actin promoter, the woodchuck post-transcriptional regulatory element, and the bovine growth hormone polyA. Inserts were verified by sequencing. Virions were added to neurons at 5 d in vitro and cells were incubated for at least 6 d before experimentation. Virion titer was adjusted to produce 75–90% infection efficiency. Plasmid-based transfections were performed as described previously (Gitler et al., 2004b), using lipofectamine 2000 (Invitrogen). EGPF and EYFP were from Clontech and TagBFP from Evrogen. SypI-EGFP viral vectors were kindly provided by Thomas Kuner (Heidelberg University, Heidelburg, Germany). Synaptophysin I-2XpHluorin expression plasmids were kindly provided by Ruud Toonen (Vrije Universitat, Amsterdam, The Netherlands).
Fluorescence Microscopy
Imaging was performed on two imaging systems. FRAP experiments used a C1si spectral confocal microscope driven by the EZC1 software package (Nikon). The scanhead was mounted on a Nikon FN1 microscope, equipped with a 1.0 NA 60× water dipping objective. The 488 nm line of a multiline 65 mW argon laser was used for excitation of EGFP, EYFP, and FM1–43; emissions were filtered with a 510–560 nm bandpass filter (Chroma); and detection was performed using the confocal's photomultiplier tube. TagBFP was excited using a 405 nm solid state diode laser, and unfiltered emissions were detected using the C1si's spectral detector box. Other measurements were performed on a Nikon TiE inverted microscope driven by the NIS-elements software package (Nikon). The microscope was equipped with a CoolSnap HQ2 14 bit CCD camera (Roper Scientific); an ×40 0.75 NA Super Fluor objective; an ×60 1.4 NA oil-immersion apochromatic objective (Nikon); a perfect-focus mechanism (Nikon), and EGFP, EYFP, and Cy3 TE-series optical filter sets (Chroma).
Semiquantitative synaptic immunofluorescence
Neurons were fixed with 4% paraformaldehyde (EMS) in PBS for 10 min, rinsed with PBS, permeabilized with 0.1% Triton X-100 in PBS for 2 min, blocked with 5% powdered milk in PBS for 1 h, rinsed, incubated with the primary antibody for 1 h, rinsed, incubated with the secondary antibody for 1 h, rinsed, and mounted in immu-mount (Thermo Fisher Scientific). To allow for semiquantitative comparison of immunostaining intensity of synapses, experimental and control groups were processed under identical conditions using the same reagents. To avoid experimenter bias, images of WT and synapsin TKO neurons were acquired and analyzed by an experimenter blind to both the experimental question and the phenotype of the neurons. Synapses were semi-automatically detected using an in-house iterative algorithm based on serially decreasing thresholds as described previously (Bergsman et al., 2006) and implemented in NIS-elements. Fluorescence values for each synapse were obtained from an area of 2 × 2 pixels located on its center of mass and an image average was generated. Because intensity values of the controls can vary from session to session, a normalization value was determined from the control experiments of each session, and this value was used to normalize all images acquired during that session. In the case of experiments in which we assessed the effect of EGFP-synapsin IIa on the intensity of the Syb2 signal, colocalization of the two signals was visually verified during the analysis process.
Synapse width analysis
Synapse width was measured by drawing a line starting in the axon and through the synapse punctum, and fitting it using a Gaussian function as follows (Orenbuch et al., 2012):
 |
where xc is the center of the punctum maximum, y0 is the fluorescence of the axon, w2 is the variance of the Gaussian, and A its amplitude. The full-width at half-maximum (FWHM) is as follows:
 |
Fitting was performed by the least-squares error method using Origin (OriginLab). Independent images were acquired from cells grown on various coverslips obtained from at least three different cultures. A mean FWHM was determined for each image, and then a grand average was calculated for each condition. Image acquisition and analysis of the width of Syb2 in WT and synapsin TKO neurons were performed using a double-blind protocol. We note that the variance of a Gaussian should not be confused with the area of a synapse, since the apparent area is affected by the Gaussian's amplitude while the variance is not.
Vital vesicle imaging
Neurons were loaded with FM1–43 (10 μm) by its application for 2 min in hyperkalemic saline, followed by wash in FM1–43 containing normal saline for 5 min (Ryan et al., 1993). Subsequent washing was with normal saline for 5 min followed by 5 min in normal saline supplemented with 1 mm ADVASEP-7 (Kay et al., 1999). Unloading was by the application of hyperkalemic saline for 5 min. Experiments were performed at room temperature in normal saline to which APV (50 μm) and DNQX (10 μm) were added to prevent network activity propagation. Background-corrected FM1–43 loading was measured in sister WT and TKO cultures, and was normalized in each imaging session by the average value measured in WT neurons. The unloading percentage of each synapse was calculated as follows:
 |
where ΔF is the fluorescence lost during unloading, and F0 is the background-corrected loading intensity.
Alkaline trapping (Sankaranarayanan and Ryan, 2001) of Synaptophysin I-2XpHluorin was performed by depolarizing transfected neurons with hyperkalemic saline in the presence of bafilomycin A1 (0.5 μm) for 5 min. The total vesicle population was visualized by subsequent application of NH4Cl saline. The fraction of the resting pool is as follows:
 |
where Ftrap is the fluorescence intensity during alkaline trapping, F0 is the intensity of the same synapse before depolarization, and Ftotal is the fluorescence during the application of NH4Cl saline.
FRAP
FRAP measurements were performed using a Zoom-FRAP protocol implemented on the EZC1 software package on the confocal microscope. Each bleaching session consisted of the acquisition of five prebleaching images at 1/5 Hz at 5% power, followed by sequentially zooming-in by a factor of 25.6 onto three selected synapses, each scanned twice with the laser set to 20% power, resulting in a theoretical 2600-fold increase in power. Bleaching conditions were chosen to produce a depth of bleach of 75–95%, as recorded during the first postbleaching image, without significantly affecting neighboring synapses. Recovery was monitored by successively acquiring 5-plane z-stacks at the original power and magnification (Δz = 0.6 μm), manually adjusting for focus drift as necessary. Imaging frequency was 12 images at 1/5 Hz and 30 images at 1/15 Hz, and for long recovery sessions (see Fig. 4), also 10 images at 1/90 Hz followed by 7 images at 1/150 Hz. Fluorescence intensity values were obtained from the bleached synapses as well as from five additional unbleached ones. In each case, the plane in each z-stack that was maximally in focus was chosen for extraction of data. To correct for imaging-related bleaching, the fluorescence value measured at each bleached synapse was normalized by the average intensity of the nonbleached synapses. The imaging-related bleaching of SypI-EGFP was 12 ± 4% at the end of a 41 min imaging session, while for FM1–43 bleaching was of 26 ± 3% in 8.5 min. No correlation was found between the rate of FRAP and either the depth of bleach or the initial fluorescence value. Therefore, for each synapse we scaled to 1 the average value during the prebleach stage, and to 0 the value measured immediately after bleaching. The recovery traces of the three synapses in each experiment were averaged, and a grand average was calculated for each condition. Experiments were performed on at least three independent cultures. No systematic differences were observed within each group for data obtained during different imaging sessions. FRAP experiments were performed in the presence of APV (50 μm) and DNQX (10 μm) to prevent propagation of network activity.
Figure 4.
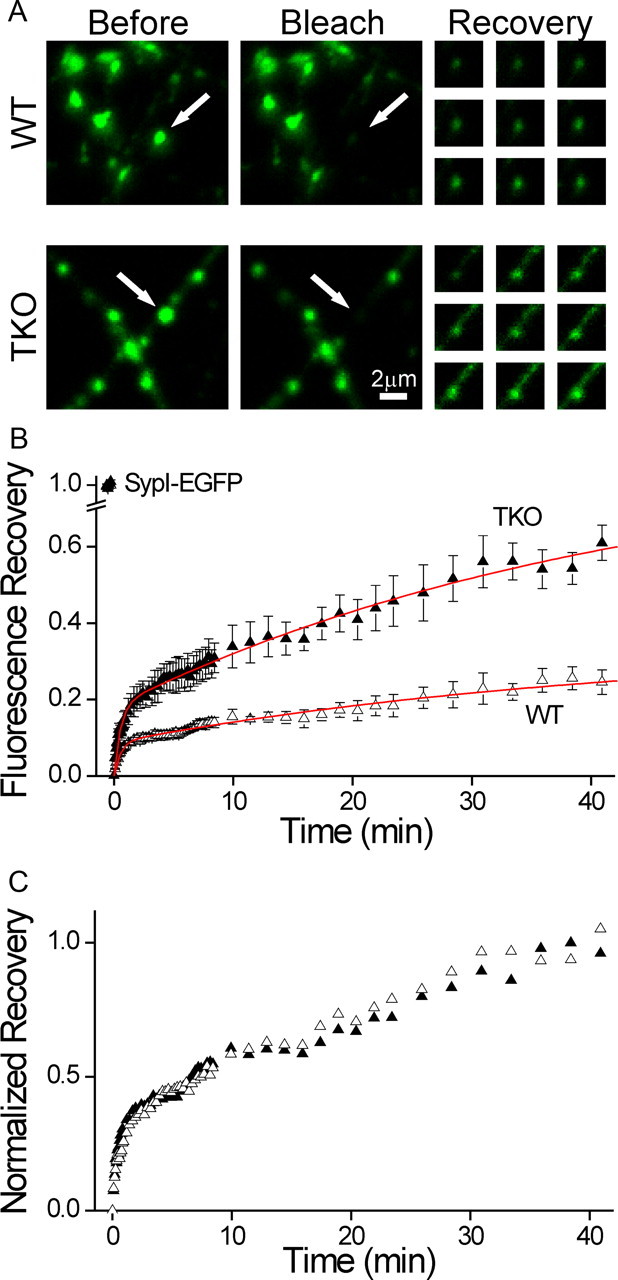
The fraction of mobile synaptic vesicles is determined by the synapsins. Mobility of synaptic vesicles was probed by FRAP of SypI-EGFP. A, Representative FRAP experiments in WT (top) and synapsin TKO (bottom) neurons. Shown is a field including several synaptic puncta before (left) and immediately after (middle) bleaching of a single punctum (white arrow). The fluorescence of the affected synapse is reduced to >85% of its original value, while neighboring synapses are unaffected. The recovery of fluorescence is shown at right 1, 6, 11.5, 16, 20.5, 26, 31, 36, and 41 min after bleaching, with the contrast homogeneously increased to improve visibility. Recovery in the synapsin TKO neuron is noticeably faster. B, Average time course of FRAP experiments in WT (open triangles) and TKO (solid triangles) synapses (mean ± SEM). Intensity data are normalized by the initial fluorescence (leftmost symbols), and lowest value is shifted to 0. Sampling was performed at intervals of 5, 15, 90, and 150 s. The data are corrected by changes in the intensity of unbleached synapses in distant areas of the image. Red lines represent global fitting of the data with a double exponential function, where the time constants are shared. Recovery in TKO neurons is more extensive. C, Same data as in B, normalized by the average of the three end points. Recovery proceeds with equal kinetics, as evidenced also by the global fit lines in B.
Fitting of FRAP traces (Fig. 4) was performed by least-squares global fitting (Origin) using the following formula:
 |
where A1 and A2 are amplitudes and τ1 and τ2 are shared time constants.
Electron microscopy
Cultured neurons were fixed and processed for electron microscopy as described previously (Darcy et al., 2006b). Embedded samples were sectioned (thickness, 70 nm) and collected on formvar-coated slot grids. Sections were viewed using a Hitachi 7100 transmission electron microscope and images were acquired using a 2048 × 2048 CCD camera (Gatan). For analysis of vesicle organization along axons, we randomly selected regions of a section and identified areas containing discrete synaptic terminals which had a defined postsynaptic target and a length of flanking axon running parallel to the plane of the section. Electron micrographs of the same target region were collected for all available sections. To allow us to make a comparative analysis of vesicle distribution in the axon for WT versus TKO, we first determined the average vesicle cluster boundary in WT neurons by measuring the distance from the center of the active zone to the point where vesicle separation exceeded 200 nm in the longitudinal axis of the axon (550 ± 77 nm, n = 22 synapses from three coverslips). This distance was then marked in all aligned synapse–axon series (WT and TKO), and the average number of vesicles per section was determined for 200 nm bins extending away from the synapse. We also examined the vesicle profile within the presynaptic terminal for each synapse using a representative central section which included the defined vesicle cluster and a flanking length of axon. The longitudinal distance between each vesicle and the active zone center was then measured and plotted in 150 nm bins, extending out to the defined synaptic boundary. Micrographs were aligned and reconstructed using Xara Xtreme and Reconstruct (Synapse Web; K. M. Harris, http://synapses.clm.utexas.edu/).
Statistics
Mean ± SEM values are reported throughout. Comparisons were performed by the Student's t test after confirming a normal distribution by the Shapiro–Wilk normality test or with Mann–Whitney's nonparametric test otherwise. Multiple comparisons of normally distributed data were performed using one-way ANOVA followed by Tukey's post hoc tests. Comparison of FRAP traces were performed using repeated-measures ANOVA. Difference in variance was tested using Levene's test for equal variance. Results of the test were deemed significant at a confidence level of 0.05. Statistical analysis was performed with Origin or SPSS (IBM).
Results
Deletion of the synapsins alters the distribution of synaptic vesicles
To gain an insight into the organization of vesicles in presynaptic terminals of synapsin TKO neurons, we examined immunofluorescence staining for the synaptic vesicle protein Syb2 (Takamori et al., 2006, Fig. 1A) and compared it to that observed in WT preparations. We began by examining the maximal signal intensity at the center of each synaptic punctum, a measure which represents the density of vesicles in the immediate area of the active zone (n = 75/>10,000 images/synapses for both genotypes; Fig. 1B). We found that in synaptic puncta of TKO neurons the Syb2 intensity was significantly lower (72.2 ± 3.0% as compared with 100 ± 4.1% in WT neurons, p < 0.001 for comparison of image averages, Student's t test). This finding is consistent with the lower vesicle count observed within the presynaptic terminal of TKO neurons (Gitler et al., 2004a; Siksou et al., 2007). Examination of the spatial distribution of Syb2 revealed that the synaptic puncta in TKO and WT neurons were morphologically distinct, with the puncta of TKO neurons appearing elongated compared with the compact appearance in WTs. To objectively quantify this observation, we extracted the width of the synaptic puncta from Gaussian functions that were fitted onto longitudinal line profiles of their fluorescence. The synaptic puncta of TKO neurons (n = 23/715) were significantly wider than those of WT neurons (n = 21/674, p = 0.0002, Fig. 2A,B), indicating that deletion of the synapsins perturbs synaptic structure. The effect was specific to synaptic vesicles, since puncta of the active-zone protein Bassoon were unaffected by deletion of the synapsins (p = 0.155; WT, n = 14/372; TKO, n = 13/354; Fig. 2A,B) (Rosahl et al., 1995). Repeating the experiment with SypI-EGFP (Fig. 2C) allowed us to examine vesicle distribution in live neurons. In agreement with the analysis of Syb2, the width of SypI-EGFP puncta was significantly larger (p < 0.001, Student's t test) in TKO (1.35 ± 0.03 μm, n = 8/157) versus WT (1.12 ± 0.02 μm, n = 13/326) neurons. To probe the stability of synaptic puncta over time, we calculated Δw/w0, the fractional change in the width of SypI-EGFP puncta in images acquired 10 min apart (Fig. 2D). On average, the width of the puncta was stable in both WT (n = 13/321, p = 0.23, single-sample Student's t test comparison to 0) and TKO (n = 8/253, p = 0.98) neurons. However, we observed that the variance of Δw/w0 in TKO neurons was significantly larger (SD = 0.21 vs 0.36 for WT and TKO, respectively; p < 0.001, Levene's test for equal variance), suggesting that the width of TKO synapses fluctuates more. Our observations therefore support the idea that deletion of the synapsins destabilizes the SV clusters, allowing redistribution of the vesicles from the vicinity of the active zone into the axon.
Figure 1.

Synaptic vesicle density is reduced in presynaptic terminals of synapsin TKO neurons. A, Syb2immunofluorescence image of cultured WT (left) and synapsin TKO (right) neurons. TKO synapses are dimmer than WT synapses. B, Cumulative probability plots of the normalized intensity of synaptobrevin synaptic puncta of WT and synapsin TKO neurons. The intensity was determined at the center of mass of each synapse and was normalized by the average per-image value of WT synapses in each experimental session.
Figure 2.
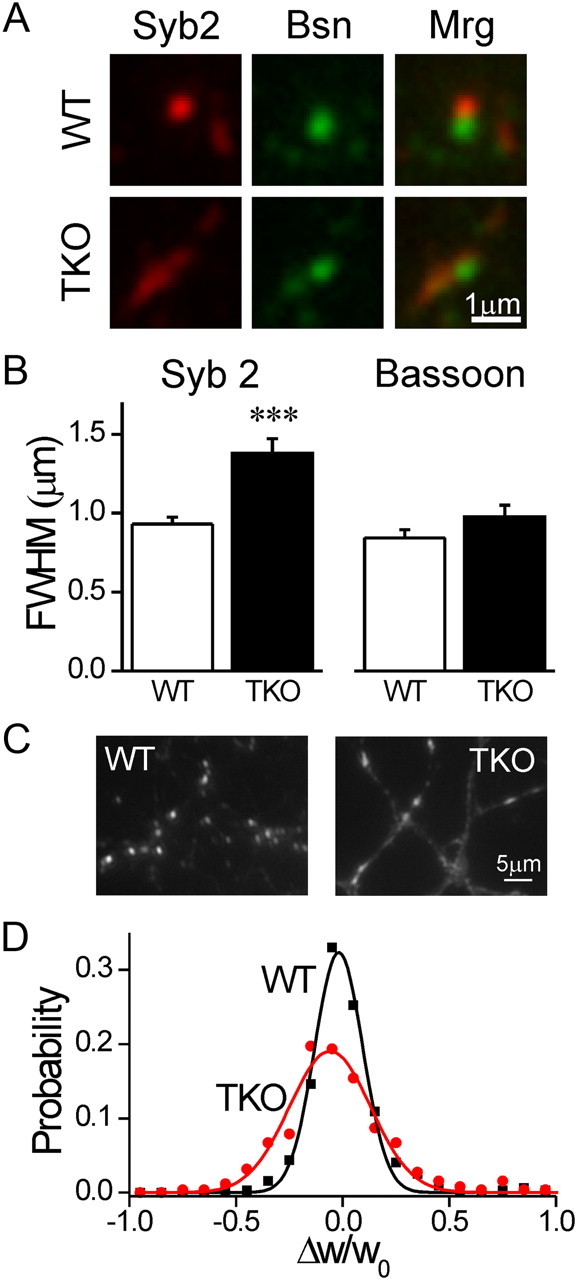
Synaptic vesicle clusters are dispersed in synapsin TKO neurons. A, Immunostaining forSyb2 (red) and Bassoon (Bsn, green) in WT (top) and synapsin TKO (bottom) neurons. Merge (Mrg) images appear at right. Bassoon staining, representing the active zone, appears similar in both genotypes, while Syb2 staining, representing the vesicle cluster, is spread out. B, Average FWHM width of Syb2 (left) and Bassoon (right) puncta (mean ± SEM). C, Representative images of SypI-EGFP puncta in WT (left) and synapsin TKO (right) neurons. Notice that in TKO neurons SypI-EGPF is distributed along longer segments of the axon and that the axon is more readily seen. D, Fractional change in the width of puncta over a period of 10 min. The distribution of Δw/w0 values, representing the change in synaptic width, indicates that while synapses of both genotypes tended to maintain their dimensions, the variability in TKO puncta was larger, even considering that they were wider to begin with. ***p < 0.001, Student's t test.
Ultrastructural analysis reveals vesicle redistribution into axons of synapsin TKO neurons
Previous investigations of the ultrastructure of synapsin KO synapses have reported a reduction in the number of vesicles in the presynaptic terminal in the vicinity of the active zone (Li et al., 1995; Rosahl et al., 1995; Gitler et al., 2004a; Siksou et al., 2007). However, these studies did not examine whether vesicles were lost or instead redistributed, an idea that would be consistent with the lower fluorescence intensity and broadened spatial distribution of synaptic puncta in TKO neurons (Figs. 1, 2). We addressed this issue by carrying out a quantitative ultrastructural analysis of the longitudinal distribution of synaptic vesicles along the main axis of the axon, specifically within synapses and their flanking axonal segments (n = 22 and n = 16, from WT and TKO cultures, three coverslips each, Fig. 3). In agreement with our light microcopy analysis (Fig. 1) and with previous reports (Gitler et al., 2004a; Siksou et al., 2007), the number of vesicles located within 150 nm of the center of the active zone was significantly lower (down to 64.4%) in TKO neurons (p = 0.02; Fig. 3D). In both groups, the number of vesicles decreased with increasing distance from the center of the active zone (Fig. 3D). However, in TKO synapses the decrease was markedly shallower, so that at distances >400 nm the vesicle numbers were higher than in WT neurons. Consecutive longitudinal serial-section analysis of the axon confirmed that the number of vesicles in the TKO axon was significantly higher (averaged across 10 bins, each 200 nm wide: WT, 0.49 ± 0.07 vesicles and TKO, 1.80 ± 0.16 vesicles; Mann–Whitney test; p < 0.02; Fig. 3D). Thus, while synaptic vesicles in WT synapses are tightly clustered near the active zone, TKO synapses have a broader spatial profile with fewer vesicles adjacent to the active zone and more in perisynaptic regions. This observation supports the aforementioned hypothesis that the loss of synapsins destabilizes the synaptic vesicle cluster, resulting in vesicle movement away from synaptic sites into intersynaptic axonal segments. Our findings are consistent with recent results showing that vesicles are constitutively transported between synapses (Darcy et al., 2006a; Fernandez-Alfonso and Ryan, 2008; Staras et al., 2010) and we propose here that a larger fraction of synaptic vesicles belongs to the superpool in TKO versus WT neurons.
Figure 3.

Ultrastructural examination of dispersal of synaptic vesicles in neurons lacking synapsins. A, Longitudinal sections through presynaptic contacts and flanking axonal segments (light blue) abutting upon postsynaptic targets (light orange) in WT and synapsin TKO neurons from hippocampal cultures. The synaptic contacts are aligned at the left of the image. The centers of synaptic vesicles are marked by black dots; clustering of vesicles is evident. B, Three-dimensional reconstruction of the synapses shown in A, with vesicles represented by brown spheres. C, Further examples of synapse–axon reconstructions for WT and TKO neurons. The green line represents the center of the active zone. Distance markers at a spacing of 200 nm are shown starting at 550 nm from the center of the active zone, which marks the average boundary of the vesicle cluster in WT synapses. Notice the larger presence of vesicles in the axons of synapsin TKO neurons. D, Vesicle density within the presynaptic terminal (left) and along the axon (right, mean ± SEM). Compared with WT, the average number of vesicles near the active zone in TKO neurons is significantly lower, but at distances larger than ∼400 nm from the center of the active zone, this relationship is reversed. The number of vesicles in the axon is consistently higher in synapsin TKO neurons. *p < 0.02, Mann–Whitney test in both graphs. The schematics above the plots indicate the regions of presynaptic terminal and axon being examined.
Synapsins immobilize synaptic vesicles
To provide a direct readout of vesicle mobility, we measured FRAP of SypI-EGFP (Fig. 4). Considering that on average the quantity of vesicles in the synaptic puncta is stable (Fig. 2D), the extent of fluorescence recovery in FRAP experiments is indicative of the relative (and not absolute) fraction of vesicles in the puncta that are replaced with vesicles transported from adjacent unbleached areas (Darcy et al., 2006a). After the bleach pulse, the fluorescence of SypI-EGFP recovered over a time scale of tens of minutes in a biphasic manner (Fig. 4B), which is comparable to that reported previously (Darcy et al., 2006a). Recovery was significantly more pronounced in synapsin TKO neurons (n = 5/14) than in WT neurons (n = 6/17, p < 0.001, repeated-measures ANOVA; Fig. 4B). This result illustrates that a larger fraction of the vesicles in the synaptic puncta of TKO neurons was replaced, resulting in a significantly higher end-point value, and indicating that the vesicles in TKO synapses are more mobile. The higher mobility could result from a change either in the transport rate of the individual vesicles or in the proportion of mobile versus immobile vesicles. To differentiate between these possibilities, we renormalized the recovery plots by the average of the last three time points. The plots fully overlapped (p = 0.84, Fig. 4C), indicating that the transport rate of mobile vesicles is equivalent in both groups and probably reflects inherent properties of axonal transport (Scott et al., 2011). Based on our results we conclude that the synapsins immobilize a subgroup of vesicles within the synaptic puncta. To estimate the mobile fraction of vesicles in WT and TKO neurons, we extrapolated both datasets assuming a double exponential model (using Eq. 5 in Materials and Methods) with shared (global) time constants (Fig. 4B, red lines; R2 = 0.99). The fit yielded the shared time constants τ1 = 0.52 ± 0.03 min and τ2 = 42.9 ± 5.8 min, and the coefficients A1 = 0.087 ± 0.003 and A2 = 0.258 ± 0.023 for WT and A1 = 0.181 ± 0.004 and A2 = 0.668 ± 0.056 for TKO synapses. Based on the sum of the two coefficients we estimate the fraction of mobile vesicles as 34 ± 3% in WT neurons versus 85 ± 6% in TKO neurons.
Synapsin is transported independently of synaptic vesicles
The restriction of vesicle mobility by synapsins could stem from the formation of stable synapsin–vesicle complexes within the presynaptic terminal. However, a model of synaptic tenacity, wherein synaptic structure is maintained dynamically in the face of continuous replacement of its components, has been recently proposed (Tsuriel et al., 2006, 2009). To differentiate between these possibilities, we contrasted the mobility of both components. Exogenous EYFP-synapsin Ia clearly colocalizes with markers of SVs (Fig. 5A; Gitler et al., 2004b), illustrating its synaptic localization. In agreement with the dynamic view of the synapsin–SV interactions, we found that EYFP-synapsin Ia recovery is significantly more pronounced than that of SypI-EGFP in WT neurons (p < 0.001; Fig. 5B,C). Comparison of the movement rate of the mobile fractions of synapsin Ia and synaptophysin I by end normalizing the recovery traces revealed no difference between them (Fig. 5D), arguing against a model in which synapsin recovery is more substantial due to faster transport. These results suggest that the mobile fraction of synapsin in the presynaptic puncta is substantially larger than that of the vesicles implying that synapsin is replaced independently of the vesicles.
Figure 5.
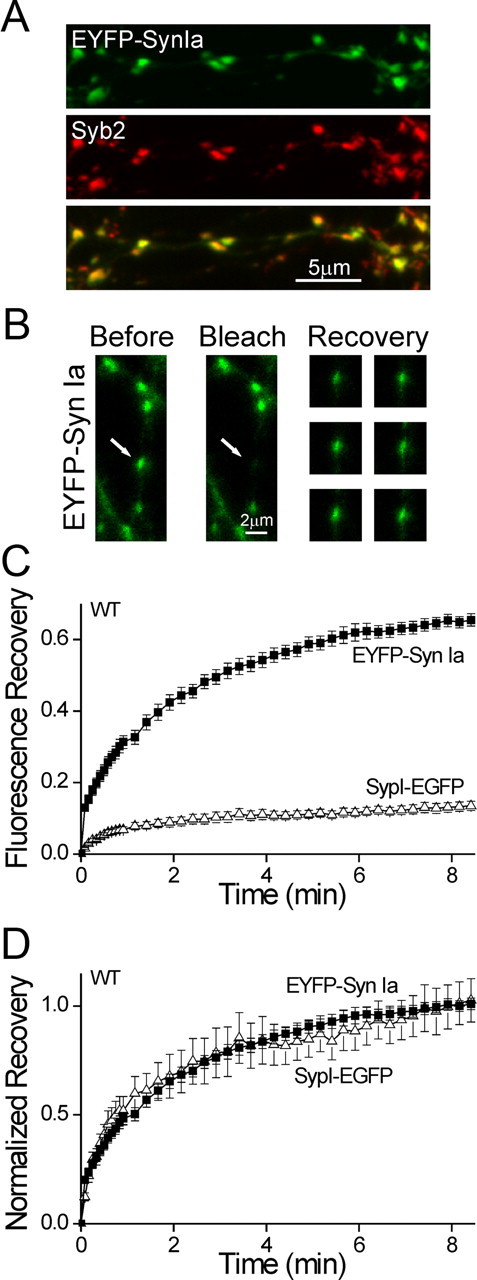
Intersynaptic transport of synapsin is faster than that of the vesicles. A, EYFP-Synapsin Ia (green) expressed in a WT neuron immunostained for Syb2 (red). Exogenous synapsin Ia localized to synapses, indicating that FRAP measures the intersynaptic transport of synapsin. B, Representative FRAP experiment in a WT neuron expressing EYFP-synapsin Ia. A single punctum (white arrow) is bleached, without affecting neighboring synapses. The recovery of fluorescence is shown at right 90, 165, 255, 330, 420, and 510 s after bleaching. The contrast of the recovery images was increased homogeneously to enhance visibility. C, Comparison of time course of FRAP of EYPF-synapsin Ia (EYPF-Syn Ia; solid squares) and SypI-EGFP (open triangles) in WT neurons (mean ± SEM). A shorter time period is shown than in Figure 4. Recovery of synapsin is markedly more extensive, indicating that synapsin Ia is transported independently of the vesicles. D, Same data as in C, normalized by the average of the three end points. Recovery proceeds with equal kinetics, indicating that the rate of movement of the mobile fractions of both labels is similar.
Synapsins interact differently with recycling and resting vesicle pools
Previous work has concluded that synapsins do not play a significant role in vesicle immobilization based on the similarity of FRAP of FM1–43 loaded vesicles in the neuromuscular junction of WT and TKO mice (Gaffield and Betz, 2007). Our finding that the synapsins drastically affect vesicle mobility (Fig. 4) is in apparent contradiction with this study. We reasoned that the difference could arise from the fact that different pools of vesicles were labeled in the two studies: while FM1–43 labels specifically the recycling pool (Betz and Bewick, 1992), SypI-EGFP labels all vesicles. To address this hypothesis directly, we directly compared FRAP of FM1–43 and of SypI-EGFP in our system (Fig. 6). We found that in WT neurons, recovery of FM1–43 (n = 11/23) was significantly larger than that of SypI-EGFP (n = 22/65, p = 0.001; Fig. 6A), suggesting that in WT neurons recycling vesicles are more mobile than nonrecycling (resting) ones (Kamin et al., 2010). In contrast, no difference was observed between recovery of FM1–43 (n = 12/30) and of SypI-EGFP (n = 13/37) in TKO neurons (p = 0.31; Fig. 6B), implying that synapsins are the agent immobilizing the resting vesicles. Moreover, consistent with results in mouse neuromuscular junction, no difference was observed for recovery of FM1–43 in TKO and WT neurons (p = 0.75; Fig. 6C), implying that the synapsins do not play a role in confining recycling vesicles. We thus conclude that the synapsins specifically restrict the movement of resting SVs, and that since SypI-EGFP visualizes all vesicles, FRAP of SypI-EGFP represents an overestimation of the mobility of the resting pool. These results are also consistent with the observed width of FM1–43 and SypI-EGFP puncta in WT and TKO neurons (Fig. 6D). The width of the FM1–43 puncta was similar (p = 0.86) in WT (n = 5/244) and TKO (n = 6/270) neurons. Both were wider (p = 0.001) than the SypI-EGFP puncta in WT (n = 13/321) neurons, but similar (p = 0.8) in width to the SypI-EGFP puncta in TKO neurons (n = 13/253). Because the resting and recycling pools together compose the total pool, its apparent FWHM (which does not equate with its area; see Materials and Methods, Eq. 1) is intermediate to that of its two components, implying that the distribution of the resting pool in WT neurons is even narrower. Our observations are therefore consistent with the idea that the mobility of vesicles contributes to their longitudinal distribution.
Figure 6.
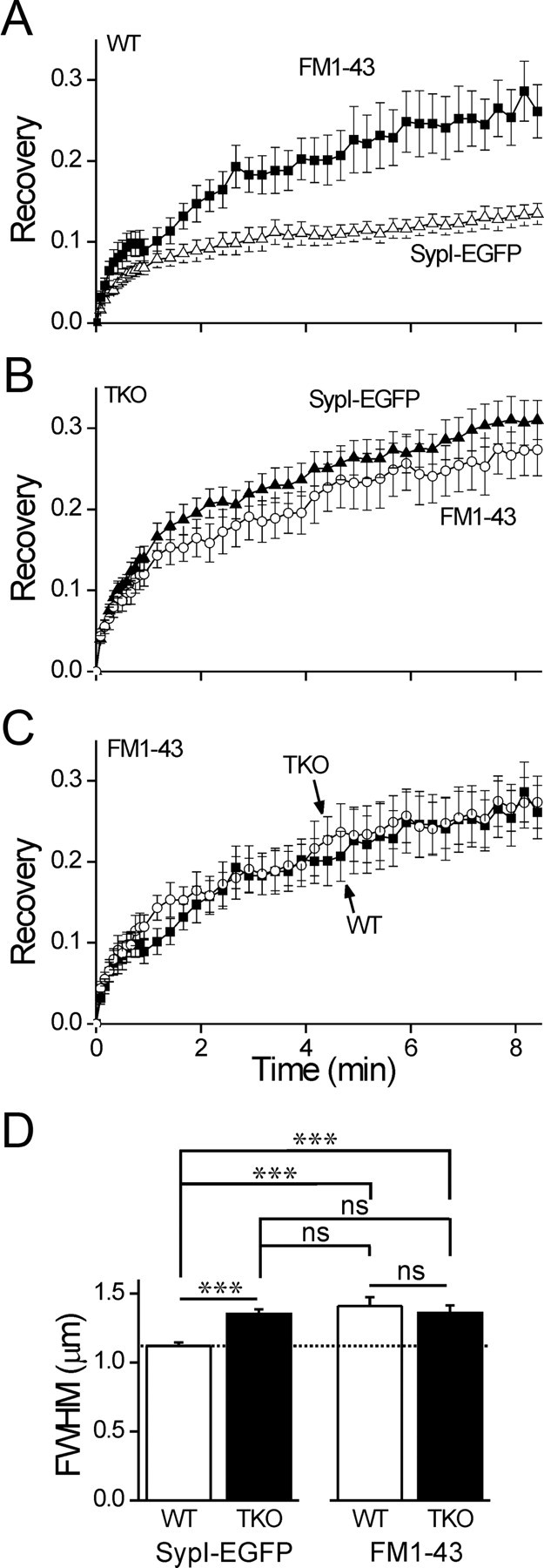
Specific immobilization of the resting pool of vesicles by synapsin. A, Comparison of the time course of FRAP of recycling vesicles loaded with FM1–43 (solid squares) and of the total pool of vesicles as visualized by SypI-EGFP (open triangles) in WT neurons (mean ± SEM). A shorter time period is shown than in Figure 4. B, FRAP of FM1–43 (open circles) and SypI-EGFP (solid triangles) in TKO neurons. C, FRAP of FM1–43 in WT (solid squares) and TKO (open circles) neurons. D, Width analysis of FM1–43 loaded vesicles in WT and TKO neurons, compared with width of SypI-EGFP label. The width of SypI-EGFP in WT neurons (dashed line) is narrower than that of the other groups, all of which are similar in width. ns, p > 0.05; ***p < 0.001, one-way ANOVA, using Tukey's post hoc test.
Synapsins do not define resting/recycling status of vesicles
Based on our observation that the synapsins interact specifically with SVs in the resting pool, it might be expected that deletion of the synapsins should increase the proportion of recycling pool vesicles at the expense of the resting pool. This could happen due to two different reasons: either the synapsins directly define the status of the SVs as resting or recycling, or a larger fraction of the resting vesicles is lost into the axon of TKO neurons due to their higher mobility. To assess this possibility, we first measured the size of the recycling pool near the active zone using hyperkalemic loading of FM1–43 (Ryan et al., 1993). The peak FM1–43 intensity of synapses in TKO neurons (n = 29/925) was significantly weaker than in WT ones (n = 29/964, p < 0.001; Fig. 7A,B) (Gabriel et al., 2011), much like the decrease in the total vesicle population (Fig. 1). The extent of FM1–43 destaining was similar for both groups (WT, n = 24/790; TKO, n = 21/663; p = 0.38; Fig. 7A,C) (Gabriel et al., 2011), illustrating that loss of the synapsins does not affect the propensity of the recycling vesicles to be released. Our results using FM1–43 argue against a compensatory increase in the size of the recycling pool. However, because synapsin may participate in endocytosis (Bloom et al., 2003), which could affect FM1–43 loading, we also performed experiments to probe the recycling pool in an endocytosis-independent manner. For this we used the alkaline trapping method (Sankaranarayanan and Ryan, 2001; Fig. 7D) in neurons expressing Synaptophysin I-2XpHluorin (Granseth et al., 2006). The relative fraction of the recycling vesicles within the presynaptic terminals of cultured hippocampal neurons has been estimated to be ∼0.5 on average (Fernandez-Alfonso and Ryan, 2008; Kim and Ryan, 2010). Our findings reveal a comparable fraction, which was equivalent for WT (n = 11/225) and TKO synapses (n = 19/412, p = 0.77; Fig. 7E,F). Our results demonstrate that the synapsins do not participate in the allocation of vesicles into specific functional pools (but see Akbergenova and Bykhovskaia, 2010), contrary to the hypothesis stated above. From this conclusion it follows that the resting and recycling pools scale in relation to one another in a synapsin-independent manner.
Figure 7.
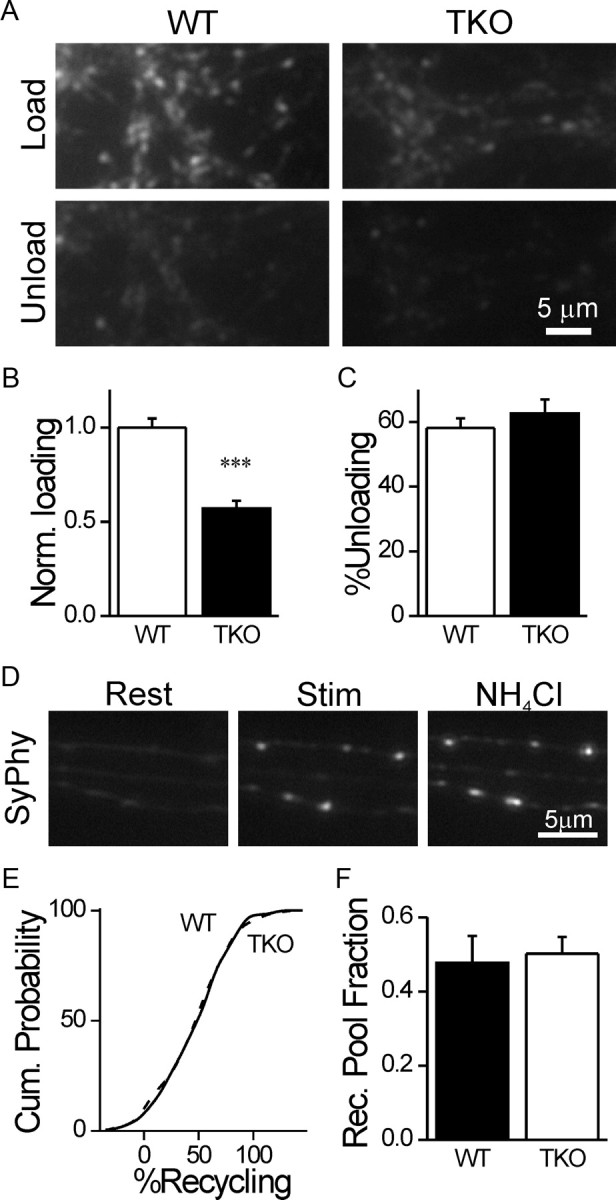
Subdivision of vesicles into resting and recycling pools is unaffected by the synapsins. A, Synapses loaded with FM1–43 (top) in WT (left) and synapsin TKO (right) neurons. Notice the brighter signal in WT neurons. The same fields are shown after unloading (bottom). B, Normalized intensity of FM1–43 intensity in loaded terminals (mean ± SEM), as shown in A. C, Percentage of FM1–43 unloaded by hyperkalemic stimulation. D, Representative images of synapses of WT neurons expressing Synaptophysin I-2XpHluorin at rest (left), after the recycling pool is revealed by hyperkalemic stimulation in the presence of bafilomycin A (middle), and after the total pool is visualized by alkalinization of all vesicles by NH4Cl saline (right). E, Cumulative probability plots of the fraction of the recycling pool out of the total population for all synapses analyzed. The plots overlap. F, Average of the mean values obtained from independent experiments. No difference is observed between WT and TKO neurons. ***p < 0.001, Student's t test.
Roscovitine increases vesicle mobility
Our results suggest that the synapsins do not determine the division of vesicles between the resting and recycling pools. In contrast, altering the balance between the activity of the kinase cdk5 and the phosphatase calcineurin has been shown to do so; inhibiting cdk5 increases the relative size of the recycling pool, while knocking down calcineurin has the opposite effect (Kim and Ryan, 2010). Because we found that resting and recycling vesicles differ in their mobility (Fig. 6), we reasoned that changing the ratio between these pools should alter the total vesicle mobility. To test this hypothesis, we measured FRAP of SypI-EGFP in neurons treated with roscovitine, a cdk5 inhibitor. We found that application of roscovitine (100 μm) to WT neurons (n = 13/39) 30 min before FRAP significantly enhanced vesicle mobility (p = 0.002; Fig. 8A). In contrast, in TKO neurons (n = 10/30), where the resting and recycling vesicles exhibit equal mobility (Fig. 6), roscovitine had no effect (p = 0.73; Fig. 8B).
Figure 8.
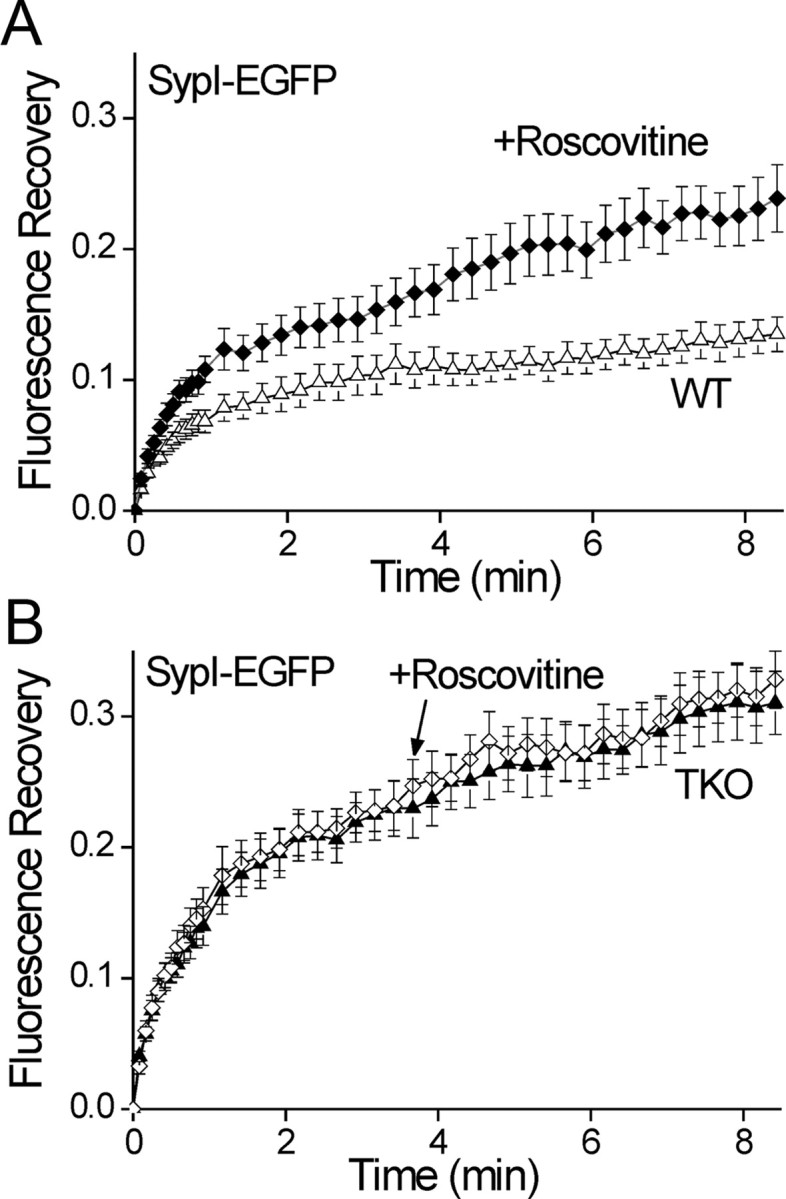
Roscovitine significantly enhances vesicle mobility. A, Comparison of the time course of FRAP of SypI-EGFP-labeled vesicles in WT neurons preincubated for 30 min with 100 μm roscovitine (solid diamonds) and control neurons (open triangles, mean ± SEM). Roscovitine significantly increased the fraction of mobile vesicles. B, as in A but in TKO neurons treated (open diamonds) or untreated (solid triangles) with roscovitine. No effect of roscovitine on the FRAP time course is evident.
Expression of synapsin IIa rescues the quantity, distribution, and mobility of synaptic vesicles
Our results show that deletion of the synapsins decreases the size of SV clusters and increases the overall SV mobility, but this could be the result of secondary compensatory processes unrelated to the synapsins. To test this possibility, we specifically examined the capability of exogenous synapsin IIa, the only isoform known to rescue synaptic depression in TKO hippocampal neurons (Gitler et al., 2008), to rescue SV distribution and mobility. We found that expression of EGFP-synapsin IIa in synapsin TKO neurons (n = 19/>6000) produced a significant (p = 0.0037) increase in the mean Syb2 fluorescence (Fig. 9A,B) as compared with uninfected TKO neurons (n = 25/>6000). Moreover, the width of the puncta in infected neurons (Fig. 9C; n = 10/177) was significantly reduced (p < 0.001) compared with uninfected ones (n = 10/169), indicating tighter clustering of the SVs. Finally, FRAP of SypI-EGFP was significantly decreased in neurons coinfected with synapsin IIa tagged with the blue fluorescent protein TagBFP (n = 10/29, p = 0.008, repeated measures ANOVA followed by Tukey's post hoc analysis; Fig. 9D,E) as compared with TKO ones (n = 13/37). Therefore, synapsin IIa could rescue the number, localization and immobilization of vesicles. Expression of EGFP-synapsin IIa in WT neurons, where native synapsins are present, significantly increased the fluorescence intensity of Syb2 (n = 19/>6000 as compared with n = 25/>6000 controls, p < 0.001; Fig. 9F,G), indicating that synapsin IIa could further augment the vesicle clusters. In contrast, it did not affect the width of the synaptic puncta (n = 21/548 as compared with n = 21/599 controls, p = 0.25; Fig. 9H). Finally, TagBFP-synapsin IIa did not accelerate FRAP of SypI-EGFP (n = 10/30, p = 0.51, repeated-measures ANOVA; Fig. 9J) as compared with control WT neurons (n = 22/65). Thus, the normal complement of synapsin IIa is sufficient to determine the form of the synaptic vesicle cluster and the fraction of vesicles that remain mobile within it.
Figure 9.

Synapsin IIa rescues the immobilization and clustering of vesicles in synapsin TKO neurons. A, Cumulative plot of Syb2 immunofluorescence in synaptic puncta of TKO neurons expressing EGPF-synapsin IIa compared with uninfected neurons. B, Average of the means of Syb2 signal calculated for independent images (mean ± SEM). EGFP-synapsin IIa increases the intensity of Syb2 staining. G, The data were normalized by the mean intensity observed in synapses of WT neurons. C, Average FWHM width of the Syb2 signal in synaptic puncta in TKO neurons expressing EGFP-synapsin IIa compared with uninfected neurons. D, Representative images of synapses in TKO neurons coexpressing TagBFP-Synapsin IIa (blue, left) and SypI-EGFP (green, right). E, Effect of coexpression of TagBFP-Synapsin IIa (filled squares) on the FRAP time course of SypI-EGFP in TKO neurons (filled triangles). TagBFP-Synapsin IIa reduced the mobility of the vesicles. F–J is the same as A–E except performed in WT neurons. Synapsin IIa increased the intensity of Syb2 puncta in WT neurons, but did not affect their width, or the time course of FRAP. **p < 0.01, ***p < 0.001.
Discussion
Resting and recycling SVs are constitutively transported along the axon between adjacent synapses (Darcy et al., 2006a; Fernandez-Alfonso and Ryan, 2008; Kamin et al., 2010; Staras et al., 2010). Therefore, in the absence of an opposing mechanism, SV trafficking is expected to undermine the integrity of synapses by dispersing SVs into the axon. Contrary to this expectation, high density clustering of SVs is typically observed within the terminal (De Robertis and Bennett, 1955; Palay, 1956; Siksou et al., 2007), suggesting that proactive maintenance of SV organization must take place. Several classes of proteins have been implicated in organizing the presynaptic vesicle cluster, including active zone proteins (Mukherjee et al., 2010); cell adhesion proteins (Stan et al., 2010); actin, which may corral SVs from outside clusters (Bloom et al., 2003; Sankaranarayanan et al., 2003); and endocytic proteins, which may establish a vesicle-binding intervesicular matrix (Pechstein and Shupliakov, 2010; Shupliakov et al., 2011). Here we examined the hypothesis that the synapsins bind SVs to each other, allowing their accumulation near the active zone as a reserve pool (Greengard et al., 1993). To this end, we assessed the role that the synapsins play in determining presynaptic structure and intersynaptic vesicle mobility. Specifically, we examined whether they cluster SVs near the active zone by immobilizing them. We found that the synapsins specifically immobilize resting pool SVs. In neurons lacking synapsins the mobility of resting SVs equalizes to that of the recycling ones. Consequently, SV transport into the axon is enhanced at the expense of retention of SVs within the terminal. Despite the enhancement in the mobility of resting vesicles, the relative division of SVs between resting and recycling pools in the synapse is preserved. Therefore, we infer that a separate mechanism regulates the interplay between these pools.
The original synapsin hypothesis assumed that the synapsins control intrasynaptic mobility of SVs from the vesicle cluster to the active zone. However, in the mouse neuromuscular junction (NMJ), intrasynaptic movement of FM1–43 loaded vesicles was unaffected by deletion of the synapsins (Gaffield and Betz, 2007), arguing against such a role. Conversely, our results show that deleting the synapsins or overexpressing synapsin IIa substantially affects the quantity, distribution, and intersynaptic mobility of vesicles. How can this apparent contradiction be explained? First, we note that the two studies refer to different vesicle populations. FM1–43 exclusively visualizes the recycling pool (Betz and Bewick, 1992; Rizzoli and Betz, 2004), whereas our results implicate the synapsins in immobilizing the resting pool (Fig. 6). Specifically, we observed that the mobility of the total vesicle population, as visualized by SypI-EGFP, is significantly increased in the absence of the synapsins (Fig. 4), but that the mobility of FM1–43 loaded recycling vesicles remains unchanged (Fig. 6). Another possible explanation for the different conclusions relating to regulation of SV mobility by the synapsins is that the term “SV mobility” encompasses two different phenomena. Although probably related, the properties of intersynaptic and intrasynaptic vesicle mobility differ. For example, intersynaptic mobility is actin dependent (Darcy et al., 2006a), whereas intrasynaptic mobility is not (Jordan et al., 2005; Gaffield and Betz, 2006, 2007). Finally, differences between animal models may contribute to the observed variability. Contrary to results in the frog NMJ, synapsin deletion in Drosophila affects intrasynaptic mobility (Denker et al., 2011a). To conclude, we show that intersynaptic mobility of resting vesicles in small CNS synapses is controlled by the synapsins. Whether they similarly affect intrasynaptic mobility of resting vesicles remains an open question.
What are the implications of increasing intersynaptic mobility of resting SVs? Longitudinal examination of synaptic contacts by electron microscopy implies that the synapsins play a crucial role in maintaining well defined and compact vesicle clusters (Fig. 3). Specifically, in the absence of synapsins the edges of the vesicle clusters become indistinct because fewer vesicles remain near the active zone while their abundance in the flanking axons increases substantially. Quantitative analysis of the width of Syb2 and bassoon puncta supports this observation, illustrating that the distribution of SVs around the active zone is wider in synapsin TKO neurons (Fig. 2). This critical observation has probably evaded detection because the axonal segments adjacent to the synapses were not preserved in previous ultrastructural studies (Li et al., 1995; Rosahl et al., 1995; Takei et al., 1995; Samigullin et al., 2004; Gitler et al., 2004a; Siksou et al., 2007). In conclusion, the synapsins serve as a key conservative mechanism (but not an exclusive one, see above) that clusters SVs next to the active zone. In this study we directly probed only the effect of synapsin IIa (Fig. 9), which is the single synapsin isoform known to affect the kinetics of synaptic depression (Gitler et al., 2008); the involvement of other isoforms in regulating vesicle mobility remains to be examined.
Previous studies have alternatively suggested that the synapsins stabilize the SVs themselves (Rosahl et al., 1995; Sun et al., 2006), and that vesicle scavenging reduces the vesicle population in synapsin II KO neurons (Coleman et al., 2008), based on the overall reduction in the total quantity of synaptic vesicle markers (Rosahl et al., 1995; Gitler et al., 2004a; Bogen et al., 2006, 2009). An intriguing possibility is that the prevalence of SVs in the axon (Fig. 3) may contribute to such a process. Our study does not directly address vesicle destabilization, leaving this question open for future examination. Nevertheless, we note that the observed widening of the synaptic puncta in synapsin TKO neurons (Fig. 2) and the higher density of SVs in the axons (Fig. 3) is sufficient to account for the decrease in the peak fluorescence of the puncta (Fig. 1), even if most SVs are preserved.
It has been suggested that the recycling vesicles eventually mature and become part of the resting pool, and that as part of this maturation process their mobility is reduced (Denker and Rizzoli, 2010; Kamin et al., 2010). Here we report that deletion of the synapsins changes the fraction of resting vesicles that are mobile between synapses, but surprisingly, it does not change the division of the total population of vesicles into resting and recycling pools as measured using alkaline trapping of Synaptophysin I-2XpHluorin (Fig. 7). This implies that balance between the resting and recycling pools is unrelated to the synapsins, and that intersynaptic mobility and readiness for release are independent properties of the SVs. Cdk5 and calcineurin have been implicated in modulating this balance (Kim and Ryan, 2010). In agreement, we report that inhibiting cdk5 enhances SV mobility in WT neurons (Fig. 8), consistent with an increase in the relative size of the recycling pool at the expense of the resting pool. Synapsin is a target for cdk5 (Matsubara et al., 1996). Because roscovitine had no effect on vesicle mobility in synapsin TKO neurons, it is tempting to suggest synapsin as a mediator of the cdk5 effect. However, it is equally possible that the enhancement in SV mobility caused by deletion of the synapsins occludes the effect that cdk5 inhibition may exert. The identity of the cdk5 target therefore remains an open question.
According to our results, the synapsins may recruit extrasynaptic SVs to the synapse's resting pool by immobilizing laterally traveling vesicles (Fig. 10), and in this manner increment the total number of SVs in the terminal. A synapsin-independent balance between the recycling and resting pools then produces a comparable increment in the recycling pool. Inversely, when the synapsins are deleted, resting vesicles become as mobile as the recycling ones. Under these conditions, other conservative mechanisms preserve some of the SVs near the active zone. Consequently, the size of the local SV cluster decreases, concurrently with an increase in the number of SVs in the axon. The functional implications of deleting the synapsins have been described extensively previously(Cesca et al., 2010), and include a marked acceleration of synaptic depression, consistent with specific loss of vesicles that are not immediately ready for release. In this context, we also point out that previous work indicates that the synapsins do not participate in defining the size of the readily releasable pool, at least in several excitatory neurons (Gitler et al., 2004a; Sun et al., 2006), suggesting that the balance between this critical pool and the recycling pool which contains it is also subject to a different molecular control mechanism.
Figure 10.

Model for restriction of resting pool mobility by synapsin and its effect on vesicle distribution. Vesicles are divided approximately equally into the resting (green) and recycling (red) pool by a synapsin-independent mechanism. In this manner, the fraction of recycling vesicles of the total population is similar in WT (top) and synapsin TKO (bottom) neurons, although the number of vesicles within the presynaptic terminal is smaller by ∼30% in the TKO terminals. The mobility of most resting vesicles is restricted by the synapsins (blue circle), while the mobility of ∼30% of the recycling vesicles is restricted by a different mechanism or protein (black circle). Altogether, ∼65% of the vesicles are immobile. In the absence of the synapsins, the immobilization of the resting vesicles is lost, and the mobile fraction of vesicles increases to ∼85% (∼15% immobile). Due to the increased mobility, SVs are dispersed into the axon, causing the boundaries of the synapse to become indistinct and increasing the number of SVs in the axon by a factor of 3.
Do resting and recycling SVs differ at the molecular level? Vesicles slowly interchange between these pools (Denker and Rizzoli, 2010), making it conceivable that switchable molecular/functional tags exist. Our results imply that the synapsins are capable of differentiating between these pools. Further support for this idea emerges from the recent report that these two pools harbor different SNAREs (Hua et al., 2011). A related question concerns the functional relevance of the resting pool. Our results and those of others are consistent with this pool being a store for scaling the recycling pool. Interestingly, such recruitment could be controlled by signaling cascades (Kim and Ryan, 2010), providing means for local and temporal modulation. On a larger scale, the superpool of extrasynaptic SVs could serve as an additional depot accessible to individual synapses (Staras et al., 2010). Finally, the reserve/resting pool of vesicles has been proposed to serve as a reservoir of synaptic vesicle proteins (Denker et al., 2011b) providing additional support to the notion of the presynaptic terminal being a semi-autonomous entity located far from the cell body. The implications of these observations and novel ideas remain unclear, providing substantial opportunities for future investigation.
Footnotes
This work was supported by Grants 452/06 of the Israel Science Foundation (D.G.), 916-114.1/2006 from the German–Israeli Foundation for Scientific Research and Development (D.G.), 3-0000–6217 from the Chief Scientist of the Ministry of Health of Israel (D.G.), the Marc Rich Foundation for Culture and Welfare, Lucerne, Switzerland (D.G.), WT084357MF from the Wellcome Trust (K.S.), and BB/F018371 from the BBSRC (K.S.).We thank Thomas Kuner for the Synaptophysin I-EGFP viral construct, Ruud Toonen for the Synaptophysin I-2XpHluorin expression vector, Jürgen Kleinschmidt for kindly sharing the AAV expression vectors, Yona Lichtenfeld for expert processing of samples for electron microscopy, and Jens Eilers and Yael Amitai for thoroughly commenting on the manuscript.
References
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Akbergenova Y, Bykhovskaia M. Synapsin regulates vesicle organization and activity-dependent recycling at Drosophila motor boutons. Neuroscience. 2010;170:441–452. doi: 10.1016/j.neuroscience.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsman JB, Krueger SR, Fitzsimonds RM. Automated criteria-based selection and analysis of fluorescent synaptic puncta. J Neurosci Methods. 2006;152:32–39. doi: 10.1016/j.jneumeth.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- Bloom O, Evergren E, Tomilin N, Kjaerulff O, Löw P, Brodin L, Pieribone VA, Greengard P, Shupliakov O. Colocalization of synapsin and actin during synaptic vesicle recycling. J Cell Biol. 2003;161:737–747. doi: 10.1083/jcb.200212140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogen IL, Boulland JL, Mariussen E, Wright MS, Fonnum F, Kao HT, Walaas SI. Absence of synapsin I and II is accompanied by decreases in vesicular transport of specific neurotransmitters. J Neurochem. 2006;96:1458–1466. doi: 10.1111/j.1471-4159.2005.03636.x. [DOI] [PubMed] [Google Scholar]
- Bogen IL, Haug KH, Roberg B, Fonnum F, Walaas SI. The importance of synapsin I and II for neurotransmitter levels and vesicular storage in cholinergic, glutamatergic and GABAergic nerve terminals. Neurochem Int. 2009;55:13–21. doi: 10.1016/j.neuint.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Coleman WL, Bill CA, Simsek-Duran F, Lonart G, Samigullin D, Bykhovskaia M. Synapsin II and calcium regulate vesicle docking and the cross-talk between vesicle pools at the mouse motor terminals. J Physiol. 2008;586:4649–4673. doi: 10.1113/jphysiol.2008.154666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darcy KJ, Staras K, Collinson LM, Goda Y. Constitutive sharing of recycling synaptic vesicles between presynaptic boutons. Nat Neurosci. 2006a;9:315–321. doi: 10.1038/nn1640. [DOI] [PubMed] [Google Scholar]
- Darcy KJ, Staras K, Collinson LM, Goda Y. An ultrastructural readout of fluorescence recovery after photobleaching using correlative light and electron microscopy. Nat Protoc. 2006b;1:988–994. doi: 10.1038/nprot.2006.146. [DOI] [PubMed] [Google Scholar]
- Denker A, Rizzoli SO. Synaptic vesicle pools: an update. Front Synaptic Neurosci. 2010;2:135. doi: 10.3389/fnsyn.2010.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker A, Bethani I, Kröhnert K, Körber C, Horstmann H, Wilhelm BG, Barysch SV, Kuner T, Neher E, Rizzoli SO. A small pool of vesicles maintains synaptic activity in vivo. Proc Natl Acad Sci U S A. 2011a;108:17177–17182. doi: 10.1073/pnas.1112688108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker A, Kröhnert K, Bückers J, Neher E, Rizzoli SO. The reserve pool of synaptic vesicles acts as a buffer for proteins involved in synaptic vesicle recycling. Proc Natl Acad Sci U S A. 2011b;108:17183–17188. doi: 10.1073/pnas.1112690108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Robertis ED, Bennett HS. Some features of the submicroscopic morphology of synapses in frog and earthworm. J Biophys Biochem Cytol. 1955;1:47–58. doi: 10.1083/jcb.1.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Alfonso T, Ryan TA. A heterogeneous “resting” pool of synaptic vesicles that is dynamically interchanged across boutons in mammalian CNS synapses. Brain Cell Biol. 2008;36:87–100. doi: 10.1007/s11068-008-9030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel T, García-Pérez E, Mahfooz K, Goñi J, Martínez-Turrillas R, Pérez-Otaño I, Lo DC, Wesseling JF. A new kinetic framework for synaptic vesicle trafficking tested in synapsin knock-outs. J Neurosci. 2011;31:11563–11577. doi: 10.1523/JNEUROSCI.1447-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffield MA, Betz WJ. Imaging synaptic vesicle exocytosis and endocytosis with FM dyes. Nat Protoc. 2006;1:2916–2921. doi: 10.1038/nprot.2006.476. [DOI] [PubMed] [Google Scholar]
- Gaffield MA, Betz WJ. Synaptic vesicle mobility in mouse motor nerve terminals with and without synapsin. J Neurosci. 2007;27:13691–13700. doi: 10.1523/JNEUROSCI.3910-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffield MA, Rizzoli SO, Betz WJ. Mobility of synaptic vesicles in different pools in resting and stimulated frog motor nerve terminals. Neuron. 2006;51:317–325. doi: 10.1016/j.neuron.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ. Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci. 2004a;24:11368–11380. doi: 10.1523/JNEUROSCI.3795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Xu Y, Kao HT, Lin D, Lim S, Feng J, Greengard P, Augustine GJ. Molecular determinants of synapsin targeting to presynaptic terminals. J Neurosci. 2004b;24:3711–3720. doi: 10.1523/JNEUROSCI.5225-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Cheng Q, Greengard P, Augustine GJ. Synapsin IIa controls the reserve pool of glutamatergic synaptic vesicles. J Neurosci. 2008;28:10835–10843. doi: 10.1523/JNEUROSCI.0924-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Groh A, de Kock CP, Wimmer VC, Sakmann B, Kuner T. Driver or coincidence detector: modal switch of a corticothalamic giant synapse controlled by spontaneous activity and short-term depression. J Neurosci. 2008;28:9652–9663. doi: 10.1523/JNEUROSCI.1554-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Schweizer FE, Kao HT, Czernik AJ, Greengard P, Augustine GJ. Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci. 1998;1:29–35. doi: 10.1038/229. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Benfenati F, Doussau F, Nairn AC, Czernik AJ, Augustine GJ, Greengard P. Structural domains involved in the regulation of transmitter release by synapsins. J Neurosci. 2005;25:2658–2669. doi: 10.1523/JNEUROSCI.4278-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Z, Leal-Ortiz S, Foss SM, Waites CL, Garner CC, Voglmaier SM, Edwards RH. v-SNARE composition distinguishes synaptic vesicle pools. Neuron. 2011;71:474–487. doi: 10.1016/j.neuron.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan R, Lemke EA, Klingauf J. Visualization of synaptic vesicle movement in intact synaptic boutons using fluorescence fluctuation spectroscopy. Biophys J. 2005;89:2091–2102. doi: 10.1529/biophysj.105.061663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamin D, Lauterbach MA, Westphal V, Keller J, Schönle A, Hell SW, Rizzoli SO. High- and low-mobility stages in the synaptic vesicle cycle. Biophys J. 2010;99:675–684. doi: 10.1016/j.bpj.2010.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay AR, Alfonso A, Alford S, Cline HT, Holgado AM, Sakmann B, Snitsarev VA, Stricker TP, Takahashi M, Wu LG. Imaging synaptic activity in intact brain and slices with FM1–43 in C. elegans, lamprey, and rat. Neuron. 1999;24:809–817. doi: 10.1016/s0896-6273(00)81029-6. [DOI] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. CDK5 serves as a major control point in neurotransmitter release. Neuron. 2010;67:797–809. doi: 10.1016/j.neuron.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, Jensen V, Zheng D, McNamara JO, Greengard P. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci U S A. 1995;92:9235–9239. doi: 10.1073/pnas.92.20.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara M, Kusubata M, Ishiguro K, Uchida T, Titani K, Taniguchi H. Site-specific phosphorylation of synapsin I by mitogen-activated protein kinase and Cdk5 and its effects on physiological functions. J Biol Chem. 1996;271:21108–21113. doi: 10.1074/jbc.271.35.21108. [DOI] [PubMed] [Google Scholar]
- Mukherjee K, Yang X, Gerber SH, Kwon HB, Ho A, Castillo PE, Liu X, Südhof TC. Piccolo and bassoon maintain synaptic vesicle clustering without directly participating in vesicle exocytosis. Proc Natl Acad Sci U S A. 2010;107:6504–6509. doi: 10.1073/pnas.1002307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenbuch A, Shulman Y, Lipstein N, Brumer E, Bechar A, Lavy Y, Vasileva M, Kahn J, Barki-Harrington L, Kuner T, Gitler D. Inhibition of exocytosis and endocytosis blocks activity dependent redistribution of synapsin. J Neurochem. 2012;120:248–258. doi: 10.1111/j.1471-4159.2011.07579.x. [DOI] [PubMed] [Google Scholar]
- Palay SL. Synapses in the central nervous system. J Biophys Biochem Cytol. 1956;2:193–202. doi: 10.1083/jcb.2.4.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Zucker RS. A general model of synaptic transmission and short-term plasticity. Neuron. 2009;62:539–554. doi: 10.1016/j.neuron.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechstein A, Shupliakov O. Taking a back seat: synaptic vesicle clustering in presynaptic terminals. Front Synaptic Neurosci. 2010;2:143. doi: 10.3389/fnsyn.2010.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. The structural organization of the readily releasable pool of synaptic vesicles. Science. 2004;303:2037–2039. doi: 10.1126/science.1094682. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- Samigullin D, Bill CA, Coleman WL, Bykhovskaia M. Regulation of transmitter release by synapsin II in mouse motor terminals. J Physiol. 2004;561:149–158. doi: 10.1113/jphysiol.2004.073494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Calcium accelerates endocytosis of vSNAREs at hippocampal synapses. Nat Neurosci. 2001;4:129–136. doi: 10.1038/83949. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Atluri PP, Ryan TA. Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat Neurosci. 2003;6:127–135. doi: 10.1038/nn1002. [DOI] [PubMed] [Google Scholar]
- Scott DA, Das U, Tang Y, Roy S. Mechanistic logic underlying the axonal transport of cytosolic proteins. Neuron. 2011;70:441–454. doi: 10.1016/j.neuron.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupliakov O, Haucke V, Pechstein A. How synapsin I may cluster synaptic vesicles. Semin Cell Dev Biol. 2011;22:393–399. doi: 10.1016/j.semcdb.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Siksou L, Rostaing P, Lechaire JP, Boudier T, Ohtsuka T, Fejtová A, Kao HT, Greengard P, Gundelfinger ED, Triller A, Marty S. Three-dimensional architecture of presynaptic terminal cytomatrix. J Neurosci. 2007;27:6868–6877. doi: 10.1523/JNEUROSCI.1773-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan A, Pielarski KN, Brigadski T, Wittenmayer N, Fedorchenko O, Gohla A, Lessmann V, Dresbach T, Gottmann K. Essential cooperation of N-cadherin and neuroligin-1 in the transsynaptic control of vesicle accumulation. Proc Natl Acad Sci U S A. 2010;107:11116–11121. doi: 10.1073/pnas.0914233107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staras K, Branco T, Burden JJ, Pozo K, Darcy K, Marra V, Ratnayaka A, Goda Y. A vesicle superpool spans multiple presynaptic terminals in hippocampal neurons. Neuron. 2010;66:37–44. doi: 10.1016/j.neuron.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Bronk P, Liu X, Han W, Südhof TC. Synapsins regulate use-dependent synaptic plasticity in the calyx of Held by a Ca2+/calmodulin-dependent pathway. Proc Natl Acad Sci U S A. 2006;103:2880–2885. doi: 10.1073/pnas.0511300103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, Wieland F, Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Takei Y, Harada A, Takeda S, Kobayashi K, Terada S, Noda T, Takahashi T, Hirokawa N. Synapsin I deficiency results in the structural change in the presynaptic terminals in the murine nervous system. J Cell Biol. 1995;131:1789–1800. doi: 10.1083/jcb.131.6.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuriel S, Geva R, Zamorano P, Dresbach T, Boeckers T, Gundelfinger ED, Garner CC, Ziv NE. Local sharing as a predominant determinant of synaptic matrix molecular dynamics. PLoS Biol. 2006;4:e271. doi: 10.1371/journal.pbio.0040271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuriel S, Fisher A, Wittenmayer N, Dresbach T, Garner CC, Ziv NE. Exchange and redistribution dynamics of the cytoskeleton of the active zone molecule bassoon. J Neurosci. 2009;29:351–358. doi: 10.1523/JNEUROSCI.4777-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]