

Abstract
In an attempt to combine the ability of indolobenzazepines (paullones) to inhibit cyclin-dependent kinases (Cdks) and that of platinum-group metal ions to interact with proteins and DNA, ruthenium(II) and osmium(II) arene complexes with paullones were prepared, expecting synergies and an increase of solubility of paullones. Complexes with the general formula [MIICl(η6-p-cymene)L]Cl, where M = Ru (1, 3) or Os (2, 4), and L = L1 (1, 2) or L2 (3, 4), L1 = N-(9-bromo-7,12-dihydroindolo[3,2-d][1]-benzazepin-6(5H)-yliden-N′-(2-hydroxybenzylidene)azine and L2 = N-(9-bromo-7,12-dihydroindolo[3,2-d][1]benzazepin-6-yl)-N′-[3-hydroxy-5-(hydroxymethyl)-2-methylpyridin-4-yl-methylene]azinium chloride (L2*HCl), were now investigated regarding cytotoxicity and accumulation in cancer cells, impact on the cell cycle, capacity of inhibiting DNA synthesis and inducing apoptosis as well as their ability to inhibit Cdk activity. The MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay yielded IC50 values in the nanomolar to low micromolar range. In accordance with cytotoxicity data, the BrdU assay showed that 1 is the most and 4 the least effective of these compounds regarding inhibition of DNA synthesis. Effects on the cell cycle are minor, although concentration-dependent inhibition of Cdk2/cyclin E activity was observed in cell-free experiments. Induction of apoptosis is most pronounced for complex 1, accompanied by a low fraction of necrotic cells, as observed by annexin V–fluorescein isothiocyanate/propidium iodide staining and flow cytometric analysis.
Keywords: Paullones, Indolobenzazepines, Cyclin-dependent kinases, Ruthenium, Osmium, Arene complexes
Graphical abstract
Ruthenium and osmium arene complexes with modified paullone ligands strongly inhibit cancer cell growth. Antiproliferative activity correlates with inhibition of DNA synthesis and induction of apoptosis, but seems to be independent of Cdk inhibition, given the moderate potency of inhibiting Cdk2/cyclin E and the lack of pronounced cell cycle effects.
Highlights
► Ru and Os arene complexes with Paullone ligands strongly inhibit cancer cell growth. ► Cytotoxicity correlates with inhibition of DNA synthesis and induction of apoptosis. ► Cdk2/cyclin E inhibition and cell cycle effects insufficiently explain cytotoxicity.
1. Introduction
The search for an alternative to platinum anticancer agents is a major motivation for continuing investigations concerning the antitumor properties of other transition metal-based compounds. Considering the resistance of many tumors to cisplatin, oxaliplatin or carboplatin and the adverse effects of these drugs [1,2], great expectations are associated with the antitumor activity and lower general toxicity of certain ruthenium compounds. Beside ruthenium also osmium with similar chemical properties is under investigation, mostly yielding cytotoxic effects in cancer cell lines comparable to ruthenium analogues [3]. NAMI-A, a compound aimed at metastasis inhibition, and KP1019 are examples of promising ruthenium complexes under clinical investigation [4]. Major advantages of ruthenium are slow ligand exchange kinetics, activation by reduction and ability to use iron transporter mechanisms [5]. Interaction with DNA has been supposed; but given the extensive protein binding of compounds such as KP1019 [6], protein targets are much more likely to be relevant in vivo. Furthermore, ways of cellular accumulation are still being discussed [7,8].
Indolobenzazepines, also known as paullones, were first identified as inhibitors of cyclin-dependent kinases (Cdk) by Kunick and co-workers [9] and are since under investigation regarding not only their Cdk-inhibition potency but also their effects on glycogen synthase kinase-3 [8] and mitochondrial malate dehydrogenase [10]. An underivatized lactam unit and an electron-withdrawing substituent, such as bromine, favor Cdk-inhibitory activity [11] and have, at least in some cases, favorable effects on cytotoxicity as well [12]. Insufficient aqueous solubility and bioavailability of paullones have hampered their further development as anticancer agents. By creating paullones able to bind to ruthenium(II) and osmium(II) arene moieties, we expected to reduce the encountered problems markedly. Moreover, synergistic effects and the differing targets of metals and ligands could be an advantage for inhibiting cancer cell growth.
Indolobenzazepines with the general formula [MIICl(η6-p-cymene)L]Cl (L = L1 or L2; M = Ru or Os) (Fig. 1) have been synthesized and characterized previously [13]. These substances have shown their potency in a cytotoxicity test in three human cancer cell lines, with IC50 values in the lower micromolar range. Hydrolysis behavior and reactivity to 5′-GMP were also reported. High cytotoxic activity was the reason for further studies on their impact on human cancer cells. Because of the known Cdk-inhibitory activity of the metal-free paullones, inhibition of Cdk2/cyclin E was also investigated in a cell-free assay with the metal complexes. Effects on the cell cycle were quantified by flow cytometry, and the metal accumulation in the cells, inhibition of DNA synthesis and induction of apoptosis were compared to cytotoxic potency.
Fig. 1.
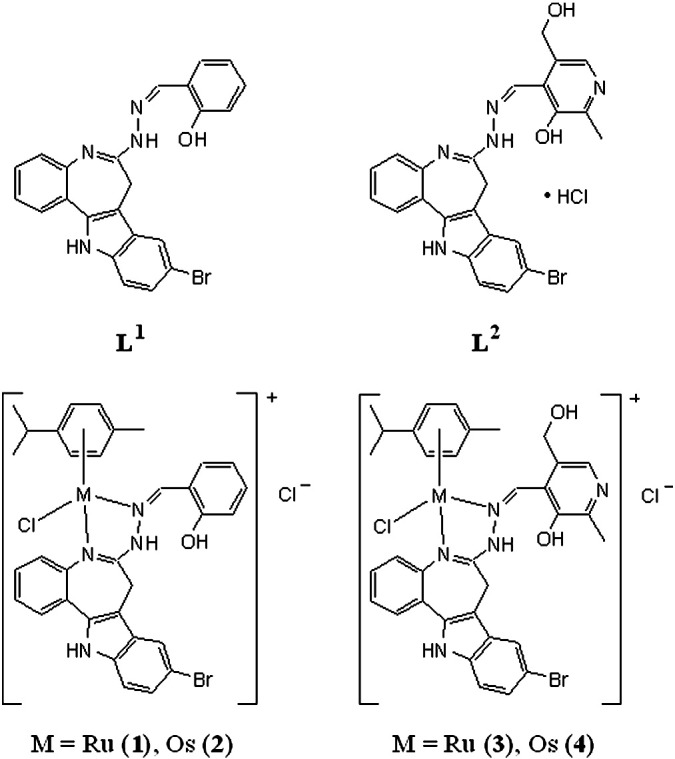
Structures of indolobenzazepine derivatives L1 and L2 and their ruthenium and osmium arene complexes.
2. Experimental section
2.1. Compounds
Compounds 1–4 were prepared as described previously [13]. For all experiments, the compounds were first dissolved in DMSO and then diluted in medium/buffer as appropriate. Flavopiridol was kindly provided by Sanofi-Aventis.
2.2. Cell lines and culture conditions
CH1 (ovarian carcinoma, human) cells were donated by Lloyd R. Kelland (CRC Centre for Cancer Therapeutics, Institute of Cancer Research, Sutton, U.K.). SW480 (colon adenocarcinoma, human) and A549 (non-small cell lung cancer, human) cells were kindly provided by Brigitte Marian (Institute of Cancer Research, Department of Medicine I, Medical University of Vienna, Austria). Prostate carcinoma cell line LNCaP, mammary gland carcinoma cell line T47D as well as the gastric carcinoma cell line N87 were purchased from the American Type Culture Collection (ATCC). Cells were grown without antibiotics in 75-cm2 culture flasks (Iwaki/Asahi Technoglass) as adherent monolayer cultures in minimal essential medium (MEM) (for CH1, SW480 and A549 cells) or in RPMI 1640 medium (for LNCaP, N87 and T47D cells), both media supplemented with 10% heat-inactivated fetal bovine serum and 4 mM l-glutamine, but only MEM supplemented with 1 mM sodium pyruvate and 1% non-essential amino acids (from 100 × ready-to-use stock) (all purchased from Sigma-Aldrich) without antibiotics. Cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air.
2.3. Cytotoxicity in cancer cell lines
Cytotoxicity in the cell lines mentioned above was determined by the colorimetric MTT assay (MTT = 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide, purchased from Sigma-Aldrich). Cells were harvested from culture flasks by trypsinization and seeded in 100 μL aliquots into 96-well microculture plates (Iwaki/Asahi Technoglass) in the following densities, in order to ensure exponential growth of untreated controls throughout the experiment: 1.5 × 103 (CH1), 2.5 × 103 (SW480), 4.0 × 103 (A549), 6.0 × 103 (N87 and T47D) and 1.0 × 104 (LNCaP) viable cells per well. Cells were allowed for 24 h to settle and resume exponential growth in drug-free MEM, followed by the addition of dilutions of the test compounds in aliquots of 100 μL/well in the same medium (eventually containing not more than 0.5% DMSO). After continuous exposure for 96 h, the medium was replaced by 100 μL/well RPMI 1640 medium plus 20 μL/well solution of MTT in phosphate-buffered saline (5 mg/mL) (all purchased from Sigma-Aldrich). After incubation for 4 h, medium/MTT mixtures were removed, and the formazan precipitate formed by viable cells was dissolved in DMSO (150 μL/well). Optical densities at 550 nm were measured with a microplate reader (Tecan Spectra Classic), using a reference wavelength of 690 nm to correct for unspecific absorption. The quantity of viable cells was expressed as percentage of untreated controls, and 50% inhibitory concentrations (IC50) were calculated from concentration–effect curves by interpolation. Evaluation is based on at least three independent experiments, each comprising three replicates per concentration level.
2.4. Kinase assay
The activities of recombinant Cdk2/cyclin E expressed in and isolated from Sf21 insect cells were determined by a radioassay [14] with minor modifications, using histone H1 as the substrate for phosphorylation. Briefly, MOPS-buffered assay mixtures containing the test compound (and a maximum of 1% DMSO), the kinase/cyclin complex, histone H1 and 0.4 μCi (γ-32P)ATP per sample were incubated for 10 min at 30 °C. Aliquots of the solution were spotted onto phosphocellulose squares, which had been washed 3 times with 0.75% phosphoric acid followed by acetone. The dried squares were measured in scintillation vials by beta counting (Perkin Elmer Tri-Carb 2800TR; software: Quanta Smart). Results were obtained in duplicates in at least two independent experiments.
2.5. Cell cycle analyses
The impact of the compounds on the cell cycle was studied by flow-cytometric analysis of DNA contents of cells stained with propidium iodide. Briefly, 1 million A549 cells were seeded into Petri dishes and allowed to recover for 24 h. Cells were then exposed for 24 h to the test compounds dissolved in a medium containing a maximum of 0.5% DMSO. Control and treated cells were collected, washed with PBS (phosphate-buffered saline), fixed in 70% ice-cold ethanol, and stored at − 20 °C. To determine cell cycle distribution, cells were transferred in physiological saline (0.9% w/v aqueous NaCl solution) into PBS, incubated with 10 μg/ml RNAse A for 30 min at 37 °C, followed by treatment with 5 μg/ml propidium iodide (PI) for 30 min. Fluorescence of 10 000 cells was measured with a FACS Calibur instrument (Becton Dickinson). The resulting DNA histograms were quantified by using the Cell Quest Pro software (Becton Dickinson).
2.6. DNA synthesis
Cellular DNA synthesis was quantified by the BrdU Cell Proliferation Assay Kit from Millipore. BrdU (5-bromo-2′-deoxyuridine) is incorporated into DNA instead of thymidine and serves as an indicator of DNA synthesis activity of the cells in a colorimetric immunoassay. For this purpose, 2 × 105 A549 cells were seeded into 96-well plates and allowed to recover for 24 h. Cells were then exposed to the test compounds for 24 h and incubated with BrdU for 6 h afterwards. For detection by anti-BrdU monoclonal antibody, cells were previously treated with fixing and denaturizing reagents followed by washing steps according to the manufacturer's instructions and finally incubated with a goat anti-mouse IgG peroxidase conjugate. Transformation of the TMB (3,3′,5,5′-tetramethylbenzidine) substrate was measured spectrophotometrically at 450/550 nm.
2.7. Cellular accumulation
Studies on cellular accumulation of the compounds were performed according to the method described previously [15]. Briefly, SW480 cells were seeded in 6-well plates in densities of 3 × 105 cells per well in aliquots of 2.5 mL complete culture medium. Accumulation experiments and corresponding adsorption/desorption controls were located on the same plate. Plates were kept at 37 °C for 24 h prior to addition of the compounds. Cells were incubated with the compounds in concentrations of 10 μM for 2 h at 37 °C. Afterwards, the medium was removed, cells were washed three times with PBS, lysed with 0.5 mL sub-boiled HNO3 per well for 1 h at room temperature, and ruthenium was quantified by ICP-MS (inductively coupled plasma mass spectrometry) in aliquots of 400 μL diluted to a total volume of 8 mL and internally standardized with indium (0.5 ppb). The adsorption/desorption blank was subtracted from the corresponding cellular accumulation sample. Results are based on three independent experiments, each consisting of three replicates.
Metal concentrations were determined by an ICP-MS instrument (Agilent 7500ce, Waldbronn, Germany), equipped with a CETAC ASX-520 autosampler and a MicroMist nebulizer, at a sample accumulation rate of approx. 0.25 mL/min. Indium and ruthenium standards were obtained from CPI International (Amsterdam, The Netherlands). Standards were prepared in matrices matching the sample matrix with regard to internal standard and concentration of the acid. Nitric acid (pro analysi) was purchased from Fluka (Buchs, Switzerland) and further purified in a quartz sub-boiling point distillation unit. All samples and dilutions were prepared with Milli-Q water (18.2 MΩcm). Concentrations were determined by means of the isotopes 115In and 102Ru.
2.8. Annexin V/propidium iodide assay
This assay was performed in order to determine induction and progress of apoptosis. This method was described by Aubry et al. [16] and allows for distinguishing early and late apoptosis as well as necrosis. In early apoptosis, phosphatidylserine flips from the inner side of the plasma membrane to the cell surface, having a Ca2 +-dependent affinity to Annexin V–FITC conjugate which can be measured fluorometrically at an emission wavelength of 530 nm upon excitation at a wavelength of 488 nm. Staining with PI, having an emission wavelength of 612 nm upon excitation at 488 nm, on the other hand, requires a loss of cell membrane integrity and therefore only works in the advanced apoptotic stage or in necrotic cells.
For this assay, 2 × 105 SW480 cells per well were seeded into 6-well plates and allowed to recover for 24 h. Cells were then exposed to different concentrations of test compounds for 48 h. The supernatant and cells which were detached by trypsinization were transferred into FACS tubes, centrifuged, and the supernatant was discarded. After resuspension in 0.5 mL of binding buffer, cells were incubated with 1 μL Annexin V–FITC from Bio Vision. After 5 min, propidium iodide with an end concentration of 1 μg/mL was added. Fluorescence was immediately measured by flow cytometry using a FACS Calibur instrument (Becton Dickinson), using FL1 channel for Annexin V-FITC and FL2 channel for PI staining. Resulting dot plots were quantified by Cell Quest Pro software (Becton Dickinson).
3. Results
3.1. Cytotoxicity in cancer cell lines
Cytotoxicity of the compounds was assessed by means of a colorimetric microculture assay (MTT assay) in six human cancer cell lines. The calculated IC50 values are listed in Table 1, and the corresponding concentration–effect curves are depicted in Fig. 2. Generally, the ovarian cancer cell line CH1 and the colon cancer cell line SW480 are invariably more sensitive, with IC50 values ranging from 0.67 to 3.3 μM and from 0.64 to 4.1 μM, respectively, whereas the non-small cell lung cancer cell line A549 and the prostate cancer cell line LNCaP are less sensitive, with IC50 values ranging from 3.1 to 10 μM and from 2.3 to 16 μM, respectively. The IC50 values are in all investigated cell lines in the lower micromolar to submicromolar range. The following structure–activity relationships can be deduced from these data: ruthenium complexes are in general more active than the osmium analogues. Ruthenium complex 1 (with L1) is in all cell lines at least 1.5 times (and up to 4.8 times) more active than its osmium analogue 2. The same applies to the complexes with L2, of which ruthenium complex 3 shows at least 1.7 times (and up to 4.4 times) higher cytotoxicity, depending on the cell line, than the analogous osmium complex 4. Ruthenium complex 1 is 1.9 to 7.3 times more cytotoxic, based on a comparison of IC50 values, than complex 3, and osmium complex 2 is 2.1 to 6.7 times more cytotoxic than 4, indicating that L1 yields more potent complexes than L2, irrespective of the chosen metal.
Table 1.
Cytotoxicity of ruthenium and osmium arene-based indolobenzazepine complexes in six human cancer cell lines.
| Compound | IC50 (μM) |
|||||
|---|---|---|---|---|---|---|
| A549 | SW480 | CH1 | LNCaP | N87 | T47D | |
| 1 | 3.1 ± 0.4 | 0.64 ± 0.14 | 0.67 ± 0.11 | 2.3 ± 0.4 | 1.1 ± 0.1 | 0.80 ± 0.10 |
| 2 | 4.6 ± 0.9 | 1.7 ± 0.1 | 1.6 ± 0.3 | 5.3 ± 1.3 | 1.6 ± 0.3 | 3.5 ± 0.3 |
| 3 | 6.0 ± 0.3 | 1.8 ± 0.3 | 1.5 ± 0.2 | 5.2 ± 1.5 | 2.5 ± 0.7 | 5.8 ± 0.3 |
| 4 | 10 ± 2 | 4.1 ± 1.0 | 3.3 ± 0.2 | 16 ± 1 | 11 ± 3 | 12 ± 1 |
Fig. 2.

Concentration–effect curves of ruthenium (1, 3) and osmium (2, 4) arene-based indolobenzazepine complexes in the human cancer cell lines A549 (A), SW480 (B), CH1 (C), LNCaP (D), T47D (E) and N87 (F), as obtained by the MTT assay (continuous exposure for 96 h).
3.2. Inhibition of Cdk activity
Since paullones are known as inhibitors of cyclin-dependent kinases [9], inhibitory potencies of the ruthenium and osmium arene complexes with L1 and L2 were studied in a cell-free setting. In particular, measurements of kinase activity of recombinant Cdk2/cyclin E complexes after exposure to 1–4 and to flavopiridol (Fp) as a positive control were performed by using (γ-32P)ATP and histone H1 as the substrate for phosphorylation. In general, ruthenium complexes 1 and 3 show a higher inhibitory potency on Cdk2/cyclin E than their osmium congeners 2 and 4. At a concentration of 10 μM, ruthenium complexes 1 and 3 yield 43% and 37% inhibition, which is about twice as high as the effect exerted by osmium congeners 2 and 4. A 50% inhibition of Cdk2/cyclin E requires concentrations of up to 40 μM (or even higher in the case of 2) (Fig. 3). Correlation with cytotoxic potencies is rather weak overall, but closest at the intermediate concentration of 10 μM.
Fig. 3.
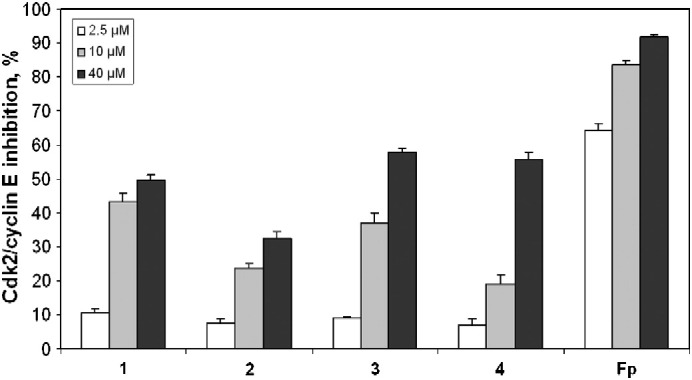
Concentration-dependent inhibition of Cdk2/cyclin E activity (means ± standard deviations) by complexes 1–4 in vitro. Flavopiridol (Fp) was used as a positive control.
3.3. Cell cycle effects
Given the capacity of inhibiting Cdk activity, an impact on the cell cycle of proliferating cells might be expected from these compounds. Therefore, changes in cell cycle distribution induced by 1–4 were studied in exponentially growing A549 cells treated with these compounds in varying concentrations for 24 h, then stained with propidium iodide and analyzed for their DNA content by flow cytometry. The compounds 1–4 have only weak effects on the cell cycle within the concentration range tested (Fig. 4). A slight increase of the G0/G1 fraction and a decrease of the S phase fraction could be observed up to a concentration of 40 μM of complexes 1 and 2. Reduced numbers of cells in G2/M phase compared to the control are visible at low concentrations of these compounds (2.5 μM and 10 μM). In the case of complexes 3 and 4, the cell fraction in G0/G1 phase is slightly increased only at the lowest (2.5 μM) and/or the medium concentration (10 μM) of the compounds.
Fig. 4.

Concentration-dependent impact of 1 (A), 2 (B), 3 (C) and 4 (D) on the cell cycle distribution of A549 cells after exposure for 24 h. DNA content of PI-stained cells was analyzed by flow cytometry.
3.4. Incorporation of BrdU
The inhibitory potency of the ruthenium and osmium complexes on DNA synthesis was determined by the BrdU assay. All four compounds inhibit BrdU incorporation into DNA of A549 non-small cell lung cancer cells within 24 h. Although the compounds have little effect on the cell cycle, a clear reduction of DNA synthesis could be observed (Fig. 5). Ruthenium complexes 1 and 3 are again somewhat more effective than the corresponding osmium complexes 2 and 4, in accordance with the structure–activity relationships revealed in the MTT assay. A concentration of 5 μM resulted in nearly 50% and 30% inhibition of BrdU incorporation by 1 and 3, respectively, whereas the effects of 2 and 4 are still modest. In any case, a strong reduction of DNA synthesis requires concentrations higher than 5 μM. A concentration of 20 μM, however, is sufficient for diminishing BrdU incorporation to values below 15% for all compounds.
Fig. 5.

Impact of complexes 1–4 on DNA synthesis in A549 cells, as determined by the BrdU incorporation assay after 24 h exposure to the indicated concentrations (μM) of the compounds.
3.5. Cellular accumulation
Cellular accumulation of complexes 1 and 3 was studied in the colon carcinoma cell line SW480. The cells were incubated at 37 °C for 2 h with 10 μM of the respective compound, and cellular metal contents were then determined by ICP-MS measurement, revealing that cellular amounts of ruthenium are one third lower after exposure to 1 (2.0 ± 0.3 fmol/cell) than those after treatment with 3 (3.0 ± 0.2 fmol/cell). These results do not correlate with cytotoxicity (compare Fig. 2b). Results obtained with the osmium analogues are not shown because of poor reproducibility; the most likely reason is uncontrolled loss of analyte due to the strongly oxidizing conditions during cell digestion.
3.6. Induction of apoptosis/necrosis
SW480 colon carcinoma cells were treated with complexes 1–4 for 48 h with concentrations between 5 and 40 μM, and cells were then collected for annexin V–FITC and propidium iodide staining. Exemplarily, dot plots of cell populations treated with 5 μM of each compound from one representative experiment are shown in Fig. 6. Complex 1 shows the strongest impact on cell viability, only 15% cells remain viable, whereas cells in early and late apoptosis amount to 72% in total. Complex 2 shows a much more moderate impact on cell viability, indicated by 63% viable cells and only 31% apoptotic cells. The same applies for complex 3, yielding a slightly lower amount of viable cells (56%) and a slightly higher amount of apoptotic cells (35%). Complex 4 is the least potent compound and has hardly any impact on the cells at a concentration of 5 μM. Percentages of necrotic cells remain generally low (with a maximum of 14% in the case of 1).
Fig. 6.

Flow cytometric quantification of apoptotic and necrotic SW480 cells upon exposure to 5 μM of ruthenium and osmium arene complexes with indolobenzazepine ligands (exposure time 48 h): control (A), 1 (B), 2 (C), 3 (D) and 4 (E). Viable cells appear in the lower left quadrant, necrotic cells in the top left quadrant, early apoptotic cells in the lower right quadrant and late apoptotic cells in the top right quadrant. (Plots from one representative experiment; numbers inserted indicate the percentages of cells in the respective quadrant.)
The concentration dependence of apoptosis/necrosis induction is illustrated in Fig. 7, and the corresponding values are listed in Table 2. They provide further evidence for the differences in cytotoxic potencies of the compounds, correlating with those observed in the MTT assay. Whereas 5 μM of compound 1 is sufficient for near-maximum effect, even 40 μM of compound 4 is insufficient for comparable effects. Compounds 2 and 3 require concentrations of 20 μM to induce 57% and 61% apoptosis, respectively, taking intermediate positions. Furthermore, compounds 2–4 induce higher proportions of necrotic cells relative to those undergoing apoptosis, making compound 1 the one with the most favorable properties.
Fig. 7.

Concentration–effect curves of ruthenium and osmium arene-based indolobenzazepine complexes regarding apoptosis/necrosis induction in SW480 cells after 48 h exposure, as obtained by flow cytometric quantification (means ± standard deviations): 1 (A), 2 (B), 3 (C) and 4 (D).
Table 2.
Distribution of viable, apoptotic and necrotic SW480 cells (means ± standard deviations) after 24 h exposure to ruthenium and osmium arene-based indolobenzazepine complexes, as obtained by flow cytometric quantification.
| Concentration (μM) |
Cell fraction (%) |
||||
|---|---|---|---|---|---|
| Viable | Early apoptosis | Late apoptosis | Necrosis | ||
| Control | 0 | 99 ± 0 | < 1 | < 1 | < 1 |
| 1 | 5 | 16 ± 2 | 43 ± 2 | 28 ± 4 | 13 ± 4 |
| 10 | 15 ± 2 | 41 ± 5 | 28 ± 5 | 16 ± 5 | |
| 20 | 7 ± 1 | 43 ± 2 | 35 ± 3 | 15 ± 2 | |
| 40 | 8 ± 3 | 37 ± 7 | 38 ± 2 | 18 ± 2 | |
| 2 | 5 | 62 ± 7 | 24 ± 6 | 9 ± 2 | 5 ± 1 |
| 10 | 31 ± 5 | 31 ± 8 | 13 ± 3 | 24 ± 7 | |
| 20 | 13 ± 1 | 37 ± 4 | 20 ± 4 | 30 ± 3 | |
| 40 | 8 ± 1 | 24 ± 3 | 33 ± 4 | 34 ± 3 | |
| 3 | 5 | 53 ± 3 | 23 ± 2 | 14 ± 2 | 9 ± 1 |
| 10 | 15 ± 2 | 32 ± 3 | 35 ± 2 | 18 ± 3 | |
| 20 | 15 ± 6 | 43 ± 4 | 18 ± 2 | 24 ± 2 | |
| 40 | 11 ± 3 | 23 ± 8 | 23 ± 3 | 43 ± 2 | |
| 4 | 5 | 96 ± 2 | 3 ± 2 | 1 ± 0 | 1 ± 1 |
| 10 | 88 ± 2 | 10 ± 1 | 1 ± 0 | 1 ± 1 | |
| 20 | 83 ± 2 | 12 ± 2 | 3 ± 1 | 2 ± 1 | |
| 40 | 50 ± 6 | 19 ± 2 | 13 ± 2 | 18 ± 3 | |
4. Discussion
Binding paullone ligands to ruthenium(II) and osmium(II) arene moieties led to a considerable improvement of solubility compared to the uncomplexed compounds, enabling biological studies. A comparison with previous results for Sadler's ruthenium complex with ethylenediamine (instead of the paullone ligand), [(η6-p-cymene)RuII(en)Cl](PF6), (IC50 values of 7.1, 3.5 and 4.4 μM in A549, SW480 and CH1 cells, respectively) under the same experimental conditions [17] reveals that the presence of the paullone ligand causes a 2.3- to 6.6-fold (complex 1) and a 1.2- to 2.9-fold (complex 3) increase in cytotoxicity, depending on the cell line.
In general, complexes with L1 show stronger cytotoxic effects than those with L2 in all human carcinoma cell lines tested. In the most sensitive cell lines SW480 and CH1, IC50 values of complex 1 are in the nanomolar range, whereas in the least sensitive cell lines A549 and LNCaP IC50 values are in the low micromolar range.
Whereas differences between ruthenium and osmium analogues appeared to be rather negligible in a previous report [13], the results in an extended panel of six cell lines reported here give a clearer indication that ruthenium is the slightly more favorable central metal than osmium for cytotoxic activity. As a third-row transition metal ion, OsII might be expected to be relatively inert compared to the second-row ion RuII. While fast exchange of the chlorido ligand and partial loss of the arene ligand was observed for all four complexes, a different number of cymene and cymene-free paullone species was detected for ruthenium and osmium complexes, but remarkably metal-paullone bonds remained intact in water/DMSO mixtures. The previous observation that the ruthenium complexes form N7 adducts with 5′-GMP, whereas osmium analogues do not under the same conditions, suggests a higher reactivity of the former to biological target molecules and may provide an explanation for the different cytotoxic potencies, which were not so evident in our previous studies [13]. In this context, covalent DNA binding cannot be excluded as a mode of action of this type of compounds, similar to simple ruthenium(II)-arene complexes lacking a biologically active co-ligand [18], but it seems unlikely that the above-mentioned increased potency mediated by the presence of a (sterically demanding) paullone ligand (see first paragraph of Discussion) is related to the formation of DNA adducts. A certain extent of DNA intercalation might be conceivable (compare the results with a related indolobenzazepine complex [19]), but the compounds are structurally not particularly predestined for this kind of interaction, leaving protein interactions as a more likely cause of the high antiproliferative activities of paullone-based ruthenium(II) and osmium(II) complexes.
Activity of Cdk2/cyclin E, envisaged as a potential protein target, is concentration-dependently inhibited by all four compounds, again strongest by complex 1, which shows at 10 μM about 50% of the inhibitory activity of the well-known Cdk inhibitor flavopiridol. Inhibitory potency on Cdk1/cyclin B, which is responsible for the G2/M transition, was not tested because previous studies with a related osmium–paullone complex showed a much lower inhibition of Cdk1/cyclin B than of Cdk2/cyclin E [19]. Furthermore, the lack of strong cell cycle effects, in particular the absence of a distinct G2 arrest, argued against further studies in that direction. Overall, the results presented here suggest that Cdks are not the crucial target of the complexes. Probably, the derivatization at the lactam unit of the paullones is the reason for the decrease in inhibitory potency, in accordance with the structure–activity relationships described by Kunick and coworkers [11].
Complex 1 is also most potent in the inhibition of DNA synthesis, as indicated by reduced BrdU incorporation into newly synthesized DNA. Overall, the reduction of DNA synthesis, requiring concentrations considerably higher than 5 μM, can hardly be interpreted as a direct interference with processes of the S phase. Rather the lack of a distinct S phase arrest suggests that the compounds exert their antiproliferative effects by inhibiting cell cycle progression more or less irrespective of the cell cycle phase, thereby also lowering the number of cells entering the phase of DNA synthesis.
Binding to 5′-GMP in the cell-free setting suggests the possibility of DNA interactions, at least for the ruthenium complexes, but cannot explain the cytotoxic potency of the osmium analogues. Moreover, other ruthenium complexes such as KP1019 are known to avidly bind proteins, both extra- and intracellular [20], lowering the probability that DNA interaction is relevant for their antitumor activity in vivo.
5. Conclusion
Cell biological activities of ruthenium/osmium complexes with modified paullone (indolobenzazepine) ligands derived from known Cdk inhibitors were characterized in human cancer cell lines in vitro. Apart from the beneficial effect on aqueous solubility, the presence of the paullone ligands seems to be favorable for biological activity as well. All of these compounds inhibit cancer cell growth in low micromolar concentrations and induce apoptotic cell death (to a lower extent also necrosis). The capacity of Cdk inhibition could be demonstrated in the cell-free setting, but is rather unlikely to be decisive for the antiproliferative activity of the complexes studied here, given the weak effects on cell cycle progression. Further investigations will be required to clarify the actual basis for their mechanism of action.
Abbreviations
- BrdU
Bromodeoxyuridine
- Cdk
Cyclin-dependent kinase
- FACS
Fluorescence-activated cell sorting
- FITC
Fluorescein isothiocyanate
- Fp
Flavopiridol
- GMP
Guanosine monophosphate
- IC50
50% inhibitory concentration
- ICP-MS
Inductively coupled plasma mass spectrometry
- IgG
Immunoglobulin G
- KP1019
Indazolium trans-[tetrachloridobis(1H-indazole)ruthenate(III)]
- MEM
Minimal Essential Medium
- MOPS
3-(N-Morpholino)propanesulfonic acid
- MTT
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
- NAMI-A
Imidazolium trans-[tetrachlorido(dimethyl sulfoxide)(imidazole)-ruthenate(III)]
- PBS
Phosphate-buffered saline
- PI
Propidium iodide
- TMB
3,3′,5,5′-Tetramethylbenzidine
Acknowledgments
We are indebted to the Austrian Science Fund (FWF) for financial support (project no. P20897-N19). G. Schmetterer (Institute of Physical Chemistry, University of Vienna) is gratefully acknowledged for providing the radiochemical facilities for kinase experiments. V. Dirsch and D. Schachner (Department of Pharmacognosy, University of Vienna) are gratefully acknowledged for providing the FACS instrument and for the technical instructions, respectively.
Contributor Information
Michael A. Jakupec, Email: michael.jakupec@univie.ac.at.
Bernhard K. Keppler, Email: bernhard.keppler@univie.ac.at.
References
- 1.Hamers F.P.T., Gispen W.H., Neijt J.P. Eur. J. Cancer. 1991;27:372–376. doi: 10.1016/0277-5379(91)90549-s. [DOI] [PubMed] [Google Scholar]
- 2.Godwin A.K., Meister A., O'Dwyer P.J., Huang C.S., Hamilton T.C., Anderson M.E. Proc. Natl. Acad. Sci. U. S. A. 1992;89:3070–3074. doi: 10.1073/pnas.89.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorcier A., Ang W.H., Bolaño S., Gonsalvi L., Juillerat-Jeannerat L., Laurenczy G., Peruzzini M., Phillips A.D., Zanobini F., Dyson P.J. Organometallics. 2006;25:4090–4096. [Google Scholar]
- 4.Sava G., Bergamo A. In: Platinum and Other Heavy Metal Compounds in Cancer Chemotherapy. Bonetti A., Leone R., Muggia F.M., Howell S.B., editors. Humana Press; New York: 2009. pp. 57–66. [Google Scholar]
- 5.Clarke M.J., Zhu F., Frasca D.R. Chem. Rev. 1999;99:2511–2534. doi: 10.1021/cr9804238. [DOI] [PubMed] [Google Scholar]
- 6.Hartinger C.G., Zorbas-Seifried S., Jakupec M.A., Kynast B., Zorbas H., Keppler B.K. J. Inorg. Biochem. 2006;100:891–904. doi: 10.1016/j.jinorgbio.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Ang W.H., Dyson P.J. Eur. J. Inorg. Chem. 2006;20:4003–4018. [Google Scholar]
- 8.Reedijk J. Platinum Metals Rev. 2008;52:2–11. [Google Scholar]
- 9.Zaharevitz D., Gussio R., Leost M., Senderowicz A.M., Lahusen T., Kunick C., Meijer L., Sausville E.A. Cancer Res. 1999;59:2566–2569. [PubMed] [Google Scholar]
- 10.Knockaert M., Wieking K., Schmitt S., Leost M., Grant K.M., Mottram J.C., Kunick C., Meijer L. J. Biol. Chem. 2002;277:25493–25501. doi: 10.1074/jbc.M202651200. [DOI] [PubMed] [Google Scholar]
- 11.Schultz C., Link A., Leost M., Zaharevitz D.W., Gussio R., Sausville E.A., Meijer L., Kunick C. J. Med. Chem. 1999;42:2909–2919. doi: 10.1021/jm9900570. [DOI] [PubMed] [Google Scholar]
- 12.Primik M.F., Mühlgassner G., Jakupec M.A., Zava O., Dyson P.J., Arion V.B., Keppler B.K. Inorg. Chem. 2010;49:302–311. doi: 10.1021/ic902042a. [DOI] [PubMed] [Google Scholar]
- 13.Schmid W.F., John R.O., Arion V.B., Jakupec M.A., Keppler B.K. Organometallics. 2007;26:6643–6652. [Google Scholar]
- 14.Marko D., Schätzle S., Friedel A., Genzlinger A., Zankl H., Meijer L., Eisenbrand G. Br. J. Cancer. 2001;84:283–289. doi: 10.1054/bjoc.2000.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egger A.E., Rappel C., Jakupec M.A., Hartinger C.G., Heffeter P., Keppler B.K. J. Anal. At. Spectrom. 2009;24:51–61. doi: 10.1039/B810481F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aubry J.-P., Blaecke A., Lecoanet-Henchoz S., Jeannin P., Herbault N., Caron G., Moine V., Bonnefoy J.-Y. Cytometry. 1999;37:197–204. doi: 10.1002/(sici)1097-0320(19991101)37:3<197::aid-cyto6>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 17.Grguric-Sipka S., Stepanenko I.N., Lazic J.M., Bartel C., Jakupec M.A., Arion V.B., Keppler B.K. Dalton Trans. 2009;17:3334–3339. doi: 10.1039/b822725j. [DOI] [PubMed] [Google Scholar]
- 18.Yan Y.K., Melchart M., Habtemariam A., Sadler P.J. Chem. Commun. 2005;2005:4764–4776. doi: 10.1039/b508531b. [DOI] [PubMed] [Google Scholar]
- 19.Filak L.K., Mühlgassner G., Jakupec M.A., Heffeter P., Berger W., Arion V.B., Keppler B.K. J. Biol. Inorg. Chem. 2010;15:903–918. doi: 10.1007/s00775-010-0653-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heffeter P., Böck K., Atil B., Reza Hoda M.A., Körner W., Bartel C., Jungwirth U., Keppler B.K., Micksche M., Berger W., Koellensperger G. J. Biol. Inorg. Chem. 2010;15:737–748. doi: 10.1007/s00775-010-0642-1. [DOI] [PMC free article] [PubMed] [Google Scholar]