

Abstract
A decrease in capillary density due to an increase in endothelial cell apoptosis in the heart is implicated in cardiac ischemia in diabetes. The voltage-dependent anion channel (VDAC) plays a crucial role in the regulation of mitochondrial metabolic function and mitochondria-mediated apoptosis. This study is designed to examine the role of VDAC in coronary endothelial dysfunction in diabetes. Endothelial cells (ECs) were more apoptotic in diabetic left ventricle of diabetic mice and mouse coronary ECs (MCECs) isolated from diabetic mice exhibited significantly higher mitochondrial Ca2+ concentration and VDAC protein levels than control MCECs. The expression of VDAC-short hairpin RNA (shRNA) not only decreased the resting mitochondrial Ca2+ concentration but also attenuated mitochondrial Ca2+ uptake in diabetic MCECs. Furthermore, the downregulation of VDAC in diabetic MCECs significantly decreased mitochondrial superoxide anion (O2−) production and the activity of the mitochondrial permeability transition pore (mPTP) opening (an indirect indicator of cell apoptosis) toward control levels. These data suggest that the increased VDAC level in diabetic MCECs is responsible for increased mitochondrial Ca2+ concentration, mitochondrial O2− production, and mPTP opening activity. Normalizing VDAC protein level may help to decrease endothelial cell apoptosis, increase capillary density in the heart, and subsequently decrease the incidence of cardiac ischemia in diabetes.
Keywords: Ca2+ overload in mitochondria, apoptosis, vascular complications, vascular rarefaction
endothelial dysfunction is a common feature of diabetic vascular complications (8). Insufficient formation and rarefaction of capillaries, as a result of endothelial dysfunction, may represent one of the most critical mechanisms involved in cardiac ischemia. Along with other investigators, we have reported that capillary density in the heart is progressively decreased in diabetes (15, 25, 37, 39). Increased endothelial cell (EC) apoptosis (39) and attenuated regeneration of new capillaries by circulating endothelial progenitor cells (23, 39) both contribute to microvascular rarefaction in diabetes. Preventing coronary EC apoptosis would restore the decrease in capillary density, improve oxygen transport to cardiac tissues, and decrease the incidence of cardiac ischemia in diabetes.
Voltage-dependent anion channel (VDAC) was identified in 1976, and three VDAC isoforms have been characterized (VDAC1, VDAC2, and VDAC3). Among these three subtypes, VDAC1 is highly expressed in most cell types and is considered a key player in mitochondria-mediated apoptosis (36, 40, 41). Although other subtypes are also related with cell apoptosis, the molecular mechanisms proposed to explain how they regulate cell apoptosis are still controversial (4, 5). VDAC is located in the outer mitochondrial membrane and is a bidirectional transporter. Under physiological conditions VDAC serves as a shuttle of ATP and other small molecules (35), whereas under pathophysiological conditions VDAC contributes to cell apoptosis at the early stages [e.g., Ca2+ overload into mitochondria (12)] as well as during later stages [such as apoptotic protein release from the mitochondria by opening the mitochondrial permeability transition pore (mPTP) or rupture of the mitochondrial outer membrane (34, 38)]. The present study was designed to investigate the pathological role of VDAC in coronary endothelial dysfunction in diabetes.
MATERIALS AND METHODS
Materials.
Medium 199 (M199) was obtained from Mediatech (Manassas, VA). Antibiotic reagents, dispase II, MitoSOX Red, Rhod-2 AM, and a LIVE Mitochondrial Transition Pore Assay Kit were purchased from Invitrogen (Carlsbad, CA). Anti-VDAC, obtained from BioVision (Mountain view, CA), anti-hexokinase II from Cell Signaling Technology (Danvers, MA), and anti-hexokinase I and anti-actin from Santa Cruz Biotechnology (Santa Cruz, CA) were used for Western blot. Collagenase II was purchased from Worthington Biochemical (Lakewood, NJ). All other chemicals were from Sigma-Aldrich (St. Louis, MO).
Animal preparation.
All investigations conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, Revised 1985). This study was approved and conducted in accordance with the guidelines established by the Institutional Animal Care and Use Committee in the University of Illinois at Chicago. Six-week-old male C57BL/6 mice were purchased from Harlan Laboratories (Madison, WI), and mice in the diabetic group received a single injection of streptozotocin (133 mg/kg, dissolved in citrate buffer, i.v.). Data were obtained from mice 4–6 wk after diabetic induction with the exception of data for apoptotic EC determination in the heart (10 wk). Plasma glucose levels were 138.5 ± 4.3 mg/dl in control mice and 570.4 ± 18.1 mg/dl in diabetic mice.
Analysis of EC apoptosis in left ventricular myocardium.
Transferase-mediated dUTP nick end-labeling (TUNEL) assay was performed to detect apoptotic cells with an in situ cell detection kit (Roche). Sections of subepicardial regions of the left ventricular (LV) free wall were costained with BS-1-FITC to identify ECs. The images were photographed in sequence by a CCD camera connected to a fluorescence microscope with a ×20 objective lens. Apoptotic ECs were defined as TUNEL-positive cells colocalized with the EC marker and were counted with ImageJ software (National Institutes of Health, Bethesda, MD). Data were described as the percentage of apoptotic ECs (ratio of TUNEL-positive ECs/total ECs).
Isolation of mouse coronary vascular endothelial cells.
Mouse coronary vascular endothelial cells (MCECs) were isolated as described previously (26, 27). Briefly, dissected heart tissues were minced and incubated with M199 containing 1 mg/ml collagenase II and 0.6 U/ml dispase II for 1 h at 37°C. The digested material was filtered through sterile 40-μm nylon mesh and washed in 2% (vol/vol) FCS-M199. Subsequently, the cells were incubated with Dynabeads (Invitrogen), which were prepared as follows: beads coated with sheep anti-rat IgG were incubated with purified rat anti-mouse CD31 monoclonal antibody (1 μg/ml) at 4°C overnight and then washed with PBS containing 0.1% (wt/vol) BSA and 2 mM EDTA. The cell suspension was incubated with beads for 1 h at 4°C and then the beads attached to ECs were captured and isolated by the Dynal magnet (Invitrogen).
Western blot analysis.
Freshly isolated MCECs were used for protein extraction. Cell lysates were centrifuged at 16,000 g for 10 min at 4°C. Supernatants were used as sample protein. Samples were separated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Blots were then incubated with a primary antibody (anti-VDAC [1:2,000] or anti-actin [1:4,000]) followed by incubation with a horseradish peroxidase-conjugated secondary antibody. The immunoblots were detected with SuperSignal West Pico reagent (Thermo Fisher Scientific, Rockford, IL). Band intensity was normalized to actin controls and expressed in arbitrary units.
Mitochondrial Ca2+ concentration measurement.
Mitochondrial Ca2+ concentration ([Ca2+]mit) in MCECs was measured using a modification of previously described methods (7). Isolated MCECs were cultured in M199 (containing 5 mmol/l glucose) supplemented with 10% (vol/vol) FBS, 100 μg/ml endothelial cell growth supplement (ECGS), 100 U/ml penicillin, 100 μg/ml streptomycin, 50 mg/l d-valine, and 16 U/ml heparin. Cells were plated on glass chamber slides coated with 5% (wt/vol) gelatin. Three days after isolation, [Ca2+]mit was measured by a digital imaging fluorescence microscope. Cells on coverslips were loaded with the membrane-permeable acetoxymethyl ester form of Rhod-2 (Rhod-2-AM; 2 μmol) for 20 min in the dark at 37°C. The Rhod-2-AM-loaded cells were then superfused with physiological salt solution (PSS) for 90 min at 32°C to wash away extracellular and extramitochondrial dye. Rhod-2 fluorescence (530 nm excitation; 580 nm emission) from the cells and background fluorescence were imaged using a Nikon Eclipse Ti-E inverted fluorescence microscope. The background intensity was subtracted from the cell intensity. The resting level of [Ca2+]mit was obtained in Ca2+-PSS. The cells were then exposed to Ca2+-free PSS for 10 min, and 10 μmol cyclopiazonic acid [CPA, a sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor] was added to increase the cytosolic Ca2+. The peak of [Ca2+]mit increase (Δpeak) and the area under the curve (AUC) of the peak per individual cell were calculated (Fig. 2C).
Fig. 2.

Effect of VDAC inhibition by VDAC-short hairpin RNA (shRNA) adenovirus (Adv) (VDAC-shRNA) on mitochondrial [Ca2+]mit in MCECs. A: mouse VDAC shRNA was designed and cloned into an adenoviral vector with a U6 promoter. B: infection of mouse ECs with VDAC-shRNA significantly decreases VDAC protein levels. Western blots showing VDAC and actin protein levels (left). Actin was used as a loading control. Right columns show VDAC protein level normalized to actin. ECs infected with control Adv (Cont-Adv), n = 2; ECs infected with VDAC-shRNA, n = 2. Data are means ± SE. *P < 0.05 vs. control. C and D: measurement of [Ca2+]mit in MCECs at rest and after treatment with the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor, cyclopiazonic acid (CPA), in the absence of extracellular Ca2+. C: time course of the [Ca2+]mit measurement experiments. Resting level indicates [Ca2+]mit without treatment in the presence of Ca2+ in the extracellular media. After changing the extracellular media to Ca2+-free media, CPA (10 μmol/l) was added to test Ca2+ uptake by mitochondria. Area under the curve (AUC) and Δpeak were used to analyze Ca2+ uptake. D: summarized [Ca2+]mit data. Control ECs infected with control Adv (Cont-EC, Cont-Adv), n = 16; diabetic ECs infected with control Adv (Dia-EC, Cont-Adv), n = 19; diabetic ECs infected with VDAC-shRNA (Dia-EC, VDAC-shRNA), n = 22. Data are means ± SE. *P < 0.05 vs. Cont-EC, Cont-Adv. #P < 0.05 vs. Dia-EC, Cont-Adv.
Construction of adenoviral vectors.
To knockdown VDAC1, the following oligonucleotide (5′-3′): CACCGCAACTTCGCAGTTGGCTATACGAATATAGCCAACTGCGAAGTTGC was designed using an RNAi designer (Invitrogen). The oligonucleotide was inserted in a pENTR/U6 shuttle vector (Invitrogen) and recombined with the pAd/PL-DEST backbone vector in vitro. Replication-deficient adenovirus (Adv) particles containing the target gene or empty vector (Control-Adv) were generated in 293 cells, and single plaques were isolated and propagated to achieve high titer. Adenoviral particles were CsCl purified and quantified by a plaque titer assay. Two days after EC isolation, cells were infected with Adv at a concentration of 40 pfu/cell and incubated for 48 h.
Mitochondrial O2− concentration measurements.
Mitochondrial O2− concentration ([O2−]mit) was measured as described previously (26). Briefly, O2− was detected by preloading the cells with 5 μmol/l MitoSOX Red for 30 min before capturing images. MitoSOX Red is a fluorescent probe that is cell permeable and is rapidly and selectively targeted to the mitochondria. Once in the mitochondria, MitoSOX Red is oxidized by O2− and exhibits red fluorescence. A value of red fluorescence intensities in the mitochondria and from the background were measured and background signal was subtracted from the cell intensity.
Mitochondrial permeability transition pore activity assay.
Mitochondrial permeability transition pore (mPTP) activity in MCECs was measured using a LIVE Mitochondrial Transition Pore Assay Kit (Invitrogen). Calcein acetoxymethyl ester (calcein-AM) diffuses into the cells and accumulates in cytosolic compartments, including the mitochondria. Applying CoCl2 to the cells quenches the fluorescence from calcein in the cytosol but not in the mitochondria. The opening of the mPTP releases calcein-AM from the mitochondria and decreases the calcein-AM signal in the mitochondria. Two days after cell isolation, cells were infected with control or VDAC-short hairpin RNA (shRNA) adenovirus. The next day cells were washed with media and cultured for an additional 24 h. Cells were then stained with 1 μM calcein-AM (an indicator of mPTP activity, ex/em: 494/517), 200 nM MitoTracker Red CMXRos (to visualize mitochondrial structure, ex/em: 579/599), 1 μM Hoechst 33342 (nuclear staining, em/ex: 350/461), and 1 mM CoCl2 (to quench cytosolic calcein-AM fluorescence) for 30 min at 37°C protected from light. After staining was completed, cells were washed with Hank's balanced salt solution and the fluorescence images were taken using a Nikon Eclipse Ti-E inverted fluorescence microscope. The structure of the mitochondria was determined by MitoTracker Red signal, and the fluorescence intensity of the calcein-AM in the mitochondria was measured. The background intensity was subtracted from the cell intensity.
Statistical analysis.
Values are expressed as means ± SE. Bonferroni tests for multiple statistical comparisons and Student's t-test for unpaired samples were carried out to identify significant differences. Differences were considered to be statistically significant when P < 0.05.
RESULTS
More ECs are apoptotic in diabetic left ventricle and [Ca2+]mit and VDAC protein expression are increased in MCECs isolated from diabetic mice.
ECs in the LV were stained with BS-lectin (EC marker) (25), and apoptotic cells were detected by TUNEL assay. The percentage of apoptotic ECs (ratio of TUNEL-positive ECs/total ECs) is significantly higher in diabetic versus control mice (Fig. 1A). In addition, [Ca2+]mit and VDAC protein levels are significantly increased in MCECs from diabetic mice compared with control (Fig. 1, B and C).
Fig. 1.

Augmented endothelial cell (EC) apoptosis in the left ventricle (LV) and increased voltage-dependent anion channel (VDAC) protein level and increased mitochondrial Ca2+ concentration ([Ca2+]mit) in mouse coronary ECs (MCECs) isolated from diabetic mice. A: columns show summarized data of the percentage of apoptotic ECs (the number of apoptotic ECs divided by total number of ECs). Control (Cont), n = 5; diabetic (Dia), n = 6. Data are means ± SE. *P < 0.05 vs. control. B: representative images showing [Ca2+]mit in MCECs isolated from control and diabetic mice (left photomicrographs). Right columns show summarized [Ca2+]mit data (Rhod-2-fluorescence intensity from the cell minus background intensity). Cont, n = 25; Dia, n = 38. Data are means ± SE. *P < 0.05 vs. control. Bar = 50 μm. C: Western blots showing VDAC and actin protein levels (left). Actin was used as a loading control. Right columns show VDAC protein levels normalized by actin. Cont, n = 6; Dia, n = 6. Data are means ± SE. *P < 0.05 vs. control.
VDAC-shRNA expression decreases VDAC protein levels and restores the resting [Ca2+]mit and rate of Ca2+ uptake into mitochondria in diabetic MCECs.
To test whether the increased [Ca2+]mit in diabetic MCECs results from the increased levels of VDAC protein, we generated mouse VDAC1 (VDAC)-shRNA Adv to decrease VDAC protein levels (Fig. 2, A and B). The inhibition of VDAC in diabetic MCECs decreases not only the resting [Ca2+]mit but also Ca2+ uptake into mitochondria when cytosolic Ca2+ levels are raised by inhibiting SERCA with CPA (10 μmol/l) (Fig. 2, C and D).
VDAC-shRNA expression attenuates mitochondrial O2− production and decreases the mPTP opening activity in diabetic MCECs.
Ca2+ overload into mitochondria leads to cell apoptosis partially due to increasing mitochondrial O2− production as well as opening the mPTP, which subsequently releases pro-apoptotic molecules from the mitochondria to the cytosol. Figure 3 demonstrates that [O2−]mit in MCECs isolated from diabetic mice is significantly higher than in control mice, whereas the inhibition of VDAC in diabetic MCECs restores the increased [O2−]mit to the level observed in the control. In addition, diabetic MCECs exhibit a significant decrease in calcein-AM fluorescence in the mitochondria compared with control MCECs, suggesting that the mPTP opening is more activated at the resting level in diabetic MCECs. VDAC-shRNA expression significantly increases the calcein-AM intensity in diabetic MCECs, suggesting that the activity of the mPTP opening was decreased by VDAC-shRNA overexpression in diabetic MCECs (Fig. 4).
Fig. 3.

Effect of VDAC-shRNA on mitochondrial O2− concentration ([O2−]mit) in diabetic MCECs. A: representative images showing [O2−]mit in MCECs isolated from control and diabetic mice infected with Cont-Adv or VDAC-shRNA (left photomicrographs). Bar = 25 μm. B: summarized data of [O2−]mit. Cont-EC, Cont-Adv, n = 41; Dia-EC, Cont-Adv, n = 44; Dia-EC, shRNA-VDAC, n = 30. Data are means ± SE. *P < 0.05 vs. Cont-EC, Cont-Adv. #P < 0.05 vs. Dia-EC, Cont-Adv.
Fig. 4.

Activity of the mitochondrial permeability transition pore (mPTP) opening is significantly increased in diabetic MCECs, and VDAC-shRNA expression decreases the activity toward the control level. The fluorescence intensity of calcein-AM in mitochondria was measured to assess the activity of the mPTP opening. The decrease in calcein-AM intensity indicates the increase of the mPTP opening. Cont-EC and Cont-Adv, n = 108; Dia-EC and Cont-Adv, n = 133; Dia-EC and VDAC-shRNA, n = 98. Data are means ± SE. *P < 0.05 vs. Cont-EC, Cont-Adv. #P < 0.05 vs. Dia-EC, Cont-Adv.
Hexokinase II protein expression in diabetic MCECs is significantly lower than control.
Hexokinases (HKs) serve as endogenous VDAC regulators with inhibiting VDAC activation. Figure 5 demonstrates that hexokinase II (HK2) is significantly decreased in diabetic MCECs compared with control, whereas there is no difference in HK1 protein levels between control and diabetic MCECs.
Fig. 5.
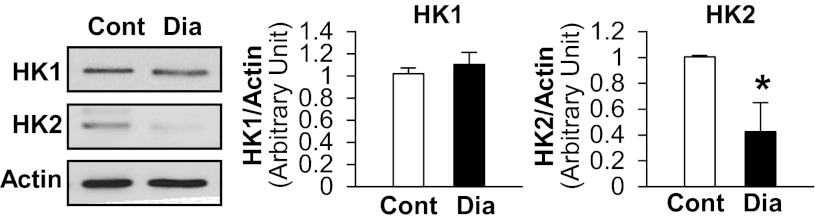
Hexokinase II (HK2), but not hexokinase I (HK1), is significantly decreased in diabetic MCECs. Western blots showing HK1, HK2, and actin protein levels (left). Actin was used as a loading control. Right columns show HK1 and HK2 protein expression levels normalized by actin. Cont, n = 4; Dia, n = 4. Data are means ± SE. *P < 0.05 vs. control.
DISCUSSION
Hyperglycemia increases VDAC1 expression in β cells (1) and in the kidney (14), whereas VDAC levels are not altered or decreased in cardiac myocytes after high-glucose treatment (19, 24). It is, however, yet to be explored whether VDAC contributes to coronary endothelial dysfunction in diabetes. Here, we demonstrate that MCECs isolated from diabetic mice exhibit a significant increase in VDAC protein level when compared with control (Fig. 1C). The experiments of Fig. 1A and our previous work showing decreased vascular density (25) are performed 10 wk after diabetic induction. In the current study, the increase of VDAC protein and [Ca2+]mit could be seen 4 wk after diabetic induction, implying that EC apoptosis in the heart may be initiated by VDAC overexpression at an early stage.
Increased [Ca2+]mit can facilitate ATP production to meet increased cellular ATP demands by activating Ca2+-sensitive enzymes in the tricarboxylic acid cycle (21, 28). However, Ca2+ overload in the mitochondria leads to apoptosis (16, 22), suggesting that maintaining [Ca2+]mit within the physiological range is crucial to keep cells functional and viable. High-glucose treatment leads to decreased [Ca2+]mit in the adipocyte (10) and cardiac myocyte (19), which may be the cause of insufficient energy production. Interestingly, [Ca2+]mit is significantly increased in diabetic coronary ECs compared with control (Fig. 1B), and the inhibition of VDAC in diabetic MCECs decreases not only the resting [Ca2+]mit level, but also mitochondrial Ca2+ uptake, when cytosolic Ca2+ levels are raised. These data suggest that in diabetic MCECs increased VDAC expression is responsible for augmented Ca2+ uptake into mitochondria and increased resting [Ca2+]mit. VDAC overexpression increases mitochondrial Ca2+ uptake by augmenting mitochondrial permeability and/or by increasing the number of contact sites between mitochondria and the endoplasmic reticulum (33). Mitochondrial Ca2+ overload sequentially triggers the release of pro-apoptotic proteins from the mitochondria to the cytosol (29, 38) and increases [O2−]mit (32).
Increased O2− production is implicated in the pathogenesis of diabetes-associated vascular complications (11, 13, 18). Figure 3 shows that VDAC inhibition significantly decreases [O2−]mit in diabetic MCECs, suggesting that overproduction of O2− is induced by Ca2+ overload in mitochondria via increased VDAC expression in diabetic coronary ECs.
An increased mPTP opening activity is the hallmark of mitochondria-induced cell apoptosis (30). The mPTP is composed of VDAC in the outer mitochondrial membrane (OMM), adenine nucleotide translocator (ANT) in the inner mitochondrial membrane, and cyclophilin D in the mitochondrial matrix (6, 17). Excess Ca2+ influx triggers the increase of ANT conductivity followed by an inward flux of protons and ions through the ANT. The increase in matrix osmolality leads to water influx, mitochondrial swelling, and apoptogenic protein release from the mitochondrial storage to the cytosol though the mPTP opening, BAX/BAK-VDAC channel, and/or ruptured OMM (3, 29, 38). There are reports showing that in an ex vivo study acute high-glucose treatment increases the mPTP opening in ECs (9, 20). We demonstrate that the mPTP opening is augmented at the resting level in diabetic MCECs compared with control MCECs, and VDAC-shRNA Adv infection significantly inhibits the mPTP opening in diabetic MCECs, implying that increased VDAC protein expression in diabetic MCECs leads to endothelial apoptosis via augmented mPTP opening.
VDAC has been reported to display the binding sites for HK, glycerol kinase, creatine kinase, as well as pro- and anti-apoptotic proteins of the Bcl-2 family (e.g., BAX, Bcl-2, Bcl-xl), and these bindings regulate the mPTP opening, which in turn modulate cell apoptosis (31, 35). Vertebrates have four isoforms of HK, and HK1 and HK2 exhibit their anti-apoptotic effect by directly binding to VDAC, followed by the channel closure (2), as well as by indirect mechanisms including interfering with the megachannel formation, which consists with VDAC and BAX (31). We found that MCECs isolated from diabetic mice have significantly decreased HK2 protein expression compared with control MCECs, suggesting that the apoptotic effect via VDAC-induced mPTP opening is enhanced due to decreased HK2 protein expression.
To summarize, diabetic MCECs exhibit a significant increase in VDAC protein expression compared with control MCECs, and VDAC inhibition restores the increased [Ca2+]mit, [O2−]mit and mPTP opening activity in diabetic MCECs to control levels. These data suggest that normalizing VDAC protein levels may decrease endothelial cell apoptosis, increase capillary density in the heart, and subsequently decrease the incidence of cardiac ischemia in diabetes.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: K.S., R.D., M.H., Y.-E.C., and A.M. performed experiments; K.S., R.D., M.H., Y.-E.C., and A.M. analyzed data; K.S., R.D., and A.M. interpreted results of experiments; K.S. and A.M. prepared figures; K.S., R.D., B.T.S., M.H., Y.-E.C., and A.M. approved final version of manuscript; R.D., B.T.S., and A.M. edited and revised manuscript; A.M. conception and design of research; A.M. drafted manuscript.
ACKNOWLEDGMENTS
This work was supported by the National Heart, Lung, and Blood Institute Grant of HL-115578 (to A. Makino).
REFERENCES
- 1.Ahmed M, Muhammed SJ, Kessler B, Salehi A. Mitochondrial proteome analysis reveals altered expression of voltage dependent anion channels in pancreatic beta-cells exposed to high glucose. Islets 2: 283–292, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J 377: 347–355, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baines CP. The mitochondrial permeability transition pore and the cardiac necrotic program. Pediatr Cardiol 32: 258–262, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9: 550–555, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301: 513–517, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Crompton M, Virji S, Doyle V, Johnson N, Ward JM. The mitochondrial permeability transition pore. Biochem Soc Symp 66: 167–179, 1999 [DOI] [PubMed] [Google Scholar]
- 7.Darios F, Muriel MP, Khondiker ME, Brice A, Ruberg M. Neurotoxic calcium transfer from endoplasmic reticulum to mitochondria is regulated by cyclin-dependent kinase 5-dependent phosphorylation of tau. J Neurosci 25: 4159–4168, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH, Vanhoutte PM. Endothelial dysfunction in diabetes. Br J Pharmacol 130: 963–974, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Detaille D, Guigas B, Chauvin C, Batandier C, Fontaine E, Wiernsperger N, Leverve X. Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes 54: 2179–2187, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Gao CL, Zhu C, Zhao YP, Chen XH, Ji CB, Zhang CM, Zhu JG, Xia ZK, Tong ML, Guo XR. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3–L1 adipocytes. Mol Cell Endocrinol 320: 25–33, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 107: 1058–1070, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gincel D, Zaid H, Shoshan-Barmatz V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J 358: 147–155, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care 19: 257–267, 1996 [DOI] [PubMed] [Google Scholar]
- 14.Gong D, Chen X, Middleditch M, Huang L, Vazhoor Amarsingh G, Reddy S, Lu J, Zhang S, Ruggiero K, Phillips AR, Cooper GJ. Quantitative proteomic profiling identifies new renal targets of copper(II)-selective chelation in the reversal of diabetic nephropathy in rats. Proteomics 9: 4309–4320, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Gross ML, Heiss N, Weckbach M, Hansen A, El-Shakmak A, Szabo A, Munter K, Ritz E, Amann K. ACE-inhibition is superior to endothelin A receptor blockade in preventing abnormal capillary supply and fibrosis of the heart in experimental diabetes. Diabetologia 47: 316–324, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 40: 553–560, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta 1787: 1402–1415, 2009 [DOI] [PubMed] [Google Scholar]
- 18.Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, Meinertz T, Griendling K, Harrison DG, Forstermann U, Munzel T. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 88: E14–E22, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, Han W, Oyeleye MO, Dillmann WH. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem 284: 547–555, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang QR, Li Q, Chen YH, Li L, Liu LL, Lei SH, Chen HP, Peng WJ, He M. Involvement of anion exchanger-2 in apoptosis of endothelial cells induced by high glucose through an mPTP-ROS-Caspase-3 dependent pathway. Apoptosis 15: 693–704, 2010 [DOI] [PubMed] [Google Scholar]
- 21.Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci USA 96: 13807–13812, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 1787: 1395–1401, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loomans CJ, de Koning EJ, Staal FJ, Rookmaaker MB, Verseyden C, de Boer HC, Verhaar MC, Braam B, Rabelink TJ, van Zonneveld AJ. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes 53: 195–199, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Lumini-Oliveira J, Magalhaes J, Pereira CV, Moreira AC, Oliveira PJ, Ascensao A. Endurance training reverts heart mitochondrial dysfunction, permeability transition and apoptotic signaling in long-term severe hyperglycemia. Mitochondrion 11: 54–63, 2011 [DOI] [PubMed] [Google Scholar]
- 25.Makino A, Platoshyn O, Suarez J, Yuan JX, Dillmann WH. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am J Physiol Cell Physiol 295: C221–C230, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 53: 1783–1794, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Makino A, Suarez J, Wang H, Belke DD, Scott BT, Dillmann WH. Thyroid hormone receptor-beta is associated with coronary angiogenesis during pathological cardiac hypertrophy. Endocrinology 150: 2008–2015, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 70: 391–425, 1990 [DOI] [PubMed] [Google Scholar]
- 29.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA 99: 1259–1263, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pangare M, Makino A. Mitochondrial function in vascular endothelial cell in diabetes. J Smooth Muscle Res 48: 1–26, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr 40: 171–182, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann NY Acad Sci 1201: 183–188, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE, Rizzuto R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol 159: 613–624, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399: 483–487, 1999 [DOI] [PubMed] [Google Scholar]
- 35.Shoshan-Barmatz V, Ben-Hail D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 12: 24–34, 2011 [DOI] [PubMed] [Google Scholar]
- 36.Tajeddine N, Galluzzi L, Kepp O, Hangen E, Morselli E, Senovilla L, Araujo N, Pinna G, Larochette N, Zamzami N, Modjtahedi N, Harel-Bellan A, Kroemer G. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 27: 4221–4232, 2008 [DOI] [PubMed] [Google Scholar]
- 37.Tasca C, Stefaneanu L, Vasilescu C. The myocardial microangiopathy in human and experimental diabetes mellitus. (A microscopic, ultrastructural, morphometric and computer-assisted symbolic-logic analysis). Endocrinologie 24: 59–69, 1986 [PubMed] [Google Scholar]
- 38.Tsujimoto Y, Shimizu S. The voltage-dependent anion channel: an essential player in apoptosis. Biochimie 84: 187–193, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Yoon YS, Uchida S, Masuo O, Cejna M, Park JS, Gwon HC, Kirchmair R, Bahlman F, Walter D, Curry C, Hanley A, Isner JM, Losordo DW. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation 111: 2073–2085, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Yuan S, Fu Y, Wang X, Shi H, Huang Y, Song X, Li L, Song N, Luo Y. Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J 22: 2809–2820, 2008 [DOI] [PubMed] [Google Scholar]
- 41.Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ 12: 751–760, 2005 [DOI] [PubMed] [Google Scholar]