

Abstract
Herpes simplex virus type 1 is a neurotropic herpesvirus that establishes latency within sensory neurones. Following primary infection, the virus replicates productively within mucosal epithelial cells and enters sensory neurones via nerve termini. The virus is then transported to neuronal cell bodies where latency can be established. Periodically, the virus can reactivate to resume its normal lytic cycle gene expression programme and result in the generation of new virus progeny that are transported axonally back to the periphery. The ability to establish lifelong latency within the host and to periodically reactivate to facilitate dissemination is central to the survival strategy of this virus. Although incompletely understood, this review will focus on the mechanisms involved in the regulation of latency that centre on the functions of the virus-encoded latency-associated transcripts (LATs), epigenetic regulation of the latent virus genome and the molecular events that precipitate reactivation.
This review considers current knowledge and hypotheses relating to the mechanisms involved in the establishment, maintenance and reactivation herpes simplex virus latency.
Keywords: herpesvirus, pathogenesis, miRNA, epigenetics, chromatin, reactivation, neurotropism
Introduction
Herpes simplex virus type 1 (HSV-1) and type 2 (HSV-2) are closely related ancient human pathogens responsible for a number of diseases of minor, moderate and severe pathology including oral and genital ulceration, virally induced blindness, viral encephalitis and disseminated infection of neonates. HSV-1 is typically transmitted during childhood, whilst HSV-2 is the cause of most genital herpes (Wald & Corey, 2007). Worldwide, the global prevalence of HSV-1 is approximately 90% with a prevalence of approximately 65% in the USA (Xu et al., 2002) and 52–67% in northern Europe (Pebody et al., 2004). HSV-2 infections are less frequent than HSV-1 infections with a prevalence of 10–20% in the USA and Europe (Wald & Corey, 2007). The success of HSV can be attributed to the establishment of lifelong persistent infection of the host, termed latency. During latent infection, no infectious virus is produced from infected cells, disease is absent from the host and transmission does not occur. This seemingly benign strategy is of critical importance to the survival strategy of the virus as infected cells maintained throughout the life of the host provide a reservoir for periodic reactivation – a process in which the virus re-enters the lytic replication programme.
HSV is a neurotropic virus and it is within sensory neurons that latent infection is established. Upon primary HSV replication at the oral or genital mucosa, the virus is able to infect the neuronal dendrites of sensory ganglia that innervate these tissues, and after retrograde microtubule-associated transport to the nerve cell body, the virus encounters a ‘choice' of gene expression programmes that dictates the fate of the neuron: lytic replication or latent infection. The latent virus genome is stably retained within sensory neurones and is characterized by repression of all viral lytic genes. In response to a variety of diverse stimuli, the virus can periodically reactivate to resume virus replication and produce infectious virus, which is transported anterograde back to the periphery to facilitate epithelial cell infection and consequent transmission.
Models for the study of HSV-1 latency and reactivation
Although humans are the natural host of HSV, many, but not all, aspects of pathogenesis and latency can be modelled in experimental animals (for a detailed consideration of this topic, the reader is referred to the following reviews; (Wagner & Bloom, 1997; Dasgupta & Benmohamed, 2011). The mouse represents the most commonly used animal model system, and following peripheral infection, there is localized replication in epithelial cells followed by axonal transport to innervating sensory ganglia. During the acute stage of disease between 3 and 10 days postinfection, infectious virus can be readily detected within sensory ganglia but this is rapidly cleared by the developing adaptive immune response resulting in the establishment of latency. Unlike the situation in humans, spontaneous reactivation and the development of recurrent lesions are rarely observed in mice (Hill et al., 1975; Feldman et al., 2002; Gebhardt & Halford, 2005). For this reason, most studies of reactivation rely on explantation and culture of latently infected ganglia or the induction of reactivation in vivo, for example, by transient induction of hyperthermia (Sawtell & Thompson, 1992), CD8+ T cell depletion (Freeman et al., 2007; Orr et al., 2007) or skin abrasion (Hill et al., 1980). The other commonly used animal model is the rabbit eye model which, following infection with certain strains of HSV-1, leads to periodic spontaneous reactivation and the detection of infectious virus in animal tears; a situation more analogous to the asymptomatic shedding of HSV that occurs in humans (Koelle & Wald, 2000; Mark et al., 2008). Efficient reactivation can also be induced in this model by iontophoresis of epinephrine adding to its utility (Kwon et al., 1981). The guinea pig represents the model of choice for studies of HSV-2 latency. In this model, reactivation occurs spontaneously resulting in the formation of readily scored lesions.
In vitro systems, although easier to work with and more controllable than in vivo models, rely on an ‘unnatural’ block in lytic cycle replication through the use of antiviral drugs or the use of replication defective mutants and are often criticized for being highly artificial systems. Nonetheless, certain aspects of latency and reactivation have been addressed in such systems. For example, Wilcox et al. (1990) developed a model involving the infection of primary neuronal cultures derived from dorsal root ganglia of embryonic rats. Cultures maintained in acyclovir for 7 days resulted in latency establishment, and virus could be reactivated using a variety of stimuli such as the removal of nerve growth factor (NGF). Latency can also be efficiently established in primary neuronal cultures with virus mutants deficient for immediate early (IE) gene expression or with mutants defective for virus DNA replication and infectious progeny release (Arthur et al., 2001; Terry-Allison et al., 2007). Using such mutants, no drugs are needed to control primary infection. By measuring promoter activation, reactivation has been reported in this system following NGF withdrawal, histone deacetylase inhibition and infected cell polypeptide 0 (ICP0) expression (Arthur et al., 2001; Terry-Allison et al., 2007). Latently, infected cultures have also been established from sensory ganglia derived from latently infected mice, and in this system, the most efficient triggers of reactivation are heat shock or treatment with dexamethasone (Halford et al., 1996). A nonneuronal tissue culture latency model has also been developed that uses recombinant viruses severely impaired for IE gene expression to infect various nonneuronal cell types including human fibroblasts (Preston et al., 1997; Samaniego et al., 1998; McMahon & Walsh, 2008). The viral genome establishes a tightly repressed quiescent state, and quiescent genomes are not responsive to many of the reactivation stimuli that can de-repress latent genomes maintained in neuronal cells and can only be efficiently reactivated by the supply of ICP0 in trans (Harris et al., 1989; Coleman et al., 2008; Ferenczy et al., 2011). In these different in vitro latency model systems, the viral genome is always maintained in a circular form as found in vivo, but only in the neuronal systems are latency-associated transcripts (LATs) expressed and can the virus be easily reactivated.
The process of productive HSV-1 infection
In most cell types, HSV-1 is a highly cytolytic virus and successful productive infection requires the efficient co-ordination of a large complement of genes coding for a diverse array of structural and nonstructural components. Expression of the HSV-1 lytic genes occurs in a temporal cascade that begins with immediate early (IE or α) genes and then proceeds through early (E or β), ‘leaky-late’ (L1 or γ-1), DNA replication and finally late (L2 or γ-2) gene expression (Honess & Roizman, 1974). Successful lytic replication is dependent on the expression of the viral IE genes within all infected cells. The application of protein synthesis inhibitors during infection of cell culture reveals the accumulation of six mRNAs, those corresponding to ICP0, ICP4, ICP22, ICP27, ICP47 and Us1.5, the IE genes (Honess & Roizman, 1974; Roizman et al., 2007). These genes are further defined by the presence of the TAATGARAT VP16-response elements (VRE) within their cognate promoters. VP16 is the main transcriptional activator of IE genes and is a late, structural component of the virus tegument layer, a complex network of proteins, which resides between the viral envelope and capsid (Wysocka & Herr, 2003; Roizman et al., 2007). Upon fusion and de-envelopment of the infecting virus, VP16 is released into the cytoplasm. The viral capsid is then transported to the nuclear membrane along the microtubule network and docks to the nuclear pore facilitating the release of viral DNA into the nucleus. Within the cytoplasm, VP16 is able to bind host cell factor-1 (HCF-1) protein that contains a nuclear localization sequence, and as a result of this association, VP16 is transported to the nucleus (Boissiere et al., 1999). Within the nucleus, the two proteins form a trimeric complex with the homeodomain protein Octamer binding protein-1 (Oct-1) on the consensus sequence VREs present in each IE promoter (Wysocka & Herr, 2003; Kristie, 2007). The potent VP16 activation domain in conjunction with the essential co-activator functions provided by HCF-1 then stimulates IE transcription and the HSV-1 lytic gene cascade is initiated. VP16 is however not essential for IE gene expression as mutants lacking the VP16 transactivation function can enter the lytic cycle, although with much reduced efficiency, particularly following low multiplicity infection. Hence, VP16 serves to increase the specific infectivity of virus DNA by enhancing IE gene transcription. This model of VP16 activation of IE gene expression is well understood (O'Hare, 1993; Wysocka & Herr, 2003; Kristie, 2007) but models in which it may fail are implicated and debated with regard to the establishment of neuronal latency. As such, these will be discussed in detail later in this review.
Following successful interaction of the VP16/Oct-1/HCF complex with virus DNA, the activation and accumulation of the HSV IE proteins serve to drive the transcription of viral genes whilst subverting host gene expression. Indeed, with the exception of ICP47 (an major histocompatability complex (MHC) class 1 evasion gene), all of the IE proteins are involved in gene regulation. Two of these proteins, ICP0 and ICP4, are both major transactivators of gene expression. ICP0 does not bind DNA and is a promiscuous transactivator of gene expression (Everett, 1984, 2000; O'Hare & Hayward, 1985; Cai & Schaffer, 1992). Although ICP0 is not essential for virus replication, HSV-1 mutants lacking ICP0 are driven towards a quiescent infection, resembling latency, following low multiplicities of infection (Everett et al., 2004). ICP0 is a multifunctional protein that associates with a large number of cellular proteins involved in transcription, cell cycle regulation, DNA repair and the interferon response (Everett, 2000; Hagglund & Roizman, 2004). ICP0 harbours an E3 ubiquitin ligase RING finger domain (Everett, 2000) and has been demonstrated to degrade cellular proteins including constituents of nuclear ND10 bodies; structures that otherwise restrict HSV infection in the absence of ICP0 (Everett, 1984; Van Sant et al., 2001; Boutell et al., 2002; Cuchet et al., 2011). ICP0 also disrupts the CoREST/REST repressor complex leading to dissociation of HDACs1/2 (Gu et al., 2005) and interacts with HDACs 4, 5 and 7 (Lomonte et al., 2004) implying an important role for ICP0 in antagonizing innate cellular repression of incoming viral genomes including histone-mediated gene silencing at the earliest stages of infection (Everett, 2000; Hagglund & Roizman, 2004; Everett et al., 2006). Available evidence is therefore supportive of the view that a major function of ICP0 is to create an environment that is favourable for virus transcription by directing the destruction of cellular repressors. These observations in addition to the ability of ICP0 to de-repress tightly silenced genomes in tissue culture models of quiescence (Harris et al., 1989; Stow & Stow, 1989; Preston et al., 1997; Samaniego et al., 1998; Coleman et al., 2008; Ferenczy et al., 2011) has implicated ICP0 in the earliest events of genome de-repression and has led to the attractive hypothesis that this protein could play a key role in the initiation of reactivation from latency.
Further IE genes provide numerous and important functions involved in promoting lytic infection and evading host immunity. Such functions include the inhibition of host mRNA translation by disruption of the RNA splicing machinery and promoting nuclear export of viral mRNAs by ICP27 (reviewed in Smith et al., 2005) and evasion of CD8+ T cell recognition via ICP47-mediated TAP inhibition (Früh et al., 1995; Hill et al., 1995). Collectively, the ‘purpose’ of the IE genes is to develop an active and robust environment in which to efficiently express the viral lytic programme. The collection of genes that comprise the early and late phases of replication can be broadly conceptualized as enzymes required for DNA replication and structural proteins required for assembly of infectious virus, respectively. These distinctions are necessarily concise and incomplete, but all together represent the classification of the > 80 genes encoded by the virus for productive replication.
Virus transcription during latency
In stark contrast to the lytic programme, the only transcripts readily detectable during latency are the LATs (Fig. 1) (Wagner & Bloom, 1997). LATs are a set of co-linear RNAs transcribed from a locus within the repeat regions flanking the unique long region of the viral genome (Stevens et al., 1987). Their transcription leads to the production of an 8.3 kb ‘minor LAT’ primary transcript, which is then spliced to produce an unusually stable 2.0 kb intron which is further spliced to produce an additional stable 1.5 kb intron (Zabolotny et al., 1997). Together, these two RNA species are termed the ‘major LATs’. The nomenclature of these RNAs reflects their abundance and ease of detection within experimental systems. The inefficiently debranched stable lariat structure of the major LATs produced during splicing probably explains their abundance (Farrell et al., 1991; Wu et al., 1996, 1998; Krummenacher et al., 1997; Rødahl & Haarr, 1997). In contrast, the 8.3 kb minor LAT transcript and its 6.3 kb exon are difficult to reliably detect without more sensitive in situ hybridization (ISH) or RT-PCR methods (Rock et al., 1987; Deatly et al., 1988; Zwaagstra et al., 1990; Devi-Rao et al., 1991; Arthur et al., 1993). The scarcity of these RNAs may potentially reflect a low level of expression or/and rapid processing within the nucleus, but the latter theory is more likely. A number of recent studies have identified the presence of primary microRNA (miRNA) sequences throughout the HSV-1 and HSV-2 genomes, with the vast majority localized to the LAT locus (Cui et al., 2006; Tang et al., 2008, 2009; Umbach et al., 2008, 2009, 2010; Jurak et al., 2010). Excision of six such sequences from the 6.3 kb LAT exon of HSV-1 likely contributes to the rapid turnover of minor LAT.
Fig 1.
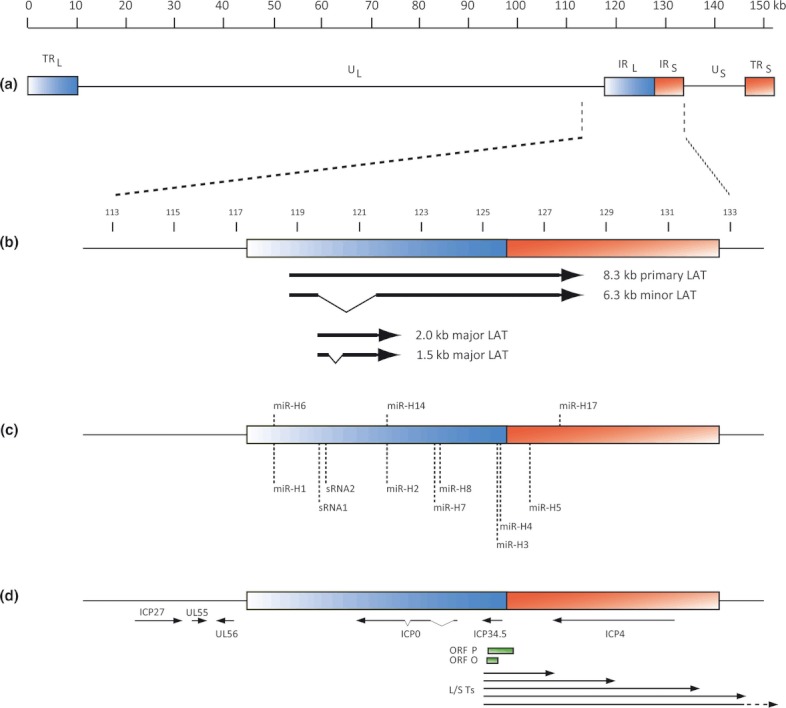
The genomic region of the HSV-1 LATs. (a) The prototypic organization of the HSV-1 genome. The 152 kb genome consists of the unique long (UL) and unique short (US) sequences, flanked by inverted repeat sequences termed the terminal and internal long and short repeats (TRL, IRL, TRS and IRS). (b) Enlarged view of the internal repeats displaying the coding location of the LAT RNA species. All LATs are processed from the 8.3 kb primary transcript, which is transcribed from a single promoter within the IRL. Above size markers represent kb. (c) Small noncoding RNAs expressed from within the LATs. With the exception of miR-H1, H6, H14 and H17, all displayed microRNA and sRNA are likely to be processed from the primary LAT transcript. (d) Lytic cycle virus genes encoded adjacent to or overlapping the LAT region. The L/ST transcripts represent a family of transcripts that are expressed late during productive infection from HSV-1 mutants lacking functional ICP4 (Yeh & Schaffer, 1993). Note that all genes displayed within the repeat sequences in b–d are diploid.
The primary research focus with regard to the LATs has aimed towards understanding their role during latent infection. To date, this is still an area of quite some debate, but via the use of various small animal and tissue culture models, gross phenotypes and mechanisms of action are becoming apparent.
Functions of the LATs
For a gene under such scrutiny concerning latent infection, the first perhaps surprising observation is that the LATs are not essential for latency establishment, maintenance or reactivation (Javier et al., 1988; Sedarati et al., 1989; Steiner et al., 1989). However, the most consistent in vivo phenotype associated with LAT-negative virus mutants has been a reduction in the efficiency of virus reactivation. This suggests that LATs must play a direct role during reactivation, but this theory has since been challenged by careful analysis of the murine latent cell reservoir by single cell contextual analysis (CXA) of viral DNA (Thompson & Sawtell, 1997). This study demonstrated that the LAT-negative viruses employed only established latency in 1/3 as many neurons as wild-type (WT) infection and that this correlated with decreased recovery of virus from mouse trigeminal ganglia following hyperthermic stress (a reactivation stimulus). Furthermore, hyperthermic stress applied during acute infection with LAT-negative viruses could yield similar numbers of latently infected neurons as WT virus, and under these preconditions, recovery of reactivated virus was equivalent (Thompson & Sawtell, 1997). Whilst it is likely that different infection models and specific methodologies affect the efficiency of latency establishment and reactivation (Perng et al., 2001; our unpublished observations), these data do provide a compelling argument that observed deficits in reactivation of LAT deficient mutants are the result of ablation of a LAT-encoded function responsible for enhancing the efficiency of latency establishment. Despite this, other studies have observed poor reactivation competence of LAT-negative virus when latent virus DNA loads are comparable to WT virus both in mice (Leib et al., 1989; Hoshino et al., 2009) and rabbits (O'Neil et al., 2004), suggesting that LATs influence latency during multiple phases of infection. However, with the appreciation that the latent virus DNA copy number within individual neurons can vary by greater than three orders of magnitude (Sawtell, 1997), whole ganglion quantification of virus DNA loads may not be an accurate reflection of the frequency of infected cells, an important variable that will be discussed later in this review.
How LATs actually mediate these gross effects in vivo is of clear importance to the strategy of persistence for HSV. A number of functions have been attributed to the LATs, but their mechanisms of action have not been forthcoming. Despite reports stating otherwise (Thomas et al., 1999; Henderson et al., 2009; Jaber et al., 2009), it is not generally accepted that the LATs are translated to a functional protein in vivo. However, with the recent description of LAT-encoded miRNAs (Fig. 1) (Cui et al., 2006; Tang et al., 2008, 2009; Umbach et al., 2008, 2009, 2010; Jurak et al., 2010), a bigger picture of the latent HSV transcriptome is becoming apparent.
Repression of lytic gene expression
The expression of lytic cycle genes by the virus is detrimental to the progression of infection into latency. It would therefore be advantageous that expression of lytic transcripts could be curtailed by the virus in order to facilitate latency establishment. This idea necessitates the existence of an ‘active’ path into latency in a population of neurons, and experimental data suggest LATs may indeed exert this function. In 1997, two studies demonstrated the increased abundance of lytic gene transcripts during acute (Garber et al., 1997) and latent infection (Chen et al., 1997) of murine TG with a LAT deletion mutant. Although at low abundance, the levels of lytic cycle transcripts detected during latency were elevated by 5–10-fold with a LAT deletion mutant (Chen et al., 1997). It is tempting to speculate that a LAT-negative mutant could be at a greater propensity of spontaneous reactivation within murine TG, especially given the increased levels of TK mRNA, an early transcript dependent on the presence of ICP4 for expression (Chen et al., 1997). Subsequently, the production of mouse neuronal cell lines expressing various forms of the LAT locus continued to provide evidence that LAT could exert a repressive effect on IE mRNA levels (Mador et al., 1998). In the same study, the authors demonstrated that productive HSV replication was reduced in cell lines encoding the LAT locus during low multiplicity infection, but not in cell lines with a deletion in the promoter driving LAT transcription. However, this may not be the whole story. Giordani et al. (2008) have recently provided evidence that within a rabbit trigeminal ganglia model of infection, expression of the LATs leads to an enhanced accumulation of lytic transcripts during latency, directly conflicting conclusions drawn from investigations in the mouse. Further work will be required to ascertain the influence of different virus strains, routes of infection and specific animal model towards these conclusions, but the potential disagreement between animals is pertinent given their use in modelling human disease.
Currently, the majority of these aforementioned data support a role for the LATs in mediating the downregulation of genes required for lytic replication, but none of these studies provide a mechanistic explanation for this effect. It had been noted that the 2.0 kb LAT was encoded in the opposite orientation and overlapped the coding sequence of the virus IE gene ICP0, suggesting a potential regulatory role between maintenance and reactivation during latency. However, an antisense mechanism for the repression of ICP0 expression was discounted after investigation showed that provision of the 2.0 kb in trans failed to prevent accumulation of ICP0 mRNA or protein (Burton et al., 2003). Additionally, LAT sequences mapping to just the first 1.5 kb of minor LAT primary transcript are sufficient for efficient reactivation in animal models, and this region does not include the overlapping sequence between LAT and ICP0 (Perng et al., 1996).
Since the discovery of RNA interference roughly a decade ago, it has been swiftly recognized that DNA viruses encode the capacity to regulate gene expression via small noncoding RNAs (sRNAs). In particular, virus-encoded miRNAs are found in the majority of herpesviruses studied to date (Umbach & Cullen, 2009; Jurak et al., 2011) and have been implicated in the regulation of several key processes such as latent/lytic cycle control, immune evasion and cell survival (Boss et al., 2009). Not only do these regulatory sRNA genes raise the coding capacity of these viruses without large increases in genome length, but also by achieving their effects without coding for proteins, small RNA-based gene products remain intrinsically nonimmunogenic. This latter concept puts these molecules in a position of great value to viruses that rely on persistence and immune evasion to complete their infection cycles. HSV-1 and HSV-2 have been shown to encode 16 and 18 miRNAs, respectively, and there is a concentration of miRNAs within or adjacent to the LAT locus. In the case of HSV-1, six miRNAs (miR-H2, H3, H4, H5, H7 and H8) are encoded within the 8.3 kb primary LAT (Fig. 1). MiR-H1 and H6 are located just upstream of the LAT transcription start site, are abundant during productive infection and represent overlapping miRNAs with miR-H6 being transcribed in the opposite orientation to LAT (Cui et al., 2006; Umbach et al., 2008; Jurak et al., 2010). In addition to being expressed during productive infection, miRs H1 to H8 are expressed during latency. However, there is considerable variation in the expression levels of individual miRNAs during latency, with miRs H2, H3 and H4 being the most abundant as deduced by the frequency of sequence reads obtained from deep sequencing and/or quantitative stem-loop real-time reverse transcription PCR (Umbach et al., 2008, 2010; Jurak et al., 2010, 2011; Kramer et al., 2011). The targets of only four HSV-encoded miRNAs have been defined to date and are so far restricted to virus-encoded RNAs. In 2008, Umbach and colleagues were the first to demonstrate that HSV-encoded miRNAs were capable of downregulating the translation of viral genes. By co-transfecting plasmids over-expressing individual miRNA and their predicted protein targets, HSV-1 miR-H2 and miR-H6 were shown to reduce ICP0 and ICP4 protein abundance, respectively, whilst not affecting cognate mRNA levels (Umbach et al., 2008). Studies following this demonstrated that HSV-2 miR-H2 can inhibit the expression of ICP0 and HSV-2 miR-H3 and H4 can downregulate the expression of the neurovirulence factor ICP34.5 (Tang et al., 2008, 2009). LAT-encoded miRNAs have been detected during latency in animal models as well as analysis of postmortem human tissue (Tang et al., 2008, 2009; Umbach et al., 2008, 2009, 2010; Jurak et al., 2010) but their exact role in the regulation of the latency/reactivation cycle is not known. In the case of miRs targeting IE genes, a role in suppression of entry into the lytic cycle and therefore enhancement of latency establishment or maintenance is attractive and warrants further attention. It will be of particular interest to determine the impact of specific miRNA mutations/deletions on IE gene expression in the context of mutant virus infection both in vitro and in vivo. Defining the role of virus-encoded miRNAs in the natural history of HSV, infection will however prove challenging owing to the inherent limitations of the available animal model systems and a lack of information concerning potential cellular targets.
So do these studies relegate the LAT transcript to merely a large primary microRNA precursor? This is intriguingly not so. Indeed, the previously cited study by Mador et al. utilized cell lines that encoded, a region of LAT in which no miRNA proposed to target ICP0 or ICP4 is found. However, within this region, two sRNA species have recently been discovered (Peng et al., 2008). Both of these sRNAs were found capable of inhibiting productive infection in tissue culture, but to different extents. Despite showing only weak repression of virus replication by comparison with sRNA1, sRNA2 exhibited downregulation of ICP4 protein accumulation whilst not affecting cognate mRNA levels (Shen et al., 2009). At 62nt and 36nt length (sRNA1 and sRNA2, respectively), these RNAs do not appear to function as microRNA, although owing to the reduction in ICP4 protein levels in these experiments such an RNA interference mechanism may occur (Shen et al., 2009).
It is clear that small noncoding RNAs such as miRNA are likely to be central to the regulatory effort of the virus towards the regulation of lytic gene expression during latency or its establishment.
Promotion of cell survival
Another major observation attributed to the expression of the LATs is the inhibition of cell death in response to virus infection. HSV-1 and HSV-2 encode a number of genes that interfere with the induction of apoptosis during lytic replication, including glycoproteins gD and gJ, as well as protein kinase Us3, ICP27 and ICP10 (Leopardi et al., 1997; Jerome et al., 1999; Zhou et al., 2000; Perkins et al., 2002). However, during latency, these gene products are presumably not expressed and therefore cannot counter triggers of apoptosis that may be encountered during latency establishment, maintenance and at the earliest stages of reactivation. A number of studies have attributed anti-apoptotic functions to the LATs. Perng et al. (2000) observed extensive apoptosis in rabbit trigeminal ganglia during the first 2 weeks of infection, utilizing TUNEL staining and antibody detection of caspase-3 cleaved poly (ADP-ribose) polymerase. Another alphaherpesvirus, bovine herpesvirus 1 (BHV-1), expresses a latency-related (LR) protein, which has a demonstrated anti-apoptosis function (Ciacci-Zanella et al., 1999; Henderson et al., 2004) and expression of this protein from HSV-1 deleted for LAT expression rescued the reactivation phenotype of this virus in rabbits and mice (Perng et al., 2002). Two further studies observed rescue of the LAT-negative reactivation phenotype utilizing the baculovirus anti-apoptosis gene product cpIAP or cellular FLICE-like inhibitory protein instead of LR BHV-1 protein (Jin et al., 2005, 2008). However, none of these three reports characterized the latent DNA loads during infection and so whether these viruses establish equivalent latency to WT virus is not clear. CXA of latently infected murine TG has shown that during LAT-negative virus infection, an increased loss of neurons occurs during establishment (Thompson & Sawtell, 2001). Additionally, both the absence (Thompson & Sawtell, 2001) and presence (Branco & Fraser, 2005) of apoptosis in murine TG have been reported. If anything is clear from these collective data, it is that the presence of a virally encoded product comprising or encoded within LAT is capable of supporting cell survival, and that this capacity is beneficial for the establishment and reactivation phenotypes of HSV. The mechanism by which this survival is mediated has been studied using various in vitro methodologies.
Inman et al. (2001) demonstrated that the sequences within the first 1.5 kb of LAT that are essential for efficient spontaneous reactivation in rabbits also map to regions of the LAT that promote survival of monkey kidney and mouse neuroblastoma cells following treatment with pro-apoptosis compounds such as etoposide and sodium butyrate. This work functionally linked the in vitro assessment of apoptosis inhibition and in vivo reactivation phenotype. This work was corroborated by a further study in HeLa and neuron-like SY5Y cells that mapped inhibition of caspase-8-mediated apoptosis to sequences within the LATs (Ahmed et al., 2002). Together, these studies agreed that anti-apoptosis activity of the LATs mapped to the 3′ end of the first exon, as well as in the 5′ end of the stable 2.0 kb LAT intron (Inman et al., 2001; Ahmed et al., 2002). These studies were furthered with the production of murine neuroblastoma cell lines expressing the first 3225 bp of LAT from the HSV latency associated promoter (LAP). These cell lines were more resistant to cold shock-induced apoptosis, leading to increased survival and reduced levels of DNA laddering compared to both WT cells and cells in which the LAP had been deleted from the LAT construct (Carpenter et al., 2007). Importantly, this study also demonstrated that cell lines with robust LAT expression were able to inhibit the cleavage (and thus activation) of caspase 3 in the absence of other virus genes. Others have also found that LAT expression promotes survival of primary neuronal cultures following NGF withdrawal from culture (Hamza et al., 2007).
Whilst these studies demonstrate a functional role in cell survival, a mechanism by which LAT could exact this outcome is still lacking. As previously described in this review, two sRNA species have been discovered within the 3′ end of the LAT exon (Peng et al., 2008). In a functional analysis, an increase in cell survival following cold shock could be observed in cells co-transfected with plasmids expressing both sRNA (Shen et al., 2009). These results appear not to be consistently replicated for each sRNA in isolation, but sRNA1 appears to mediate the majority of this activity as suggested via mutagenesis analysis (Shen et al., 2009). As previously described, the length of these RNAs indicates that they are not miRNA. Therefore, even if these RNAs were to represent bona fide effector molecules, the mechanistic action by which the LATs promote cell survival currently still eludes our understanding.
Latency establishment
HSV-1 latency establishment in sensory neurons has long been thought to be the default path of a failure to initiate IE gene expression (reviewed in Preston, 2000; Efstathiou & Preston, 2005). Consistent with this view is the fact that virus mutants severely impaired for the initiation of lytic cycle, such as mutants in one or more IE genes or VP16 (Chiocca et al., 1990; Dobson et al., 1990; Katz et al., 1990; Steiner et al., 1990; Ecob-Prince et al., 1993; Sedarati et al., 1993; Marshall et al., 2000) are able to establish latent infection in vivo. Moreover WT virus can establish latent infection in sensory ganglia that do not directly innervate the site of primary infection and where no previous IE gene expression can be detected at any time prior to the establishment of latency (Speck & Simmons, 1992; Lachmann et al., 1999). It appears that viral DNA delivery into the neuronal nucleus is the only requisite for the establishment of latency.
As described earlier, efficient lytic cycle gene expression is dependent upon the transactivating function of the VP16-induced complex that is formed by the structural tegument protein VP16, HCF-1 and Oct-1 (reviewed by Wysocka & Herr, 2003). In sensory neurons, however, the formation of the VP16-induced complex is thought to be severely impaired owing to restrictions in the availability of all its members. It has been suggested that VP16 might not be efficiently transported along axons such that insufficient amounts reach the neuronal cell body (Kristie & Roizman, 1988; Kristie et al., 1999). HCF-1 has been shown to have a distinct cellular localization in sensory neurons, where it has been detected exclusively in the cytoplasm (Kolb & Kristie, 2008) and is therefore unavailable to participate in the formation of a VP16-induced complex. Oct-1 on the other hand has been shown to be downregulated in neuronal cells (Lakin et al., 1995). Other POU domain transcriptional factors are present in neurons, such as N-Oct2, N-Oct3, Brn-2, Brn-3a or Brn-3b and although these proteins can recognize the same sequences in IE promoters, they mostly have repressive effects (Lillycrop et al., 1991; Hagmann et al., 1995; Latchman, 1999). These restrictions for the formation of the VP16-induced complex and consequently inefficient IE promoter activation are in agreement with latency being the default path of an IE gene block; however, there is evidence to support the theory that there might be other paths leading to latency establishment.
Following infection of experimental animals, HSV-1 is able to go through an acute phase of infection in the ganglia that directly innervate the site of infection and there is an early divergence of the pathways leading to either productive or latent infection (Margolis et al., 1992; Speck & Simmons, 1992; Lachmann et al., 1999). Nonetheless, a small proportion of neurones (< 1%) expresses both viral antigen and LATs during acute infection (Margolis et al., 1992; Speck & Simmons, 1992) raising the possibility that expression of lytic cycle genes may not preclude entry into latency. It has also been shown that a subpopulation of latently infected neurons harbour high numbers of viral DNA molecules that can go up to thousands of genomes per cell (Efstathiou et al., 1986; Slobedman et al., 1994; Sawtell, 1997). If these neurons are a result of abortive lytic infection post-DNA replication or the result of high viral seeding from the periphery/ganglia is currently unclear.
Studies with HSV-1 mutants lacking a functional TK gene have shown that neurones containing high latent viral DNA loads could still be achieved even in the absence of TK, demonstrating that neurons in vivo have the potential to survive very high viral inputs transported from peripheral sites (Thompson & Sawtell, 2000; Wakim et al., 2008). However, the question still remains whether the neurons that receive high numbers of genomes would have survived infection with WT virus because TK mutants are severely compromised for replication in neurons.
One possible mechanism that could account for this high loading of viral DNA would be the noncytolytic CD8+ T cell inhibition of neuronal HSV-1 replication (Khanna et al., 2004; Decman et al., 2005). This mechanism is able to arrest HSV-1 reactivation in a noncytolytic fashion owing to a block in the viral gene expression cascade induced by IFN gamma secretion and the degradation of the ICP4 protein by CD8+ T cell-derived Granzyme B (Knickelbein et al., 2008) and could explain the CD8+ T cell-dependent termination of virus replication in the absence of neuronal destruction during the late stages of acute ganglionic infection (Simmons & Tscharke, 1992). The observation that immune infiltrates composed largely of CD8+ T cells is a feature of both latently infected murine and human trigeminal ganglia (Liu et al., 1996; Theil et al., 2003) is explained by the recognition of virus antigen on rare neurones undergoing spontaneous reactivation. CD8+ T cells are therefore likely to play an important role in the control of reactivation in a manner independent to LAT-encoded miRNAs suppression of IE gene expression (Held et al., 2011). Surprisingly, the CD8+ T cells involved in this mechanism in the C57BL/6 mouse model have been found to be specific for an immunodominant glycoprotein B (gB) epitope and not IE proteins. gB is a leaky-late gene and, the fact that the reactivating neuron was recognized by a gB-specific CD8+ T cell indicates that IE and early genes are likely to be expressed prior to a CD8+ T cell-mediated block in reactivation. These data indicate that, not only are neurons able to survive high viral genome inputs, they can also survive extensive viral gene expression (Kramer et al., 1998; Ramachandran et al., 2010). This is in agreement with studies using primary neuronal cultures where upon infection with replication defective mutants, transient IE promoter activity was detected in the majority of the neurons in culture prior to the establishment of latent infection (Arthur et al., 2001). A previous study by Wilcox and colleges revealed that primary neuronal cultures infection by an ICP0 ring domain mutant resulted in significantly less neurons expressing LATs in comparison with WT virus. In these experiments, the levels of LAT expression were found to correlate with viral DNA loads revealing a deficit in the establishment of latent infection by the ICP0 mutant. This effect could not be detected with an ICP4 mutant virus or when ICP0 was supplied in trans by an adenovirus vector (Wilcox et al., 1997). These data suggest that under certain circumstances, IE ICP0 expression may function to enhance latency establishment. A more recent study using Cre reporter animals and WT HSV-1 recombinants with Cre recombinase under control of the ICP0 promoter revealed that there is a subpopulation of latently infected cells that experience ICP0 promoter activation prior to latency establishment in vivo (Proença et al., 2008). A continuation of this study has extended the range of promoters tested and has revealed that ICP4 promoter activity is also compatible with latency establishment whilst TK and VP16 promoter activation is largely incompatible (Proença et al., 2011). These data indicate that a subpopulation of latently infected cells in vivo experience IE promoter activity prior to genome silencing and the establishment of neuronal latency. Of potential relevance, here is the observation that alternative spliced forms of ICP0 have been reported during productive infection (Everett et al., 1993; Carter & Roizman, 1996), at least one of which functions as a dominant negative repressor of ICP0-mediated transactivation in transient expression assays (Weber & Wigdahl, 1992; Weber et al., 1992). Whether the results obtained from reporter mouse model systems is indicative of IE protein expression being compatible with the establishment of latency is currently unclear; however, the results obtained with this model system are difficult to reconcile with a simple default model of latency establishment and argue for the operation of posttranscriptional mechanisms to restrict lytic cycle progression.
Studies with replication defective mutants have shown that the minimal requirement for latency establishment seems to be the delivery of viral DNA to the neuronal nucleus. However, as discussed, WT viruses may follow alternate pathways into latency. Therefore, latency can be established via mechanisms that do not involve a complete block in viral gene expression and might even rely on some viral proteins/transcripts in order to efficiently establish latent infection.
The HSV genome and epigenetic regulation of gene expression
HSV-1 possesses a linear double-stranded DNA genome of 152 kb (reviewed by McGeoch et al., 2006). The virus genome is divided into two ‘unique’ regions (unique long and short or UL and US regions) and each is flanked by inverted repeats (Fig. 1). Following nuclear entry, the linear genome circularizes before the advent of viral protein production (Garber et al., 1993; Strang & Stow, 2005). During lytic infection, early genome replication is achieved by a theta replication mechanism initiated at three redundant origins of replication (two copies of oriS and one copy of oriL) but rolling circle replication later predominates, forming ‘endless’ DNA lacking termini (reviewed by Boehmer & Nimonkar, 2003; Muylaert et al., 2011). This concatemeric DNA is later cleaved to monomeric units during packaging into newly formed capsids. In contrast to lytic infection, HSV DNA isolated from murine and human latently infected tissue demonstrates a lack of genomic termini, suggesting the HSV genome is maintained as a circular episome during latency (Rock & Fraser, 1983; Efstathiou et al., 1986) and the latent viral genome is assembled into nucleosomes (Deshmane & Fraser, 1989). In contrast to members of the lymphotropic gammaherpeviruses, that establish latency within dividing cell populations, the neuronal cell tropism of HSV latency negates the requirement for virus-encoded cis- and trans-acting genome maintenance functions.
During infection, HSV DNA entering the nucleus becomes rapidly associated with histones, in a cellular response presumably aimed at silencing foreign gene expression (Fig. 2). Whilst the role of this regulation on gene expression shall be briefly considered, readers are directed to recent, thorough reviews on chromatin control of HSV infection (Knipe & Cliffe, 2008; Kutluay & Triezenberg, 2009; Bloom et al., 2010; Nevels et al., 2011). In contrast to the lymphotropic gammsherpesviruses, the HSV genome does not appear to be methylated at CpG residues during latency; thus, this form of epigenetic regulation is presumably not utilized by the virus (Dressler et al., 1987; Kubat et al., 2004b). Instead, the association of histones consistent with partial or unstable nucleosomal structure (Lacasse & Schang, 2010) is believed to mediate epigenetic control of specific viral promoters. This control is thought to be regulated by posttranslational modification (PTMs) of histones occupying key lytic and latent viral promoters (Fig. 2). Indeed, during lytic infection, activating euchromatin-like modifications such as acetylation of H3K9 (histone H3, lysine 9 from the N-terminus) and H3K14 are enriched upon lytic gene promoters, whilst repressive heterochromatin-like modifications such as di-methylation of H3K9 are underrepresented (Herrera & Triezenberg, 2004; Kent et al., 2004; Huang et al., 2006). In direct contrast, during latency, the actively transcribed LAT locus undergoes enrichment of acetylated H3K9 and H3K14 at the LAT promoter and enhancer, with these modified histones not present at the ICP0 promoter or DNA polymerase gene (Kubat et al., 2004a, b). During latency, viral lytic genes are enriched with methylated H3K27 (a marker of facultative heterochromatin) as well as methylated H3K9 (a marker of constitutive heterochromatin) (Wang et al., 2005b), demonstrating that the latent HSV genome is associated with both reversible and nonreversible repressive chromatin modifications (Cliffe et al., 2009; Kwiatkowski et al., 2009). Induction of reactivation by the explantation of latently infected mouse dorsal root ganglia results in a decrease in LAT RNA and is associated with a decrease in association with acetylated H3K9/K14 on the LAT promoter and a concomitant increase in the association of these activating histone marks on the now transcriptionally active ICP0 promoter (Amelio et al., 2006). Studies of LAT mutant viruses in the mouse have revealed a reduction in the association of both constitutive and facultative chromatin with lytic gene promoters (Wang et al., 2005b; Cliffe et al., 2009) suggesting that LATs facilitate the assembly of heterochromatin on lytic cycle promoters during latency. The ability of LATs to help maintain the epigenetic silencing of lytic gene promoters is consistent with phenotypic analyses of LAT deficient mutants, which are associated with increased lytic gene expression during latency (Chen et al., 1997; Garber et al., 1997). In contrast to the situation in the mouse, studies in rabbits have led to an opposing view on the role of LATs in the regulation of lytic cycle promoter activity. Thus, in rabbits, LATs function to keep the virus genome in a more transcriptionally active state (Giordani et al., 2008). Furthermore, a recent report using a mouse model has indicated that LATs may function to suppress the accumulation of facultative heterochromatin on the latent virus genome (Kwiatkowski et al., 2009). At the present time, it is difficult to reconcile these fundamentally opposing views of the role of LATs in epigenetic regulation of the latent virus genome. Clearly, further studies are warranted to clarify the precise role of LATs in epigenetic control of the latent virus genome and in particular to what extent results can be influenced by virus strain and animal model differences.
Fig 2.
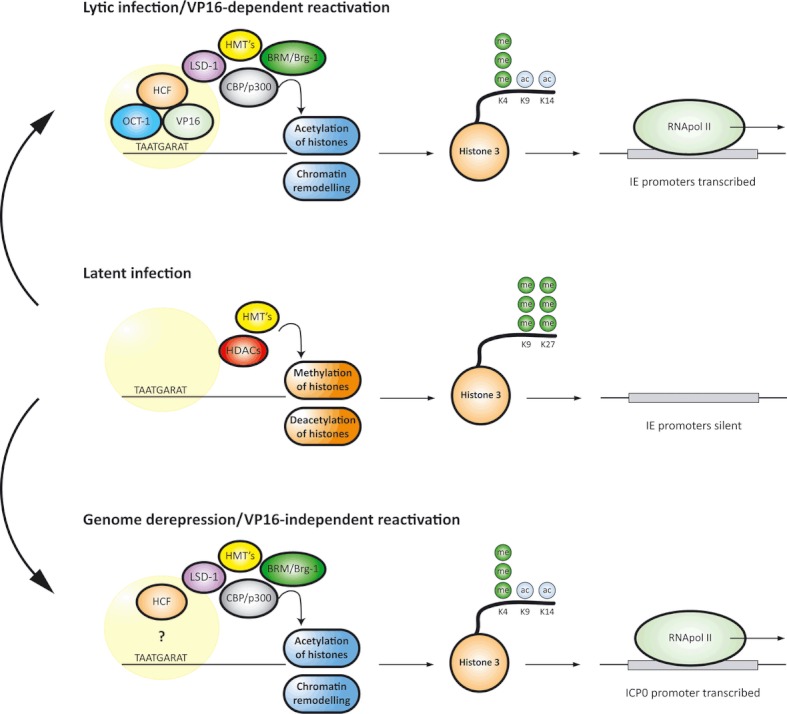
Chromatin control of HSV-1 gene expression. Two models of reactivation are shown, involving the specific activation of the virus regulatory proteins VP16 or ICP0 by cellular factors. Lytic infection/VP16-dependent reactivation: The VP16/Oct-1/HCF trimeric complex interacts with a number of co-activators including histone acetyltransferases (CBP/p300), chromatin remodelling factors (BGRF-1 and BRM), histone methyltransferases (HMT's) and lysine-specific demethylase-1 (LSD-1). Recognition of IE promoters by the VP16-induced complex results in activation of IE gene expression and prevention of repressive histone accumulation on the virus genome. Histone modifications associated with active transcription, such as H3K4me3 and H3K9ac, are associated with the virus genome during lytic infection. Latent infection: In the absence of virally encoded transactivators, HMT's and histone deacetylases (HDACs) maintain viral lytic promoters in a repressed chromatin state characterized with the accumulation of histone modifications associated with repression, such as H3K9me3, resulting in silencing of gene expression. An exception to this global silencing is the LAT locus, which is actively transcribed during latency. Genome depression/VP16-independent reactivation: In the absence of VP16, the presence of some or all cellular co-activators involved in the activation of IE promoters during lytic infection could facilitate gene expression following reversal of heterochromatin-based silencing. In this model, specific acetylation of the ICP0 promoter region would result in ICP0 gene expression, leading to further depression across the virus genome and reactivation.
The role of the CoREST/REST repressor complex in the regulation of productive and latent infection
Recruitment of the lysine-specific demethylase LSD1 to IE promoters by HCF1 has been shown to be crucial for the initiation of IE transcription during both productive infection and reactivation from latency (Liang et al., 2009). LSD1 is normally found as a component of a cellular repressor complex that includes histone deacetylase 1 or 2 (HDAC1/2) and co-repressor of REST (CoREST). In this context, the LSD1 repressor complex is recruited to specific DNA elements by repressor element-1 silencing transcription (REST) and functions as a corepressor by demethylating histone H3K4 and functions to repress neuronal gene expression in nonneuronal cells. How then does LSD1 function to stimulate viral IE gene expression? The function of LSD1 is known to be context-dependent (Lee et al., 2005; Shi et al., 2005) and under certain circumstances and when dissociated from HDAC1/2 and CoREST can function as a co-activator by the demethylation of repressive H3K9 marks. Thus, recruitment of LSD1 to IE promoters by virtue of its interaction with HCF-1 antagonizes the accumulation of repressive methylated H3K9 marks at IE promoters to facilitate IE transcription. ICP0-mediated disruption of the CoREST/REST repressor complex also plays a role in the transition from IE to early gene expression. Thus, ICP0 has been shown to interact with CoREST and functions to disrupt the cellular corepressor complex, which includes CoREST, REST and HDAC1/2 (Gu et al., 2005; Gu & Roizman, 2007, 2009). Biologically, disruption of CoRest/REST complex by ICP0 is of importance because the expression of a dominant negative CoREST protein in place of ICP0 results in improved replication of an ICP0 mutant virus (Gu & Roizman, 2007) and decreased replication of a virus mutant containing a lesion within the CoREST interaction region of ICP0 has been observed (Gu & Roizman, 2009). However, whether CoREST is directly involved in repressing HSV-1 infection in the absence of ICP0 is less clear because depletion of CoREST by ShRNA did not improve the replication of an ICP0 mutant in vitro (Everett, 2010). During latency in vivo, it is also unclear whether CoREST plays a direct role in the repression of viral gene expression and hence the maintenance of latency. Although it has been shown that a virus recombinant encoding dominant negative REST incapable of recruiting CoREST or LSD1 is more virulent than WT or control viruses (Du et al., 2010; Roizman, 2011), it is unclear whether this phenotype is owing to the suppression of recruitment of the repressor complex to the virus genome or an indirect consequence of the de-repression of cellular gene expression. Further work in this area is clearly warranted, and in particular, further studies are required to determine whether the CoREST/REST repressor complex is recruited to the virus genome during neuronal latency.
The nonuniformity of HSV latency
As has already been mentioned in this review, a striking feature of HSV latency concerns the nonuniformity of the latent state. Thus, viral DNA is not evenly distributed amongst latently infected neurons, and it is well established that the viral genome load can vary significantly, with the majority of cells harbouring 10–100 copies, whilst a smaller proportion can harbour > 1000 copies of virus DNA (Sawtell, 1997). As a consequence, analysis of latent virus DNA and in particular ChIP analyses are population weighted. The significance of this genome heterogeneity is unclear, although high latent genome copy number may be a predisposing factor for reactivation (Sawtell et al., 1998). The nonuniformity of latency also extends to the expression of LATs. Estimates of the number of neurones harbouring virus DNA by in situ PCR (Mehta et al., 1995; Maggioncalda et al., 1996) and laser capture microdissection (Chen et al., 2002; Wang et al., 2005a, b) indicate that LATs are transcribed in only a fraction (approximately 30%) of all latently infected cells. Whether the LAT promoter is ever active within cells negative by ISH is not clear, but data from our laboratory utilizing ROSA26R Cre reporter mice suggest that LAT expression occurs in a larger proportion of infected cells at some point during infection, indicative of dynamic expression of LATs during latency (Proença et al., 2008). Consistent with this view, data from LAT transgenic mice suggest that the LAT promoter can be expressed in the majority of sensory neurones (Gussow et al., 2006). Nevertheless, given evidence to date, precisely when the LATs are ‘needed’ and the role of LATs in the minority of LAT-expressing cells during latency remain pivotal questions.
To contemplate, HSV infection in terms of different neuronal populations necessitates the notion that latently infected neurons are not equivalent. Further still, the factors that pertain to this lack of equivalence are numerous and diverse. It has been shown that HSV-1 and HSV-2 preferentially establish latency and express LATs in different populations of sensory neurons identified by the expression of the cellular markers A5 and KH10, respectively (Margolis et al., 2007; Imai et al., 2009). Furthermore, whilst receptive to viral entry, A5+ neurons are intrinsically nonpermissive for productive infection and reactivation with HSV-1 but are able to support productive HSV-2 infection (Bertke et al., 2011). This intriguing observation indicates that the nonpermissiveness of A5+ neurones is largely specific for HSV-1. During mouse infection with HSV-1 mutants possessing HSV-2 LAT regions (and vice versa), these preferential sites swapped between viruses, defining the LAT as the viral determinant of this specificity (Margolis et al., 2007; Imai et al., 2009). These data demonstrate unequivocally that regulatory pathways significant to HSV infection act within individual neuronal subsets and that any concept of a homogenous population of neurons within a ganglion is a gross oversimplification. These data do not as of yet specifically address the functional bias upon replication exerted by these A5+ and KH10+ populations, but the authors do cite known differences in growth factor signalling between these cells. Specifically, A5+ neurons respond to NGF and KH10+ cells express receptors for glial cell-derived neurotrophic factor (GDNF). Given that approximately 50% of all neurones latently infected with HSV-1 are A5+ (Yang et al., 2000), it will be of particular interest to determine the characteristics of the remaining latent sites with respect to neuronal subtype and permissiveness to infection and reactivation competence. Work by others has shown that maintenance of quiescent HSV-1 infection in neuronal cultures in vitro requires NGF (Wilcox & Johnson, 1987) interaction with its high-affinity receptor, TrkA. Removal of NGF from neuronal culture resulted in recovery of productive infection, whilst only modest replication occurred after the removal of GDNF (Camarena et al., 2010). It would be of interest to explore whether A5+ neurons are rendered at all permissive to HSV-1 replication in the absence of NGF, whilst remaining viable in culture, as otherwise in the light of current data, this subtype appears to represent a ‘dead-end’ for productive HSV-1 replication. This is especially important given the interpretation of another area of nonuniformity during latent infection: the genome copy number per infected cell and the number of infected cells within a ganglion.
The implementation of CXA of viral DNA provided the first appreciation of virus genome copy number variability within latently infected cells (Sawtell, 1997). Utilizing CXA, it has been demonstrated that within a single neuron, latent HSV copy number can range from a single copy to > 1000 (Sawtell, 1997). At this time, it had already been hypothesized that latent cells bearing large HSV copy numbers may be predisposed towards reactivation relative to lower copy number cells and indeed CXA reveals that efficiently reactivating HSV-1 strains such as McKrae and 17syn+ display significantly larger average genome copy numbers compared to the poorly reactivating strain KOS (Sawtell et al., 1998). Interpreted one way, this observation could be at odds with the presence of ‘dead-end’ neuronal subtypes suggested by data mentioned previously. Any cells nonpermissive for replication could theoretically contain very high genome copy numbers if sequentially infected throughout primary disease (neurons directly innervating infection at the periphery, for example), so long as all productive replication was aborted in these cells. However, a mechanism for the onset of permissiveness of infection in such cells could render such neuronal populations highly effective reservoirs of reactivation. Another explanation for copy number variation that negates the requirement for superinfection of latently infected cells is that productive infections can be actively silenced at late stages of the HSV replication cycle. During ROSA26R reporter mouse infection, latently infected neuronal populations marked for ICP0 promoter activation represent one-third of the total infected cell reservoir (Proença et al., 2008). Follow-up data from our laboratory demonstrates the consistent presence of small numbers of neurones that have experienced early and late gene promoter activity (Proença et al., 2011). The implication of detecting latently infected cells marked for late promoter activity is that these cells are likely to have experienced and survived HSV genome replication before entering latency and thus may represent the small but constant population of high genome copy neurons detectable by CXA.
Finally, the form of heterochromatin that silences lytic gene expression during latency provides an additional layer of complexity. As previously mentioned, heterochromatin is divided into two classes: facultative and constitutive, with the former representing a ‘reversible’ mechanism of gene silencing, capable of reverting to euchromatin and the latter a more stable form of restriction associated with the centromeres and telomeres of mammalian DNA. In 2009, ChiP analyses by Cliffe et al. and Kwiatkowski et al. observed that lytic gene promoters could be found enriched for both facultative and constitutive chromatin PTMs within pooled samples of latently infected mouse tissue. These data raise the possibility that chromatin modification could be distinct not just between genomes in different cells, but also within the same cell (Bloom et al., 2010).
Reactivation from latency
Lytic cycle viral gene products are not generally detected in latently infected neurons; therefore, reactivation from latency is thought to occur in the absence of pre-existing virus proteins by cellular mechanisms. The cellular factors involved in triggering reactivation of the latent genomes are currently unknown; however, several stimuli have been found to induce reactivation. In humans, exposure to UV light, emotional stress, fever, tissue damage and immune suppression is known inducers of reactivation. In cultured neurons, several stimuli have also been identified such as: NGF deprivation (Wilcox & Johnson, 1987, 1988; Wilcox et al., 1990), the histone deacetylase inhibitor Trichostatin A (Arthur et al., 2001), forskolin (Danaher et al., 1999), inducible cyclic AMP early repressor (Colgin et al., 2001), capsaicin (Hunsperger & Wilcox, 2003b), caspase-3 activator C2-ceramide (Hunsperger & Wilcox, 2003a, b), protein kinase C activation by phorbol myristate acetate (Smith et al., 1992), transient hyperthermia (Moriya et al., 1994) or addition of dexamethesone (Halford et al., 1996). In a recent study, Camarena and colleagues examined the signalling cascade responsible for viral reactivation upon NGF withdrawal. NGF is sensed by the TrkA receptor, which in the presence of NGF activates PI3-K (phosphatidylinositol 3-kinase) p110alfa catalytic subunit. This leads to the recruitment of 3-phosphoinositide-dependent protein kinase-1 (PDK1) to the plasma membrane and phosphorylation of serine/threonine kinase Akt (Camarena et al., 2010). The authors also found that epidermal growth factor (EGF) and GDNF growth factors although able to signal through the same pathways were not able to maintain HSV latency to a similar extent as NGF and concluded that this was probably due to their different abilities to sustain AKT phosphorylation (Camarena et al., 2010). It will be of interest to know the full signalling pathway that results in genome activation, specifically, which factors are involved in triggering the first viral transcripts. Understanding which viral genes are necessary for reactivation has been a long-lasting question in HSV research. The use of viral recombinants with mutations in different genes has been a common approach to define the role of individual gene products in reactivation; however, interpretation of such data is complicated because if a mutant virus has defects in lytic replication, this will not only impact on the efficiency of latency establishment but will also impact on full virus reactivation and infectious progeny production. Reactivation is classically scored by the formation of infectious particles from the latently infected tissue or cultures. Thus, if a mutant has defects in the lytic cycle, it will also impact on the formation of viral particles following the exit from latent infection. All essential genes are therefore necessary for reactivation but not all/any may be needed for reactivation at the molecular level as defined by the initial events that lead to the switch between latency and the entry into productive cycle.
Several systems have been used to study HSV-1 reactivation from latency. The simplest and probably the most artificial is the in vitro quiescent model where viral genomes deficient for IE gene expression become tightly repressed 24–48 h postinfection (Preston et al., 1997; Samaniego et al., 1998; Coleman et al., 2008). In this system, once tight repression is established, the only HSV-1 protein able to de-repress the quiescent genomes is ICP0.
Latently, infected neuronal cultures can be reactivated by several other stimuli including other HSV proteins: such as VP16 or ICP4, which can be delivered by superinfection with adenovectors (Halford et al., 2001). Neuronal latency is therefore characterized as having a more relaxed state of repression, in at least a subpopulation of latently infected cells (Arthur et al., 2001; Terry-Allison et al., 2007). However, neuronal culture latency is generally established following an unnatural block in the viral lytic cascade of gene expression through the use of replication defective virus mutants or the inclusion of inhibitors of viral DNA replication. Such experimental methodologies therefore result in latency being established in cells that may otherwise have supported lytic cycle replication. It is currently unclear whether the ‘repressed’ state observed in such latently infected neurones resembles natural latency.
In vivo, it seems that only a small proportion of latently infected cells are competent for reactivation even following stimuli that affects the whole ganglion. A good example is the in vivo hyperthermia induced reactivation model described by Sawtell and Thompson where, on average, only 1–3 neurons per TG (approximately 1 in 2700 latently infected cells) reactivate and are antigen-positive 22-h poststimulus (Sawtell & Thompson, 1992; Thompson & Sawtell, 2010). In a similar fashion, cultures of dissociated ganglia from latently infected mice also display a low frequency of spontaneous reactivation (Halford et al., 1996).
In humans, it is clear that spontaneous reactivation leading to the shedding of virus in the genital mucosa occurs at high frequency (reviewed by Koelle & Corey, 2008; Schiffer & Corey, 2009). Thus, the frequency of asymptomatic genital HSV-2 shedding averages 25% of days, and such reactivation events have a rapid onset and are cleared within 12 h (Wald et al., 2000; Mark et al., 2008). Thus, in contrast to the situation in mouse infection models, latent neuronal infection in humans is less tightly controlled and the resultant high frequency of reactivation is a major factor in human-to-human transmission (Koelle & Corey, 2008).
Of critical importance in understanding the mechanism of virus reactivation is the responsiveness of viral promoters to triggers of virus reactivation and the identity of viral gene(s) responsible for the initiation of the lytic gene expression and exit from latency. ICP0 has always been regarded as the most likely candidate owing to its ability to target ND10 domains for degradation and its capacity to act as a promiscuous activator of gene expression (reviewed by Everett, 2000). This role is strongly supported by the fact that in the in vitro, nonneuronal quiescence model, ICP0 is the only HSV protein able to reverse the quiescent state (Harris et al., 1989; Stow & Stow, 1989; Preston et al., 1997; Samaniego et al., 1998; Coleman et al., 2008; Ferenczy et al., 2011). Furthermore, in vivo ICP0 null mutants do not efficiently reactivate following explant culture of latently infected ganglia (Cai et al., 1993; Halford & Schaffer, 2001). A key question is whether the deficits in reactivation observed for ICP0 mutants represent a failure to initiate exit from latency or failure to enter productive cycle replication following the initiation of reactivation. Studies by Thompson & Sawtell (2006) have provided evidence for the latter theory that the molecular trigger responsible for ‘primary’ reactivation from latency is not ICP0. In this study, similar latent loads were achieved in mice infected with an ICP0 mutant or WT virus and 22 h following the application of hyperthermic stress, a similar number of antigen-positive cells were found to exit from latency. Thus, following the application of an in vivo reactivation stimulus, ICP0 appears to play no important role in the initiation of reactivation from latency but is required for progression to virus production following exit from latency.
A recent publication by Thompson and Sawtell (Thompson et al., 2009) identified VP16 as the molecular trigger for reactivation following hyperthermic stress. This was an unexpected finding given that VP16 is classically defined as a late gene product and therefore optimally expressed only after the onset of virus DNA replication. Given its late temporal class, it seemed unlikely that this potent transactivator would have a pivotal role in the earliest steps of virus reactivation; yet, the VP16 promoter was found to have a distinct regulation in neurons, and de novo synthesis of VP16 was found to be necessary for efficient initiation of the viral lytic cycle in neurons, both during reactivation from latency but also during the acute phase of infection (Thompson et al., 2009). Of particular importance will be to determine the responsive elements of the VP16 promoter and identification of cellular transcription factors that mediate promoter activation in response to reactivation triggers. If VP16 is the trigger for reactivation, it would be fair to assume that its co-factors Oct-1 and HCF-1 would also be present in order to efficiently induce IE gene expression. In fact, upon ganglionic explant, the most commonly used reactivation stimulus, Oct-1 expression is induced in sensory neurons (Valyi-Nagy et al., 1991). Furthermore, HCF-1 has been shown to relocalize from the Golgi to the nucleus (Kristie et al., 1999; Kolb & Kristie, 2008) in up to half the neurons in explanted TGs and associates with viral IE promoters as early as 1-h postganglionic explant (Whitlow & Kristie, 2009). Recent data have also shown that HCF-1 recruits the lysine-specific demethylase (LSD1) to virus IE promoters to reverse repressive histone methylation marks to facilitate both lytic cycle virus gene expression and reactivation from latency (Liang et al., 2009). This new described function of VP16 correlates with its large-scale chromatin remodelling features (Tumbar et al., 1999) and potent transcriptional activator functions (reviewed in detail by Kutluay & Triezenberg, 2009). Experimental data using adenovirus vectors confirm the ability of VP16 to reactivate latent genomes and that this is dependent on its acidic transactivator domain (Halford et al., 2001). Neuronal cultures systems have shown that superinfection of latently infected cells with an IE null mutant can result in reactivation possibly via delivery of VP16 although less efficiently than with a similar mutant able to express ICP0 (Terry-Allison et al., 2007). Furthermore, a study by Jacob et al. where mutants were tested for their ability to reactivate following hyperthermia, forskolin or trichostatin A (TSA) treatments from latently infected PC12 cells revealed that only mutants lacking functional VP16 failed to reactivate (Miller et al., 2006).
Reactivation from latency is a critical phase of HSV life cycle; therefore, it is not surprising that it is highly regulated. It seems that only a small proportion of latently infected neurones are able to respond to a given reactivation stimulus at any given time. The responsiveness of latent virus genomes may be related to the nonuniform nature of the latent state. Thus, multiple factors such as latent DNA copy number, level of LAT expression, neuronal type, degree of genome repression and CD8+ T cell immunosurveillance may influence both the efficiency and mechanism by which reactivation can occur (Fig. 3). Given this knowledge, it maybe difficult to arrive at a simple model for the molecular basis of HSV latency and reactivation. An effort in understanding the nature and biological significance of the heterogeneity of latency is therefore warranted.
Fig 3.
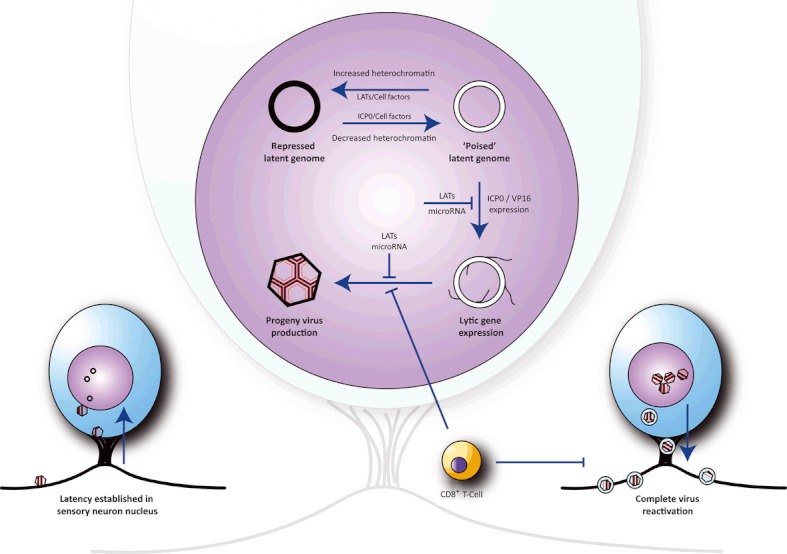
The balance between latency and productive infection/reactivation is policed by cellular and viral factors. HSV latency is established in sensory neurons innervating the sit of primary infection. During the establishment of latency, within the cell nucleus, circularized viral DNA becomes associated with heterochromatin resulting in the repression of lytic cycle gene expression. Periodically, virus reactivation and exit from latency can occur as a consequence of changes in neuronal cell physiology in response to stress. Reversal of repressive histone modifications allows transcriptional activation of one or more viral gene products responsible for initiating virus replication. Accumulation of the LATs and cognate microRNAs during latency may facilitate establishment of latency and regulate reactivation by blocking translation of the viral IE genes ICP0 and ICP4. Ganglion-resident CD8+ T cells may actively halt reactivation through noncytolytic control mechanisms. Occasionally, reactivating cells escape cellular, virus and immune mechanisms of control leading to the completion of the virus life cycle, and production of progeny virus that is then transported to the periphery resulting in either asymptomatic or symptomatic infection that can lead to transmission of reactivated virus to immunologically naïve hosts.
Acknowledgments
The authors are funded through support from the Wellcome Trust (086403/Z/08/7) and the Medical Research Council UK.
Authors' contribution
M.P.N and J.T.P are joint first authors.
Statement
Re-use of this article is permitted in accordance with the Terms and Conditions set out at http://wileyonlinelibrary.com/onlineopen#OnlineOpen_Terms
References
- Ahmed M, Lock M, Miller CG, Fraser NW. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J Virol. 2002;76:717–729. doi: 10.1128/JVI.76.2.717-729.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio AL, Giordani NV, Kubat NJ, O'neil JE, Bloom DC. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J Virol. 2006;80:2063–2068. doi: 10.1128/JVI.80.4.2063-2068.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur J, Efstathiou S, Simmons A. Intranuclear foci containing low abundance herpes simplex virus latency-associated transcripts visualized by non-isotopic in situ hybridization. J Gen Virol. 1993;74(Pt 7):1363–1370. doi: 10.1099/0022-1317-74-7-1363. [DOI] [PubMed] [Google Scholar]
- Arthur JL, Scarpini CG, Connor V, Lachmann RH, Tolkovsky AM, Efstathiou S. Herpes simplex virus type 1 promoter activity during latency establishment, maintenance, and reactivation in primary dorsal root neurons in vitro. J Virol. 2001;75:3885–3895. doi: 10.1128/JVI.75.8.3885-3895.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, Margolis TP. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J Virol. 2011;85:6669–6677. doi: 10.1128/JVI.00204-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom DC, Giordani NV, Kwiatkowski DL. Epigenetic regulation of latent HSV-1 gene expression. Biochim Biophys Acta. 2010;1799:246–256. doi: 10.1016/j.bbagrm.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehmer PE, Nimonkar AV. Herpes virus replication. IUBMB Life. 2003;55:13–22. doi: 10.1080/1521654031000070645. [DOI] [PubMed] [Google Scholar]
- Boissiere SL, Hughes T, O'Hare P. HCF-dependent nuclear import of VP16. EMBO J. 1999;18:480–489. doi: 10.1093/emboj/18.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boss IW, Plaisance KB, Renne R. Role of virus-encoded microRNAs in herpesvirus biology. Trends Microbiol. 2009;17:544–553. doi: 10.1016/j.tim.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutell C, Sadis S, Everett RD. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J Virol. 2002;76:841–850. doi: 10.1128/JVI.76.2.841-850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco FJ, Fraser NW. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J Virol. 2005;79:9019–9025. doi: 10.1128/JVI.79.14.9019-9025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton EA, Hong C-S, Glorioso JC. The stable 2.0-kilobase intron of the herpes simplex virus type 1 latency-associated transcript does not function as an antisense repressor of ICP0 in nonneuronal cells. J Virol. 2003;77:3516–3530. doi: 10.1128/JVI.77.6.3516-3530.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Schaffer PA. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J Virol. 1992;66:2904–2915. doi: 10.1128/jvi.66.5.2904-2915.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Astor TL, Liptak LM, Cho C, Coen DM, Schaffer PA. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J Virol. 1993;67:7501–7512. doi: 10.1128/jvi.67.12.7501-7512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohhr I, Chao MV. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe. 2010;8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter D, Hsiang C, Brown DJ, Jin L, Osorio N, BenMohamed L, Jones C, Wechsler SL. Stable cell lines expressing high levels of the herpes simplex virus type 1 LAT are refractory to caspase 3 activation and DNA laddering following cold shock induced apoptosis. Virology. 2007;369:12–18. doi: 10.1016/j.virol.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter KL, Roizman B. Alternatively spliced mRNAs predicted to yield frame-shift proteins and stable intron 1 RNAs of the herpes simplex virus 1 regulatory gene alpha 0 accumulate in the cytoplasm of infected cells. P Natl Acad Sci USA. 1996;93:12535–12540. doi: 10.1073/pnas.93.22.12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Kramer MF, Schaffer PA, Coen DM. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J Virol. 1997;71:5878–5884. doi: 10.1128/jvi.71.8.5878-5884.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XP, Mata M, Kelley M, Glorioso JC, Fink DJ. The relationship of herpes simplex virus latency associated transcript expression to genome copy number: a quantitative study using laser capture microdissection. J Neurovirol. 2002;8:204–210. doi: 10.1080/13550280290049642. [DOI] [PubMed] [Google Scholar]
- Chiocca EA, Choi BB, Cai WZ, DeLuca NA, Schaffer PA, DiFiglia M, Breakefield XO, Martuza RL. Transfer and expression of the lacZ gene in rat brain neurons mediated by herpes simplex virus mutants. New Biol. 1990;2:739–746. [PubMed] [Google Scholar]
- Ciacci-Zanella J, Stone M, Henderson G, Jones C. The latency-related gene of bovine herpesvirus 1 inhibits programmed cell death. J Virol. 1999;73:9734–9740. doi: 10.1128/jvi.73.12.9734-9740.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Garber DA, Knipe DM. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol. 2009;83:8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman HM, Connor V, Cheng ZS, Grey F, Preston CM, Efstathiou S. Histone modifications associated with herpes simplex virus type 1 genomes during quiescence and following ICP0-mediated de-repression. J Gen Virol. 2008;89:68–77. doi: 10.1099/vir.0.83272-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin MA, Smith RL, Wilcox CL. Inducible cyclic AMP early repressor produces reactivation of latent herpes simplex virus type 1 in neurons in vitro. J Virol. 2001;75:2912–2920. doi: 10.1128/JVI.75.6.2912-2920.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchet D, Sykes A, Nicolas A, Orr A, Murray I, Sirma H, Heeren J, Bartelt A, Everett RD. PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J Cell Sci. 2011;124:280–291. doi: 10.1242/jcs.075390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Griffiths A, Li G, Silva LM, Kramer MF, Gaasterland T, Wang XJ, Coen DM. Prediction and identification of herpes simplex virus 1-encoded microRNAs. J Virol. 2006;80:5499–5508. doi: 10.1128/JVI.00200-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaher RJ, Jacob RJ, Miller CS. Establishment of a quiescent herpes simplex virus type 1 infection in neurally-differentiated PC12 cells. J Neurovirol. 1999;5:258–267. doi: 10.3109/13550289909015812. [DOI] [PubMed] [Google Scholar]
- Dasgupta G, Benmohamed L. Of mice and not humans: how reliable are animal models for evaluation of herpes CD8(+)-T cell-epitopes-based immunotherapeutic vaccine candidates? Vaccine. 2011;29:5824–5836. doi: 10.1016/j.vaccine.2011.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatly AM, Spivack JG, Lavi E, O'Boyle DR, Fraser NW. Latent herpes simplex virus type 1 transcripts in peripheral and central nervous system tissues of mice map to similar regions of the viral genome. J Virol. 1988;62:749–756. doi: 10.1128/jvi.62.3.749-756.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decman V, Kinchington PR, Harvey SA, Hendricks RL. Gamma interferon can block herpes simplex virus type 1 reactivation from latency, even in the presence of late gene expression. J Virol. 2005;79:10339–10347. doi: 10.1128/JVI.79.16.10339-10347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmane SL, Fraser NW. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol. 1989;63:943–947. doi: 10.1128/jvi.63.2.943-947.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi-Rao GB, Goodart SA, Hecht LM, Rochford R, Rice MK, Wagner EK. Relationship between polyadenylated and nonpolyadenylated herpes simplex virus type 1 latency-associated transcripts. J Virol. 1991;65:2179–2190. doi: 10.1128/jvi.65.5.2179-2190.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson AT, Margolis TP, Sedarati F, Stevens JG, Feldman LT. A latent, nonpathogenic HSV-1-derived vector stably expresses beta-galactosidase in mouse neurons. Neuron. 1990;5:353–360. doi: 10.1016/0896-6273(90)90171-b. [DOI] [PubMed] [Google Scholar]
- Dressler GR, Rock DL, Fraser NW. Latent herpes simplex virus type 1 DNA is not extensively methylated in vivo. J Gen Virol. 1987;68:1761–1765. doi: 10.1099/0022-1317-68-6-1761. [DOI] [PubMed] [Google Scholar]
- Du T, Zhou G, Khan S, Gu H, Roizman B. Disruption of HDAC/CoREST/REST repressor by dnREST reduces genome silencing and increases virulence of herpes simplex virus. P Natl Acad Sci USA. 2010;107:15904–15909. doi: 10.1073/pnas.1010741107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecob-Prince MS, Preston CM, Rixon FJ, Hassan K, Kennedy PG. Neurons containing latency-associated transcripts are numerous and widespread in dorsal root ganglia following footpad inoculation of mice with herpes simplex virus type 1 mutant in1814. J Gen Virol. 1993;74(Pt 6):985–994. doi: 10.1099/0022-1317-74-6-985. [DOI] [PubMed] [Google Scholar]
- Efstathiou S, Preston CM. Towards an understanding of the molecular basis of herpes simplex virus latency. Virus Res. 2005;111:108–119. doi: 10.1016/j.virusres.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Efstathiou S, Minson AC, Field HJ, Anderson JR, Wildy P. Detection of herpes simplex virus-specific DNA sequences in latently infected mice and in humans. J Virol. 1986;57:446–455. doi: 10.1128/jvi.57.2.446-455.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD. Trans activation of transcription by herpes virus products: requirement for two HSV-1 immediate-early polypeptides for maximum activity. EMBO J. 1984;3:3135–3141. doi: 10.1002/j.1460-2075.1984.tb02270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays. 2000;22:761–770. doi: 10.1002/1521-1878(200008)22:8<761::AID-BIES10>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Everett RD. Depletion of CoREST does not improve the replication of ICP0 null mutant herpes simplex virus type 1. J Virol. 2010;84:3695–3698. doi: 10.1128/JVI.00021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Cross A, Orr A. A truncated form of herpes simplex virus type 1 immediate-early protein Vmw110 is expressed in a cell type dependent manner. Virology. 1993;197:751–756. doi: 10.1006/viro.1993.1651. [DOI] [PubMed] [Google Scholar]
- Everett RD, Boutell C, Orr A. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J Virol. 2004;78:1763–1774. doi: 10.1128/JVI.78.4.1763-1774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol. 2006;80:7995–8005. doi: 10.1128/JVI.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell MJ, Dobson AT, Feldman LT. Herpes simplex virus latency-associated transcript is a stable intron. P Natl Acad Sci USA. 1991;88:790–794. doi: 10.1073/pnas.88.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. P Natl Acad Sci USA. 2002;99:978–983. doi: 10.1073/pnas.022301899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenczy MW, Ranayhossaini DJ, Deluca NA. Activities of ICP0 involved in the reversal of silencing of quiescent herpes simplex virus 1. J Virol. 2011;85:4993–5002. doi: 10.1128/JVI.02265-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman ML, Sheridan BS, Bonneau RH, Hendricks RL. Psychological stress compromises CD8+ T cell control of latent herpes simplex virus type 1 infections. J Immunol. 2007;179:322–328. doi: 10.4049/jimmunol.179.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Früh K, Ahn K, Djaballah H, Sempé P, van Endert PM, Tampé R, Peterson PA, Yang Y. A viral inhibitor of peptide transporters for antigen presentation. Nature. 1995;375:415–418. doi: 10.1038/375415a0. [DOI] [PubMed] [Google Scholar]
- Garber DA, Beverley SM, Coen DM. Demonstration of circularization of herpes simplex virus DNA following infection using pulsed field gel electrophoresis. Virology. 1993;197:459–462. doi: 10.1006/viro.1993.1612. [DOI] [PubMed] [Google Scholar]
- Garber DA, Schaffer PA, Knipe DM. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J Virol. 1997;71:5885–5893. doi: 10.1128/jvi.71.8.5885-5893.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt BM, Halford WP. Evidence that spontaneous reactivation of herpes virus does not occur in mice. Virol J. 2005;2:67. doi: 10.1186/1743-422X-2-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordani NV, Neumann DM, Kwiatkowski DL, Bhattacharjee PS, McAnany PK, Hill JM, Bloom DC. During herpes simplex virus type 1 infection of rabbits, the ability to express the latency-associated transcript increases latent-phase transcription of lytic genes. J Virol. 2008;82:6056–6060. doi: 10.1128/JVI.02661-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Roizman B. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. P Natl Acad Sci USA. 2007;104:17134–17139. doi: 10.1073/pnas.0707266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Roizman B. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J Virol. 2009;83:181–187. doi: 10.1128/JVI.01940-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Liang Y, Mandel G, Roizman B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. P Natl Acad Sci USA. 2005;102:7571–7576. doi: 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussow AM, Giordani NV, Tran RK, Imai Y, Kwiatkowski DL, Rall GF, Margolis TP, Bloom DC. Tissue-specific splicing of the herpes simplex virus type 1 latency-associated transcript (LAT) intron in LAT transgenic mice. J Virol. 2006;80:9414–9423. doi: 10.1128/JVI.00530-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagglund R, Roizman B. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J Virol. 2004;78:2169–2178. doi: 10.1128/JVI.78.5.2169-2178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagmann M, Georgiev O, Schaffner W, Douville P. Transcription factors interacting with herpes simplex virus alpha gene promoters in sensory neurons. Nucleic Acids Res. 1995;23:4978–4985. doi: 10.1093/nar/23.24.4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Schaffer PA. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J Virol. 2001;75:3240–3249. doi: 10.1128/JVI.75.7.3240-3249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Gebhardt BM, Carr DJ. Mechanisms of herpes simplex virus type 1 reactivation. J Virol. 1996;70:5051–5060. doi: 10.1128/jvi.70.8.5051-5060.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Kemp CD, Isler JA, Davido DJ, Schaffer PA. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J Virol. 2001;75:6143–6153. doi: 10.1128/JVI.75.13.6143-6153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza MA, Higgins DM, Feldman LT, Ruyechan WT. The latency-associated transcript of herpes simplex virus type 1 promotes survival and stimulates axonal regeneration in sympathetic and trigeminal neurons. J Neurovirol. 2007;13:56–66. doi: 10.1080/13550280601156297. [DOI] [PubMed] [Google Scholar]
- Harris RA, Everett RD, Zhu XX, Silverstein S, Preston CM. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates latent herpes simplex virus type 2 in an in vitro latency system. J Virol. 1989;63:3513–3515. doi: 10.1128/jvi.63.8.3513-3515.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held K, Junker A, Dornmair K, Meinl E, Sinicina I, Brandt T, Theil D, Derfuss T. Expression of herpes simplex virus 1-encoded microRNAs in human trigeminal ganglia and their relation to local T-cell infiltrates. J Virol. 2011;85:9680–9685. doi: 10.1128/JVI.00874-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson G, Perng GC, Nesburn AB, Wechsler SL, Jones C. The latency-related gene encoded by bovine herpesvirus 1 can suppress caspase 3 and caspase 9 cleavage during productive infection. J Neurovirol. 2004;10:64–70. doi: 10.1080/13550280490261716. [DOI] [PubMed] [Google Scholar]
- Henderson G, Jaber T, Carpenter D, Wechsler SL, Jones C. Identification of herpes simplex virus type 1 proteins encoded within the first 1.5 kb of the latency-associated transcript. J Neurovirol. 2009;15:439–448. doi: 10.3109/13550280903296353. [DOI] [PubMed] [Google Scholar]
- Herrera FJ, Triezenberg SJ. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J Virol. 2004;78:9689–9696. doi: 10.1128/JVI.78.18.9689-9696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill TJ, Field HJ, Blyth WA. Acute and recurrent infection with herpes simplex virus in the mouse: a model for studying latency and recurrent disease. J Gen Virol. 1975;28:341–353. doi: 10.1099/0022-1317-28-3-341. [DOI] [PubMed] [Google Scholar]
- Hill TJ, Harbour DA, Blyth WA. Isolation of herpes simplex virus from the skin of clinically normal mice during latent infection. J Gen Virol. 1980;47:205–207. doi: 10.1099/0022-1317-47-1-205. [DOI] [PubMed] [Google Scholar]
- Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J, Ploegh H, Johnson D. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375:411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- Honess RW, Roizman B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J Virol. 1974;14:8–19. doi: 10.1128/jvi.14.1.8-19.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino Y, Pesnicak L, Straus SE, Cohen JI. Impairment in reactivation of a latency associated transcript (LAT)-deficient HSV-2 is not solely dependent on the latent viral load or the number of CD8(+) T cells infiltrating the ganglia. Virology. 2009;387:193–199. doi: 10.1016/j.virol.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kent JR, Placek B, Whelan KA, Hollow CM, Zeng PY, Fraser NW, Berger SL. Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. J Virol. 2006;80:5740–5746. doi: 10.1128/JVI.00169-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsperger EA, Wilcox CL. Caspase-3-dependent reactivation of latent herpes simplex virus type 1 in sensory neuronal cultures. J Neurovirol. 2003a;9:390–398. doi: 10.1080/13550280390201678. [DOI] [PubMed] [Google Scholar]
- Hunsperger EA, Wilcox CL. Capsaicin-induced reactivation of latent herpes simplex virus type 1 in sensory neurons in culture. J Gen Virol. 2003b;84:1071–1078. doi: 10.1099/vir.0.18828-0. [DOI] [PubMed] [Google Scholar]
- Imai Y, Apakupakul K, Krause PR, Halford WP, Margolis TP. Investigation of the mechanism by which herpes simplex virus type 1 LAT sequences modulate preferential establishment of latent infection in mouse trigeminal ganglia. J Virol. 2009;83:7873–7882. doi: 10.1128/JVI.00043-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman M, Perng GC, Henderson G, Ghiasi H, Nesburn AB, Wechsler SL, Jones C. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. J Virol. 2001;75:3636–3646. doi: 10.1128/JVI.75.8.3636-3646.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber T, Henderson G, Li S, Perng G-C, Carpenter D, Wechsler SL, Jones C. Identification of a novel herpes simplex virus type 1 transcript and protein (AL3) expressed during latency. J Gen Virol. 2009;90:2342–2352. doi: 10.1099/vir.0.013318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javier RT, Stevens JG, Dissette VB, Wagner EK. A herpes simplex virus transcript abundant in latently infected neurons is dispensable for establishment of the latent state. Virology. 1988;166:254–257. doi: 10.1016/0042-6822(88)90169-9. [DOI] [PubMed] [Google Scholar]
- Jerome KR, Fox R, Chen Z, Sears AE, Lee Hy, Corey L. Herpes simplex virus inhibits apoptosis through the action of two genes, Us5 and Us3. J Virol. 1999;73:8950–8957. doi: 10.1128/jvi.73.11.8950-8957.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Perng GC, Mott KR, Osorio N, Naito J, Brick DJ, Carpenter D, Jones C, Wechsler SL. A herpes simplex virus type 1 mutant expressing a baculovirus inhibitor of apoptosis gene in place of latency-associated transcript has a wild-type reactivation phenotype in the mouse. J Virol. 2005;79:12286–12295. doi: 10.1128/JVI.79.19.12286-12295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Carpenter D, Moerdyk-Schauwecker M, Vanarsdall AL, Osorio N, Hsiang C, Jones C, Wechsler SL. Cellular FLIP can substitute for the herpes simplex virus type 1 latency-associated transcript gene to support a wild-type virus reactivation phenotype in mice. J Neurovirol. 2008;14:389–400. doi: 10.1080/13550280802216510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurak I, Kramer MF, Mellor JC, van Lint AL, Roth FP, Knipe DM, Coen DM. Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J Virol. 2010;84:4659–4672. doi: 10.1128/JVI.02725-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurak I, Griffiths A, Coen DM. Mammalian alphaherpesvirus miRNAs. Biochim Biophys Acta. 2011;1809:641–653. doi: 10.1016/j.bbagrm.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz JP, Bodin ET, Coen DM. Quantitative polymerase chain reaction analysis of herpes simplex virus DNA in ganglia of mice infected with replication-incompetent mutants. J Virol. 1990;64:4288–4295. doi: 10.1128/jvi.64.9.4288-4295.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent JR, Zeng PY, Atanasiu D, Gardner J, Fraser NW, Berger SL. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J Virol. 2004;78:10178–10186. doi: 10.1128/JVI.78.18.10178-10186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna KM, Lepisto AJ, Decman V, Hendricks RL. Immune control of herpes simplex virus during latency. Curr Opin Immunol. 2004;16:463–469. doi: 10.1016/j.coi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science. 2008;322:268–271. doi: 10.1126/science.1164164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol. 2008;6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- Koelle DM, Corey L. Herpes simplex: insights on pathogenesis and possible vaccines. Annu Rev Med. 2008;59:381–395. doi: 10.1146/annurev.med.59.061606.095540. [DOI] [PubMed] [Google Scholar]
- Koelle DM, Wald A. Herpes simplex virus: the importance of asymptomatic shedding. J Antimicrob Chemother. 2000;45(Suppl T3):1–8. doi: 10.1093/jac/45.suppl_4.1. [DOI] [PubMed] [Google Scholar]
- Kolb G, Kristie TM. Association of the cellular coactivator HCF-1 with the Golgi apparatus in sensory neurons. J Virol. 2008;82:9555–9563. doi: 10.1128/JVI.01174-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MF, Chen SH, Knipe DM, Coen DM. Accumulation of viral transcripts and DNA during establishment of latency by herpes simplex virus. J Virol. 1998;72:1177–1185. doi: 10.1128/jvi.72.2.1177-1185.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MF, Jurak I, Pesola JM, Boissel S, Knipe DM, Coen DM. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology. 2011;417:239–247. doi: 10.1016/j.virol.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM. Early events pre-initiation of alphaherpes viral gene expression. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007. [PubMed] [Google Scholar]
- Kristie TM, Roizman B. Differentiation and DNA contact points of host proteins binding at the cis site for virion-mediated induction of alpha genes of herpes simplex virus 1. J Virol. 1988;62:1145–1157. doi: 10.1128/jvi.62.4.1145-1157.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM, Vogel JL, Sears AE. Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. P Natl Acad Sci USA. 1999;96:1229–1233. doi: 10.1073/pnas.96.4.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Zabolotny JM, Fraser NW. Selection of a nonconsensus branch point is influenced by an RNA stem-loop structure and is important to confer stability to the herpes simplex virus 2-kilobase latency-associated transcript. J Virol. 1997;71:5849–5860. doi: 10.1128/jvi.71.8.5849-5860.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubat NJ, Amelio AL, Giordani NV, Bloom DC. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J Virol. 2004a;78:12508–12518. doi: 10.1128/JVI.78.22.12508-12518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubat NJ, Tran RK, McAnany P, Bloom DC. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J Virol. 2004b;78:1139–1149. doi: 10.1128/JVI.78.3.1139-1149.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutluay SB, Triezenberg SJ. Role of chromatin during herpesvirus infections. Biochim Biophys Acta. 2009;1790:456–466. doi: 10.1016/j.bbagen.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DL, Thompson HW, Bloom DC. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol. 2009;83:8173–8181. doi: 10.1128/JVI.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon BS, Gangarosa LP, Burch KD, deBack J, Hill JM. Induction of ocular herpes simplex virus shedding by iontophoresis of epinephrine into rabbit cornea. Invest Ophthalmol Vis Sci. 1981;21:442–449. [PubMed] [Google Scholar]
- Lacasse JJ, Schang LM. During lytic infections, herpes simplex virus type 1 DNA is in complexes with the properties of unstable nucleosomes. J Virol. 2010;84:1920–1933. doi: 10.1128/JVI.01934-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann RH, Sadarangani M, Atkinson HR, Efstathiou S. An analysis of herpes simplex virus gene expression during latency establishment and reactivation. J Gen Virol. 1999;80(Pt 5):1271–1282. doi: 10.1099/0022-1317-80-5-1271. [DOI] [PubMed] [Google Scholar]
- Lakin ND, Palmer R, Lillycrop KA, Howard MK, Burke LC, Thomas NS, Latchman DS. Down regulation of the octamer binding protein Oct-1 during growth arrest and differentiation of a neuronal cell line. Brain Res Mol Brain Res. 1995;28:47–54. doi: 10.1016/0169-328x(94)00183-f. [DOI] [PubMed] [Google Scholar]
- Latchman DS. Regulation of DNA virus transcription by cellular POU family transcription factors. Rev Med Virol. 1999;9:31–38. doi: 10.1002/(sici)1099-1654(199901/03)9:1<31::aid-rmv231>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol. 1989;63:2893–2900. doi: 10.1128/jvi.63.7.2893-2900.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopardi R, Van Sant C, Roizman B. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. P Natl Acad Sci USA. 1997;94:7891–7896. doi: 10.1073/pnas.94.15.7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med. 2009;15:1312–1317. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillycrop KA, Dent CL, Wheatley SC, Beech MN, Ninkina NN, Wood JN, Latchman DS. The octamer-binding protein Oct-2 represses HSV immediate-early genes in cell lines derived from latently infectable sensory neurons. Neuron. 1991;7:381–390. doi: 10.1016/0896-6273(91)90290-g. [DOI] [PubMed] [Google Scholar]
- Liu T, Tang Q, Hendricks RL. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J Virol. 1996;70:264–271. doi: 10.1128/jvi.70.1.264-271.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonte P, Thomas J, Texier P, Caron C, Khochbin S, Epstein AL. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J Virol. 2004;78:6744–6757. doi: 10.1128/JVI.78.13.6744-6757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mador N, Goldenberg D, Cohen O, Panet A, Steiner I. Herpes simplex virus type 1 latency-associated transcripts suppress viral replication and reduce immediate-early gene mRNA levels in a neuronal cell line. J Virol. 1998;72:5067–5075. doi: 10.1128/jvi.72.6.5067-5075.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggioncalda J, Mehta A, Su YH, Fraser NW, Block TM. Correlation between herpes simplex virus type 1 rate of reactivation from latent infection and the number of infected neurons in trigeminal ganglia. Virology. 1996;225:72–81. doi: 10.1006/viro.1996.0576. [DOI] [PubMed] [Google Scholar]
- Margolis TP, Dawson CR, LaVail JH. Herpes simplex viral infection of the mouse trigeminal ganglion. Immunohistochemical analysis of cell populations. Invest Ophthalmol Vis Sci. 1992;33:259–267. [PubMed] [Google Scholar]
- Margolis TP, Imai Y, Yang L, Vallas V, Krause PR. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J Virol. 2007;81:1872–1878. doi: 10.1128/JVI.02110-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark KE, Wald A, Magaret AS, Selke S, Olin L, Huang ML, Corey L. Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J Infect Dis. 2008;198:1141–1149. doi: 10.1086/591913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall KR, Lachmann RH, Efstathiou S, Rinaldi A, Preston CM. Long-term transgene expression in mice infected with a herpes simplex virus type 1 mutant severely impaired for immediate-early gene expression. J Virol. 2000;74:956–964. doi: 10.1128/jvi.74.2.956-964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch DJ, Rixon FJ, Davison AJ. Topics in herpesvirus genomics and evolution. Virus Res. 2006;117:90–104. doi: 10.1016/j.virusres.2006.01.002. [DOI] [PubMed] [Google Scholar]
- McMahon R, Walsh D. Efficient quiescent infection of normal human diploid fibroblasts with wild-type herpes simplex virus type 1. J Virol. 2008;82:10218–10230. doi: 10.1128/JVI.00859-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Maggioncalda J, Bagasra O, Thikkavarapu S, Saikumari P, Valyi-Nagy T, Fraser NW, Block TM. In situ DNA PCR and RNA hybridization detection of herpes simplex virus sequences in trigeminal ganglia of latently infected mice. Virology. 1995;206:633–640. doi: 10.1016/s0042-6822(95)80080-8. [DOI] [PubMed] [Google Scholar]
- Miller CS, Danaher RJ, Jacob RJ. ICP0 is not required for efficient stress-induced reactivation of herpes simplex virus type 1 from cultured quiescently infected neuronal cells. J Virol. 2006;80:3360–3368. doi: 10.1128/JVI.80.7.3360-3368.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya A, Yoshiki A, Kita M, Fushiki S, Imanishi J. Heat shock-induced reactivation of herpes simplex virus type 1 in latently infected mouse trigeminal ganglion cells in dissociated culture. Arch Virol. 1994;135:419–425. doi: 10.1007/BF01310025. [DOI] [PubMed] [Google Scholar]
- Muylaert I, Tang KW, Elias P. Replication and recombination of herpes simplex virus DNA. J Biol Chem. 2011;286:15619–15624. doi: 10.1074/jbc.R111.233981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevels M, Nitzsche A, Paulus C. How to control an infectious bead string: nucleosome-based regulation and targeting of herpesvirus chromatin. Rev Med Virol. 2011;21:154–180. doi: 10.1002/rmv.690. [DOI] [PubMed] [Google Scholar]
- O'Hare P. The virion transactivator of herpes simplex virus. Semin Virol. 1993;4:145–155. [Google Scholar]
- O'Hare P, Hayward GS. Three trans-acting regulatory proteins of herpes simplex virus modulate immediate-early gene expression in a pathway involving positive and negative feedback regulation. J Virol. 1985;56:723–733. doi: 10.1128/jvi.56.3.723-733.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil JE, Loutsch JM, Aguilar JS, Hill JM, Wagner EK, Bloom DC. Wide variations in herpes simplex virus type 1 inoculum dose and latency-associated transcript expression phenotype do not alter the establishment of latency in the rabbit eye model. J Virol. 2004;78:5038–5044. doi: 10.1128/JVI.78.10.5038-5044.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr MT, Mathis MA, Lagunoff M, Sacks JA, Wilson CB. CD8 T cell control of HSV reactivation from latency is abrogated by viral inhibition of MHC class I. Cell Host Microbe. 2007;2:172–180. doi: 10.1016/j.chom.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebody RG, Andrews N, Brown D, et al. The seroepidemiology of herpes simplex virus type 1 and 2 in Europe. Sex Transm Infect. 2004;80:185–191. doi: 10.1136/sti.2003.005850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Vitvitskaia O, Carpenter D, Wechsler SL, Jones C. Identification of two small RNAs within the first 1.5-kb of the herpes simplex virus type 1-encoded latency-associated transcript. J Neurovirol. 2008;14:41–52. doi: 10.1080/13550280701793957. [DOI] [PubMed] [Google Scholar]
- Perkins D, Pereira EFR, Gober M, Yarowsky PJ, Aurelian L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) blocks apoptosis in hippocampal neurons, involving activation of the MEK/MAPK survival pathway. J Virol. 2002;76:1435–1449. doi: 10.1128/JVI.76.3.1435-1449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng GC, Ghiasi H, Slanina SM, Nesburn AB, Wechsler SL. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3-kilobase primary transcript. J Virol. 1996;70:976–984. doi: 10.1128/jvi.70.2.976-984.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng GC, Jones C, Ciacci-Zanella J, et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science. 2000;287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- Perng GC, Slanina SM, Ghiasi H, Nesburn AB, Wechsler SL. The effect of latency-associated transcript on the herpes simplex virus type 1 latency-reactivation phenotype is mouse strain-dependent. J Gen Virol. 2001;82:1117–1122. doi: 10.1099/0022-1317-82-5-1117. [DOI] [PubMed] [Google Scholar]
- Perng GC, Maguen B, Jin L, et al. A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. J Virol. 2002;76:1224–1235. doi: 10.1128/JVI.76.3.1224-1235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston CM. Repression of viral transcription during herpes simplex virus latency. J Gen Virol. 2000;81:1–19. doi: 10.1099/0022-1317-81-1-1. [DOI] [PubMed] [Google Scholar]
- Preston CM, Mabbs R, Nicholl MJ. Construction and characterization of herpes simplex virus type 1 mutants with conditional defects in immediate early gene expression. Virology. 1997;229:228–239. doi: 10.1006/viro.1996.8424. [DOI] [PubMed] [Google Scholar]
- Proença JT, Coleman HM, Connor V, Winton DJ, Efstathiou S. A historical analysis of herpes simplex virus promoter activation in vivo reveals distinct populations of latently infected neurones. J Gen Virol. 2008;89:2965–2974. doi: 10.1099/vir.0.2008/005066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proença JT, Coleman HM, Nicoll MP, Connor V, Preston CM, Arthur J, Efstathiou S. An investigation of HSV promoter activity compatible with latency establishment reveals VP16 independent activation of HSV immediate early promoters in sensory neurones. J Gen Virol. 2011;92:2575–2585. doi: 10.1099/vir.0.034728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Davoli KA, Yee MB, Hendricks RL, Kinchington PR. Delaying the expression of herpes simplex virus type 1 glycoprotein B (gB) to a true late gene alters neurovirulence and inhibits the gB-CD8+ T-cell response in the trigeminal ganglion. J Virol. 2010;84:8811–8820. doi: 10.1128/JVI.00496-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock DL, Fraser NW. Detection of HSV-1 genome in central nervous system of latently infected mice. Nature. 1983;302:523–525. doi: 10.1038/302523a0. [DOI] [PubMed] [Google Scholar]
- Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol. 1987;61:3820–3826. doi: 10.1128/jvi.61.12.3820-3826.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rødahl E, Haarr L. Analysis of the 2-kilobase latency-associated transcript expressed in PC12 cells productively infected with herpes simplex virus type 1: evidence for a stable, nonlinear structure. J Virol. 1997;71:1703–1707. doi: 10.1128/jvi.71.2.1703-1707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B. The checkpoints of viral gene expression in productive and latent infection: the role of the HDAC/CoREST/LSD1/REST repressor complex. J Virol. 2011;85:7474–7482. doi: 10.1128/JVI.00180-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, Knipe D, Whitley R. Herpes simplex viruses. In: Knipe D, Howley P, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fields Virology. New York: Lippincott Williams and Wilkins; 2007. pp. 2501–2601. [Google Scholar]
- Samaniego LA, Neiderhiser L, DeLuca NA. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J Virol. 1998;72:3307–3320. doi: 10.1128/jvi.72.4.3307-3320.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM. Comprehensive quantification of herpes simplex virus latency at the single-cell level. J Virol. 1997;71:5423–5431. doi: 10.1128/jvi.71.7.5423-5431.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL. Rapid in vivo reactivation of herpes simplex virus in latently infected murine ganglionic neurons after transient hyperthermia. J Virol. 1992;66:2150–2156. doi: 10.1128/jvi.66.4.2150-2156.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Poon DK, Tansky CS, Thompson RL. The latent herpes simplex virus type 1 genome copy number in individual neurons is virus strain specific and correlates with reactivation. J Virol. 1998;72:5343–5350. doi: 10.1128/jvi.72.7.5343-5350.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffer JT, Corey L. New concepts in understanding genital herpes. Curr Infect Dis Rep. 2009;11:457–464. doi: 10.1007/s11908-009-0066-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedarati F, Izumi KM, Wagner EK, Stevens JG. Herpes simplex virus type 1 latency-associated transcription plays no role in establishment or maintenance of a latent infection in murine sensory neurons. J Virol. 1989;63:4455–4458. doi: 10.1128/jvi.63.10.4455-4458.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedarati F, Margolis TP, Stevens JG. Latent infection can be established with drastically restricted transcription and replication of the HSV-1 genome. Virology. 1993;192:687–691. doi: 10.1006/viro.1993.1089. [DOI] [PubMed] [Google Scholar]
- Shen W, Sa e Silva M, Jaber T, Vitvitskaia O, Li S, Henderson G, Jones C. Two small RNAs encoded within the first 1.5 kilobases of the herpes simplex virus type 1 latency-associated transcript can inhibit productive infection and cooperate to inhibit apoptosis. J Virol. 2009;83:9131–9139. doi: 10.1128/JVI.00871-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Simmons A, Tscharke DC. Anti-CD8 impairs clearance of herpes simplex virus from the nervous system: implications for the fate of virally infected neurons. J Exp Med. 1992;175:1337–1344. doi: 10.1084/jem.175.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slobedman B, Efstathiou S, Simmons A. Quantitative analysis of herpes simplex virus DNA and transcriptional activity in ganglia of mice latently infected with wild-type and thymidine kinase-deficient viral strains. J Gen Virol. 1994;75(Pt 9):2469–2474. doi: 10.1099/0022-1317-75-9-2469. [DOI] [PubMed] [Google Scholar]
- Smith RL, Pizer LI, Johnson EM, Wilcox CL. Activation of second-messenger pathways reactivates latent herpes simplex virus in neuronal cultures. Virology. 1992;188:311–318. doi: 10.1016/0042-6822(92)90760-m. [DOI] [PubMed] [Google Scholar]
- Smith RW, Malik P, Clements JB. The herpes simplex virus ICP27 protein: a multifunctional post-transcriptional regulator of gene expression. Biochem Soc Trans. 2005;33:499–501. doi: 10.1042/BST0330499. [DOI] [PubMed] [Google Scholar]
- Speck PG, Simmons A. Synchronous appearance of antigen-positive and latently infected neurons in spinal ganglia of mice infected with a virulent strain of herpes simplex virus. J Gen Virol. 1992;73(Pt 5):1281–1285. doi: 10.1099/0022-1317-73-5-1281. [DOI] [PubMed] [Google Scholar]
- Steiner I, Spivack JG, Lirette RP, Brown SM, MacLean AR, Subak-Sharpe JH, Fraser NW. Herpes simplex virus type 1 latency-associated transcripts are evidently not essential for latent infection. EMBO J. 1989;8:505–511. doi: 10.1002/j.1460-2075.1989.tb03404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner I, Spivack JG, Deshmane SL, Ace CI, Preston CM, Fraser NW. A herpes simplex virus type 1 mutant containing a nontransinducing Vmw65 protein establishes latent infection in vivo in the absence of viral replication and reactivates efficiently from explanted trigeminal ganglia. J Virol. 1990;64:1630–1638. doi: 10.1128/jvi.64.4.1630-1638.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science. 1987;235:1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- Stow EC, Stow ND. Complementation of a herpes simplex virus type 1 Vmw110 deletion mutant by human cytomegalovirus. J Gen Virol. 1989;70(Pt 3):695–704. doi: 10.1099/0022-1317-70-3-695. [DOI] [PubMed] [Google Scholar]
- Strang BL, Stow ND. Circularization of the herpes simplex virus type 1 genome upon lytic infection. J Virol. 2005;79:12487–12494. doi: 10.1128/JVI.79.19.12487-12494.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Bertke AS, Patel A, Wang K, Cohen JI, Krause PR. An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. P Nat Acad Sci USA. 2008;105:10931–10936. doi: 10.1073/pnas.0801845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Patel A, Krause PR. Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J Virol. 2009;83:1433–1442. doi: 10.1128/JVI.01723-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry-Allison T, Smith CA, DeLuca NA. Relaxed repression of herpes simplex virus type 1 genomes in Murine trigeminal neurons. J Virol. 2007;81:12394–12405. doi: 10.1128/JVI.01068-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theil D, Derfuss T, Paripovic I, Herberger S, Meinl E, Schueler O, Strupp M, Arbusow V, Brandt T. Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. Am J Pathol. 2003;163:2179–2184. doi: 10.1016/S0002-9440(10)63575-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SK, Gough G, Latchman DS, R S, Coffin RS. Herpes simplex virus latency-associated transcript encodes a protein which greatly enhances virus growth, can compensate for deficiencies in immediate-early gene expression, and is likely to function during reactivation from virus latency. J Virol. 1999;73:6618–6625. doi: 10.1128/jvi.73.8.6618-6625.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J Virol. 1997;71:5432–5440. doi: 10.1128/jvi.71.7.5432-5440.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. Replication of herpes simplex virus type 1 within trigeminal ganglia is required for high frequency but not high viral genome copy number latency. J Virol. 2000;74:965–974. doi: 10.1128/jvi.74.2.965-974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J Virol. 2001;75:6660–6675. doi: 10.1128/JVI.75.14.6660-6675.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. J Virol. 2006;80:10919–10930. doi: 10.1128/JVI.01253-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. Therapeutic implications of new insights into the critical role of VP16 in initiating the earliest stages of HSV reactivation from latency. Future Med Chem. 2010;2:1099–1105. doi: 10.4155/fmc.10.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Preston CM, Sawtell NM. De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog. 2009;5:e1000352. doi: 10.1371/journal.ppat.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbar T, Sudlow G, Belmont AS. Large-scale chromatin unfolding and remodeling induced by VP16 acidic activation domain. J Cell Biol. 1999;145:1341–1354. doi: 10.1083/jcb.145.7.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Cullen BR. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009;23:1151–1164. doi: 10.1101/gad.1793309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Nagel MA, Cohrs RJ, Gilden DH, Cullen BR. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J Virol. 2009;83:10677–10683. doi: 10.1128/JVI.01185-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Wang K, Tang S, Krause PR, Mont EK, Cohen JI, Cullen BR. Identification of viral microRNAs expressed in human sacral ganglia latently infected with herpes simplex virus 2. J Virol. 2010;84:1189–1192. doi: 10.1128/JVI.01712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valyi-Nagy T, Deshmane S, Dillner A, Fraser NW. Induction of cellular transcription factors in trigeminal ganglia of mice by corneal scarification, herpes simplex virus type 1 infection, and explantation of trigeminal ganglia. J Virol. 1991;65:4142–4152. doi: 10.1128/jvi.65.8.4142-4152.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Sant C, Hagglund R, Lopez P, Roizman B. The infected cell protein 0 of herpes simplex virus 1 dynamically interacts with proteasomes, binds and activates the cdc34 E2 ubiquitin-conjugating enzyme, and possesses in vitro E3 ubiquitin ligase activity. P Natl Acad Sci USA. 2001;98:8815–8820. doi: 10.1073/pnas.161283098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EK, Bloom DC. Experimental investigation of herpes simplex virus latency. Clin Microbiol Rev. 1997;10:419–443. doi: 10.1128/cmr.10.3.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakim LM, Jones CM, Gebhardt T, Preston CM, Carbone FR. CD8(+) T-cell attenuation of cutaneous herpes simplex virus infection reduces the average viral copy number of the ensuing latent infection. Immunol Cell Biol. 2008;86:666–675. doi: 10.1038/icb.2008.47. [DOI] [PubMed] [Google Scholar]
- Wald A, Corey L. Persistence in the population: epidemiology, transmission. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007. [PubMed] [Google Scholar]
- Wald A, Zeh J, Selke S, Warren T, Ryncarz AJ, Ashley R, Krieger JN, Corey L. Reactivation of genital herpes simplex virus type 2 infection in asymptomatic seropositive persons. N Engl J Med. 2000;342:844–850. doi: 10.1056/NEJM200003233421203. [DOI] [PubMed] [Google Scholar]
- Wang K, Lau TY, Morales M, Mont EK, Straus SE. Laser-capture microdissection: refining estimates of the quantity and distribution of latent herpes simplex virus 1 and varicella-zoster virus DNA in human trigeminal Ganglia at the single-cell level. J Virol. 2005a;79:14079–14087. doi: 10.1128/JVI.79.22.14079-14087.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. P Natl Acad Sci USA. 2005b;102:16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber PC, Wigdahl B. Identification of dominant-negative mutants of the herpes simplex virus type 1 immediate-early protein ICP0. J Virol. 1992;66:2261–2267. doi: 10.1128/jvi.66.4.2261-2267.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber PC, Kenny JJ, Wigdahl B. Antiviral properties of a dominant negative mutant of the herpes simplex virus type 1 regulatory protein ICP0. J Gen Virol. 1992;73(Pt 11):2955–2961. doi: 10.1099/0022-1317-73-11-2955. [DOI] [PubMed] [Google Scholar]
- Whitlow Z, Kristie TM. Recruitment of the transcriptional coactivator HCF-1 to viral immediate-early promoters during initiation of reactivation from latency of herpes simplex virus type 1. J Virol. 2009;83:9591–9595. doi: 10.1128/JVI.01115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CL, Johnson EM. Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J Virol. 1987;61:2311–2315. doi: 10.1128/jvi.61.7.2311-2315.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CL, Johnson EM. Characterization of nerve growth factor-dependent herpes simplex virus latency in neurons in vitro. J Virol. 1988;62:393–399. doi: 10.1128/jvi.62.2.393-399.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CL, Smith RL, Freed CR, Johnson EM., Jr Nerve growth factor-dependence of herpes simplex virus latency in peripheral sympathetic and sensory neurons in vitro. J Neurosci. 1990;10:1268–1275. doi: 10.1523/JNEUROSCI.10-04-01268.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CL, Smith RL, Everett RD, Mysofski D. The herpes simplex virus type 1 immediate-early protein ICP0 is necessary for the efficient establishment of latent infection. J Virol. 1997;71:6777–6785. doi: 10.1128/jvi.71.9.6777-6785.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TT, Su YH, Block TM, Taylor JM. Evidence that two latency-associated transcripts of herpes simplex virus type 1 are nonlinear. J Virol. 1996;70:5962–5967. doi: 10.1128/jvi.70.9.5962-5967.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TT, Su YH, Block TM, Taylor JM. Atypical splicing of the latency-associated transcripts of herpes simplex type 1. Virology. 1998;243:140–149. doi: 10.1006/viro.1998.9036. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Herr W. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem Sci. 2003;28:294–304. doi: 10.1016/S0968-0004(03)00088-4. [DOI] [PubMed] [Google Scholar]
- Xu F, Schillinger JA, Sternberg MR, Johnson RE, Lee FK, Nahmias AJ, Markowitz LE. Seroprevalence and coinfection with herpes simplex virus type 1 and type 2 in the United States, 1988–1994. J Infect Dis. 2002;185:1019–1024. doi: 10.1086/340041. [DOI] [PubMed] [Google Scholar]
- Yang L, Voytek CC, Margolis TP. Immunohistochemical analysis of primary sensory neurons latently infected with herpes simplex virus type 1. J Virol. 2000;74:209–217. doi: 10.1128/jvi.74.1.209-217.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh L, Schaffer PA. A novel class of transcripts expressed with late kinetics in the absence of ICP4 spans the junction between the long and short segments of the herpes simplex virus type 1 genome. J Virol. 1993;67:7373–7382. doi: 10.1128/jvi.67.12.7373-7382.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabolotny JM, Krummenacher C, Fraser NW. The herpes simplex virus type 1 2.0-kilobase latency-associated transcript is a stable intron which branches at a guanosine. J Virol. 1997;71:4199–4208. doi: 10.1128/jvi.71.6.4199-4208.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Galvan V, Campadelli-Fiume G, Roizman B. Glycoprotein D or J delivered in trans blocks apoptosis in SK-N-SH cells induced by a herpes simplex virus 1 mutant lacking intact genes expressing both glycoproteins. J Virol. 2000;74:11782–11791. doi: 10.1128/jvi.74.24.11782-11791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaagstra JC, Ghiasi H, Slanina SM, Nesburn AB, Wheatley SC, Lillycrop K, Wood J, Latchman DS, Patel K, Wechsler SL. Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J Virol. 1990;64:5019–5028. doi: 10.1128/jvi.64.10.5019-5028.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]