

Abstract
A novel stereochemical substrate probe was used to assess the factors that affect the stereochemical course of nucleopalladation (cis vs. trans) in the context of an enantioselective Wacker-type reaction. We demonstrate that the enantioselectivity correlates directly with the nucleopalladation pathway, and both the neutral-donor and anionic ligands on palladium are capable of controlling selectivity for cis or trans nucleopalladation.
Keywords: amination, asymmetric catalysis, nucleopalladation, reaction mechanisms, Wacker oxidation
Palladium(II)-catalyzed oxidative functionalization of alkenes has been the focus of intense interest for decades, and Wacker-type cyclizations,[1] which enable synthesis of diverse heterocycles, are a prominent class of these reactions.[2] Substantial effort has been directed toward enantioselective applications, but successful examples (e.g., ≥ 90% ee) remain rare and often exhibit limited substrate scope.[3,4] A key challenge associated with these reactions is the possibility of cis- or trans-nucleopalladation (NP) of the alkene, because the formation of diastereomeric intermediates from these pathways could have significant consequences for the development of enantioselective transformations (Scheme 1).[3] Examples of both cis- and trans-NP pathways in catalytic reactions have been documented,[5,6] but only three enantioselective variants of these reactions have been characterized with respect to the stereochemical course of nucleopalladation. All three examples exhibited a preference for cis-NP.[4f,k,n] The possible impact of the stereochemical course of nucleopalladation on the enantioselectivity of a given asymmetric Wacker-type reaction has not been established. Here, we present a mechanistic investigation of the factors that affect the stereochemical course of nucleopalladation in the context of a recently discovered catalyst system for the enantioselective cyclization of γ-alkenyl tosylamides. Implementation of a novel stereochemical probe demonstrates that both the chiral neutral donor ligand and the anionic ligands on the palladium center are capable of controlling the stereochemical pathway for amidopalladation (AP), but only the trans-AP pathway exhibits high enantioselectivity. These data provide the first direct correlation between nucleopalladation stereoselectivity and the enantioselectivity of the transformation in question. Such insights highlight valuable considerations for the development of enantioselective reactions that involve nucleopalladation of an alkene.
Scheme 1.

Stereochemical pathways for alkene nucleopalladation.
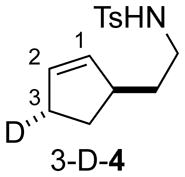
Recently, we showed that a PdII catalyst with a chiral pyridine-oxazoline (pyrox) ligand enables preparation of pyrrolidines in excellent yield and enantioselectivity (Eq. 1).[4p,7] Based on several closely related precedents, we predicted that the PdII/pyrox catalyst system would favor a cis-amidopalladation (cis-AP) mechanism.[8–11] For example, the isotopically labeled substrate 3-D-4 has been used to assess the mechanism of several different PdII catalyst systems for the aerobic, oxidative amidation of alkenes,[5d] and Pd(OAc)2/pyridine and Pd(TFA)2/(−)-sparteine were among the catalyst systems shown to afford products exclusively arising from cis-AP of the alkene.[12] In the enantioselective cyclization of γ-alkenyl tosylamides, the identity of the anionic ligand was found to have a significant impact on the reaction outcome. Replacing Pd(pyrox)(TFA)2 with Pd(pyrox)(OAc)2 gave significantly diminished yield and enantioselectivity under otherwise identical conditions (Eq. 1). The disparity of these results raised the possibility that the reactions with these catalysts might involve different AP pathways.
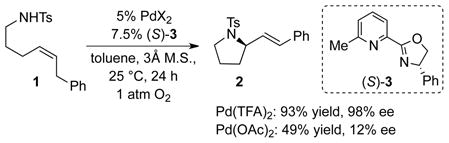 |
(1) |
Our initial attempt to probe the AP pathway for the enantioselective reaction involved the use of substrate 3-D-4 under the previously optimized conditions. However, a mixture of all four of the possible bicyclic pyrrolidines was obtained in 61% yield, favoring the trans-AP products in approximately 3:1 ratio relative to cis-AP products (Scheme 2). This result was unexpected for two reasons: first, the cis-AP pathway was anticipated to be dominant for these conditions, and second, it seemed unlikely that a highly enantioselective reaction would involve simultaneous operation of both cis- and trans-AP pathways. Analysis of the product mixture by chiral HPLC revealed poor kinetic resolution; products 3-D-5 and 5 were formed in 13% ee, and products 3-D-6 and 2-D-6 were formed in 56% ee. The relevance of these results was not entirely clear, in part, because the cyclic alkene in 3-D-4 could influence the stereochemical course of the AP step and may not be a good model for acyclic alkenes that undergo highly enantioselective cyclization.[4p]
Scheme 2.

Cyclization of substrate 3-D-4 with chiral catalyst.
To circumvent the complications associated with the use of 3-D-4 as a mechanistic probe, we prepared a novel acyclic deuterated substrate probe, 6-D-1, which is a chiral analog of substrate 1 (Scheme 3).[13] Analysis of the products formed by oxidative cyclization of 6-D-1 is more involved than the analysis of products derived from substrate 3-D-4 because both the absolute configuration of the product and the loss or retention of the deuterium atom must be accounted for (the four products A–D differ only in the absolute configuration of the stereogenic center and/or the presence or absence of the styrenyl deuterium atom at C6, Scheme 3A). Reliable results with 6-D-1 are possible because trans-styrenyl products are obtained with high selectivity over the cis isomers, and very little deuterium scrambling (≤ 5 %) occurs.[14]
Scheme 3.

(A) Mechanistic pathways for the reaction of 6-D-1 and (B) mathematical relationships used to determine the yields of products A–D.
Three independent analytical measurements were used to establish the yield of products A–D from the reaction of 6-D-1 under various conditions. First, the H/D ratio at C6 in the four styrenyl products was obtained by 1H NMR spectroscopy. This quantity established the relationship (a+d) = x(b+c), where a, b, c and d represent the percent composition of the species A–D, and x = H/D at C6. Second, the enantiomeric ratio of the products was obtained by chiral HPLC analysis. This quantity established the relationship (a+c) = y(b+d), where y = [(R)-products/(S)-products]. Third, the two sets of enantiomeric products were separated by chiral HPLC, and the H/D ratio of the enantiomerically pure products was obtained by 1H NMR spectroscopy.[15] This quantity established the a:c and b:d ratios. With these data in hand and accounting for full mass balance (a+b+c+d = 100), it was possible to solve a system of four equations and four unknowns to determine the quantities a, b, c, and d, from which the trans-AP:cis-AP selectivity was obtained from the ratio (a+b):(c+d) (Scheme 3B).[16]
Substrate 6-D-1 was subjected to the optimized chiral catalyst conditions, and the reaction proceeded in excellent yield and enantioselectivity (90% yield and 96% ee), consistent with the reactivity of 1 reported previously (cf. Eq. 1).[4p] 1H NMR spectroscopic analysis of the initial product mixture revealed a 93:7 preference for the protio products (Scheme 4). Because we had previously determined that the (S)-configuration of pyrox ligand 3 favors formation of the (R)-configuration of the pyrrolidine, the initial 1H NMR and chiral HPLC analysis were enough to conclude that product A was the major species and the trans-AP pathway was heavily favored over the cis pathway. The product ratio was established more definitively with 1H NMR analysis of the purified major enantiomer species A and C. The three measurements show that these reactions exhibit a very high selectivity for a trans-AP pathway (trans:cis-AP = 91:9). The correlation between the high enantioselectivity and high trans:cis-AP selectivity obtained from substrate 6-D-1 may be contrasted to the poor enantioselectivity and poor trans:cis-AP selectivity observed with substrate 3-D-4 (cf. Scheme 2).[17,18]
Scheme 4.

Experimental data and trans:cis-AP selectivity obtained from oxidative cyclization of substrate 6-D-1 with the optimized chiral catalyst conditions.
These results established the utility of substrate probe 6-D-1 and the protocol for product analysis to correlate the enantioselectivity with the AP pathway of the oxidative cyclization reaction. We then turned our attention to the Pd(pyrox)(OAc)2-catalyzed reaction, which proceeds with much lower enantioselectivity under conditions identical to the Pd(pyrox)(TFA)2-catalyzed reaction. The reactivity of 6-D-1 with Pd[(S)-3](OAc)2 as the catalyst was tested and, consistent with our prior results, the reaction proceeded in only 48% yield and 20% ee. 1H NMR analysis of the initial product mixture revealed a 48:52 H:D ratio. Following separation of the two enantiomeric products by chiral HPLC, analysis of the (R)-configured products revealed a 14:86 H:D ratio, while the purified (S)-configured products displayed a 96:4 H:D ratio (Scheme 5). Incorporation of the data from either of these two measurements into the system of four equations led to similar product ratios for species A, B, C and D, and the results show that the reaction strongly favored a cis-AP pathway (trans:cis-AP = 10:90), with a 9:1:51:39 ratio for A:B:C:D. While the overall reaction exhibited low enantioselectivity, consideration of the minor products A and B, which arose from trans-AP of the alkene, revealed that the trans-AP pathway was quite enantioselective (e.r. = 9:1). Thus, with this substrate and pyrox-PdII catalyst system, the trans-AP pathway proceeds with high enantioselectivity while the cis-AP pathway exhibits low enantioselectivity. These observations represent the first direct assessment of the enantioselectivity of two different NP pathways for otherwise identical reactions.
Scheme 5.

Experimental data and trans:cis-AP selectivity obtained from oxidative cyclization of substrate 6-D-1 with a Pd(pyrox)(OAc)2 catalyst system (refer to Scheme 4 for depiction of the reaction).
The results of the reactions with the chiral ligands are summarized in Table 1, entries 1 and 2. In an effort to separate the influence of the neutral-donor and anionic ligands on the stereochemical course of the AP step, we investigated the oxidative cyclization of 6-D-1 with Pd(OAc)2 and Pd(TFA)2 in the absence of an ancillary neutral-donor ligand. The results show that both PdII sources favor cis-AP of the alkene (Table 1, entries 3 and 4; see also Schemes S7–S8). The selectivity is considerably higher with Pd(OAc)2; only trace quantities of the trans-AP-derived product are detected by NMR/HPLC analysis. With Pd(TFA)2, the trans:cis-AP selectivity is 1:6, suggesting that while the TFA ligand is intrinsically more compatible with the trans-AP mechanism, it still favors cis-AP. Taken together, the data in Table 1 demonstrate that the pyrox ligand plays an important role in enforcing the trans-AP pathway with Pd(TFA)2 as the PdII source. Previous efforts to understand the factors that influence trans vs. cis-AP selectivity have implicated the carboxylate ligand as a Brønsted base to mediate Pd–amidate formation in the cis-AP pathway.[5d,19] The present findings reveal that only with the combined presence of a trifluoroacetate anionic ligand and the pyrox neutral-donor ligand is a trans-AP pathway, initiated by substitution of TFA by the substrate alkene, favored over the cis-AP pathway involving formation of a Pd–amidate.
Table 1.
Summary of amidopalladation studies with pyrox-ligated and ligand-free catalysts.
| entry | PdII (5%) | ligand (7.5%) | %yield[a] | %ee[b] | trans:cis-AP[c] |
|---|---|---|---|---|---|
| 1 | Pd(TFA)2 | (S)-3 | 90 | 96 | >9 : 1 |
| 2 | Pd(OAc)2 | (S)-3 | 48 | 20 | 1 : 9 |
| 3 | Pd(TFA)2 | None | 55 | 0 | 1 : 6 |
| 4 | Pd(OAc)2 | None | 15 | 0 | <1 : 9 |
Determined by 1H NMR analysis of the crude reaction mixture.
Determined by HPLC analysis of the purified products
See the supporting information for full disclosure of the raw data.
In summary, the design, synthesis and implementation of a novel chiral substrate probe (6-D-1) has enabled key insights into the relationship between the NP pathway and the enantioselectivity of a catalytic transformation. The ability of an ancillary neutral-donor ligand to alter the stereochemical course of NP only when a suitable anionic ligand is present highlights the challenges associated with the discovery of efficient catalysts for the asymmetric Wacker-type oxidation of alkenes. Ideally, the factors that affect the NP stereochemistry should be considered in conjunction with the exploration of chiral ancillary ligands in the future development of enantioselective reactions.
Supplementary Material
Footnotes
We thank Dr. Richard I. McDonald and Chun Pong Tam for initiating the synthesis of substrate probe 6-D-1, and Paul B. White and Dr. Charlie G. Fry for assistance with NMR spectroscopic measurements. We thank the NIH (R01 GM67163) and Organic Syntheses (ACS Division of Organic Chemistry fellowship for A.B.W.) for financial support of this work. Spectroscopic instrumentation was partially funded by the NSF (CHE-0342998, CHE-9629688, CHE-9208463) and NIH (1 S10 RR13866-01).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Henry PM. Palladium Catalyzed Oxidation of Hydrocarbons. Vol. 2. D. Reidel Publishing Co; Dordrecht, The Netherlands: 1980. [Google Scholar]
- 2.a) Zeni G, Larock RC. Chem Rev. 2004;104:2285–2309. doi: 10.1021/cr020085h. [DOI] [PubMed] [Google Scholar]; b) Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem Rev. 2007;107:5318–5365. doi: 10.1021/cr068006f. [DOI] [PubMed] [Google Scholar]; c) Minatti A, Muñiz K. Chem Soc Rev. 2007;36:1142–1152. doi: 10.1039/b607474j. [DOI] [PubMed] [Google Scholar]; d) Kotov V, Scarborough CC, Stahl SS. Inorg Chem. 2007;46:1910–1923. doi: 10.1021/ic061997v. [DOI] [PubMed] [Google Scholar]; e) Jensen KH, Sigman MS. Org Biomol Chem. 2008;6:4083–4088. doi: 10.1039/b813246a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDonald RI, Liu GS, Stahl SS. Chem Rev. 2011;111:2981–3019. doi: 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For leading primary references, see: Hosokawa T, Uno T, Inui S, Murahashi SI. J Am Chem Soc. 1981;103:2318–2323.Uozomi Y, Kato K, Hayashi T. J Am Chem Soc. 1997;119:5063–5064.Uozomi Y, Kato K, Hayashi T. J Org Chem. 1998;63:5071–5075.Arai MA, Kuraishi M, Arai T, Sasai H. J Am Chem Soc. 2001;123:2907–2908. doi: 10.1021/ja005920w.Trend RM, Ramtohul YK, Ferreira EM, Stoltz BM. Angew Chem Int Ed. 2003;42:2892–2895. doi: 10.1002/anie.200351196.Angew Chem. 2003;115:2998–3001.Hayashi T, Yamasaki K, Mimura M, Uozumi Y. J Am Chem Soc. 2004;126:3036–3037. doi: 10.1021/ja031946m.Trend RM, Ramtohul YK, Stoltz BM. J Am Chem Soc. 2005;127:17778–17788. doi: 10.1021/ja055534k.Yip KT, Yang M, Law KL, Zhu NY, Yang D. J Am Chem Soc. 2006;128:3130–3131. doi: 10.1021/ja060291x.Zhang YJ, Wang F, Zhang W. J Org Chem. 2007;72:9208–9213. doi: 10.1021/jo701469y.Tsujihara T, Shinohara T, Takenaka K, Takizawa S, Onitsuka K, Hatanaka M, Sasai H. J Org Chem. 2009;74:9274–9279. doi: 10.1021/jo901778a.He W, Yip KT, Zhu NY, Yang D. Org Lett. 2009;11:5626–5628. doi: 10.1021/ol902348t.Scarborough CC, Bergant A, Sazama GT, Guzei IA, Spencer LC, Stahl SS. Tetrahedron. 2009;65:5084–5092. doi: 10.1016/j.tet.2009.04.072.Jiang F, Wu Z, Zhang W. Tetrahedron Lett. 2010;51:5124–5126.Mai DN, Wolfe JP. J Am Chem Soc. 2010;132:12157–12159. doi: 10.1021/ja106989h.Jensen KH, Webb JD, Sigman MS. J Am Chem Soc. 2010;132:17471–17482. doi: 10.1021/ja108106h.McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. Org Lett. 2011;13:2830–2833. doi: 10.1021/ol200784y.Yang G, Shen C, Zhang W. Angew Chem Int Ed. 2012;51:9141–9145. doi: 10.1002/anie.201203693.Angew Chem. 2012;124:9275–9279.
- 5.For examples of cis-nucleopalladation, see the following leading references: 4f,g,k,n and Ney JE, Wolfe JP. Angew Chem Int Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060.Angew Chem. 2004;116:3689–3692.Hay MB, Wolfe JP. J Am Chem Soc. 2005;127:16468–16476. doi: 10.1021/ja054754v.Nakhla JS, Kampf JW, Wolfe JP. J Am Chem Soc. 2006;128:2893–2901. doi: 10.1021/ja057489m.Liu G, Stahl SS. J Am Chem Soc. 2007;129:6328–6335. doi: 10.1021/ja070424u.Muñiz K, Hovelmann CH, Streuff J. J Am Chem Soc. 2008;130:763–773. doi: 10.1021/ja075041a.Michel BW, Steffens LD, Sigman MS. J Am Chem Soc. 2011;133:8317–8325. doi: 10.1021/ja2017043.
- 6.For examples of trans-nucleopalladation, see the following leading references: 4g,o, 5c,d and Watson MP, Overman LE, Bergman RG. J Am Chem Soc. 2007;129:5031–5044. doi: 10.1021/ja0676962.Cochran BM, Michael FE. J Am Chem Soc. 2008;130:2786–2792. doi: 10.1021/ja0734997.Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J Am Chem Soc. 2009;131:15945–15951. doi: 10.1021/ja906915w.
- 7.Quinox and pyrox ligands have emerged as a versatile class of chiral ligands for PdII-catalyzed reactions with alkenes. For examples other than those described in references 4k,m,o,p and 11, see: Perch NS, Widenhoefer RA. J Am Chem Soc. 1999;121:6960–6961.Perch NS, Pei T, Widenhoefer RA. J Org Chem. 2000;65:3836–3845. doi: 10.1021/jo0003192.Zhang Y, Sigman MS. J Am Chem Soc. 2007;129:3076–3077. doi: 10.1021/ja070263u.Pathak TP, Gligorich KM, Welm BE, Sigman MS. J Am Chem Soc. 2010;132:7870–7871. doi: 10.1021/ja103472a.Jana R, Pathak TP, Jensen KH, Sigman MS. Org Lett. 2012;14:4074–4077. doi: 10.1021/ol3016989.
- 8.See reference 5d and: Ye X, Liu G, Popp BV, Stahl SS. J Org Chem. 2011;76:1031–1044. doi: 10.1021/jo102338a.
- 9.Investigation of a well-defined (bipyridine)PdII–tosylamidate complex provided fundamental insights into alkene insertion into a Pd–N(R)Ts bond (i.e., the cis-AP step); see White PB, Stahl SS. J Am Chem Soc. 2011;133:18594–18597. doi: 10.1021/ja208560h.For other studies of alkene insertion into Pd–N bonds, see the following leading references: Hanley PS, Hartwig JF. J Am Chem Soc. 2011;133:15661–15673. doi: 10.1021/ja205722f.Neukom JD, Perch NS, Wolfe JP. Organometallics. 2011;30:1269–1277.
- 10.Yang and co-workers recently reported a PdII/quinoline-oxazoline (quinox) catalyst system for enantioselective oxidative cyclization of anilides bearing tethered alkenes, and stereochemical analysis of the products revealed that the reaction involves cis-AP of the alkene (see reference 4k).
- 11.Further circumstantial evidence supporting a cis-AP mechanism comes from the use of quinox and pyrox ligands in other PdII-catalyzed reactions, such as oxidative Heck reactions, which involve cis addition of PdII and an aryl group across an alkene C=C bond. For cis-nucleopalladation reactions employing pyrox/quinox ligands, see reference 4k, 5f, and: Yoo KS, Park CP, Yoon CH, Sakaguchi S, O’Neil J, Jung KW. Org Lett. 2007;9:3933–3935. doi: 10.1021/ol701584f.
- 12.See reference 5d. (–)-Sparteine is a chiral diamine (bidentate) ligand.
- 13.Substrate 6-D-1 was synthesized from (S)-Mandelic acid in ten steps, with the first five steps having direct precedent in the literature: Alberti MN, Vassilikogiannakis G, Orfanopoulos M. Org Lett. 2008;10:3997–4000. doi: 10.1021/ol801488w.Derivatization and analysis of an advanced intermediate established the stereochemical purity of the substrate probe to be very high (>20:1, see Supp. Info. for details).
- 14.A detailed consideration of how alkene isomerization and deuterium scrambling could complicate our results is provided in the Supp. Info. (Scheme S1). However, under all catalyst conditions tested in this study, the trans-styrenyl products are obtained in >20:1 selectivity, and it was possible to remove trace quantities of the cis-styrenyl products by chromatography prior to further analysis. The extent of deuterium scrambling was characterized by 2H NMR analysis of the mixture of four products A–D (Scheme S3–S8), and only trace quantities (≤ 5%) of deuterium at the C5 position were observed in our experiments.
- 15.This quantity was corroborated by ESI-MS analysis of the enantiomerically pure products (Scheme S3–S8).
- 16.The validity of this protocol was tested by subjecting 6-D-1 to the previously reported Pd(OAc)2/pyridine oxidative cyclization conditions. The data arising from this experiment show that the reaction proceeds with very high selectivity for cis-AP of the alkene (trans:cis = 8:92; see supporting information Scheme S3), supporting the previously reported conclusions derived from the use of substrate 3-D-4 as the mechanistic probe (see reference 5d).
- 17.Cyclization of substrate 6-D-1 with the opposite antipode of the ligand, (R)-3, resulted in 93% yield, 96% ee, and a 96:4 trans:cis-AP ratio (see the supporting information Scheme S5). The similar trans:cis-AP ratios obtained with ligands (S)-3 and (R)-3 in the reactions of 6-D-1 show that any kinetic isotope effect associated with β-hydride elimination from the C6 position has minimal impact on the AP pathway.
- 18.We previously reported DFT calculations that implicated high enantioselectivity for a cis-AP pathway (see ref. 4p). The present results suggest that at least one other cis-AP pathway exists that is lower in energy than the one presented in ref. 4p. DFT calculations to explore this issue and to analyze the trans-AP mechanism are currently under investigation.
- 19.For other discussion of factors contributing to cis- vs, trans-nucleopalladation, see refs. 3, 4g, 5c and the following: Keith JA, Henry PM. Angew Chem Int Ed. 2009;48:9038–9049. doi: 10.1002/anie.200902194.Angew Chem. 2009;121:9200–9212.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.