

Abstract
The classic model of GPCR activation proposed that all agonists induce the same active receptor conformation. However, research over the last decade has shown that GPCRs exist in multiple conformations, and that agonists can stabilize different active states. The distinct receptor conformations induced by ligands result in distinct receptor–effector complexes, which produce varying levels of activation or inhibition of subsequent signalling cascades. This concept, referred to as ligand-directed signalling or biased agonism has important biological and therapeutic implications. Opioid receptors are Gi/o GPCRs and regulate a number of important physiological functions, including pain, reward, mood, stress, gastrointestinal transport and respiration. A number of in vitro studies have shown biased agonism at the three opioid receptors (µ, δ and κ); however, in vivo consequences of this phenomenon have only recently been demonstrated. For the µ and δ opioid receptors, the majority of reported ligand selective behavioural effects are observed as differential adaptations to repeated drug administration. In terms of the κ opioid receptor, clear links between ligand-selective signalling events and specific in vivo responses have been recently characterized. Drugs for all three receptors are either already used or are being developed for clinical applications. There is clearly a need to better characterize the specific events that occur following agonist stimulation and how these relate to in vivo responses. This understanding could eventually lead to the development of tailor-made pharmacotherapies where advantageous drug effects can be selectively targeted over adverse effects.
Keywords: GPCR, reward, pain, functional selectivity, receptor biology
Introduction
GPCRs are the most abundant receptor class in the human genome (Lagerstrom and Schioth, 2008), and as such, these receptors regulate diverse biological functions. Given their importance in physiological processes, they are the most commonly targeted receptor class for pharmacological therapies (Ma and Zemmel, 2002). Classical receptor theory had postulated that GPCRs existed in equilibrium between an inactive (R) and an active (R*) state, and that upon binding, all agonists equally promoted the same subsequent receptor regulation and signalling cascades (for review, see Kenakin, 2004). However, in the past 15 years, numerous studies have challenged this idea, and the current view is that the receptor can exist in multiple states, and that agonists can initiate selective receptor conformations, which in turn engage distinct signalling and receptor regulatory responses (Kenakin, 2011; Reiter et al., 2012). This concept has been referred to in a number of ways including: ligand-directed signalling, functional selectivity, biased agonism, ligand-biased efficacy, collateral efficacy and stimulus trafficking (Galandrin et al., 2007; Rajagopal et al., 2010; Vaidehi and Kenakin, 2010).
Divergent functional responses from ligand-bound GPCRs can be modulated at a number of different levels. Traditionally, it was thought that activation of G-proteins was the primary way by which GPCRs signalled. This G-protein-dependent signalling is mediated through Gα and Gβγ subunits, and includes regulation of adenylate cyclase, phospholipases, multiple kinases and ion channels. More recently, it has been shown that GPCRs can also mediate G-protein-independent signalling, through proteins such as β-arrestins and PDZ-containing proteins (Magalhaes et al., 2012). β-Arrestin regulated responses are by far the best characterized, and it appears that not only do β-arrestins mediate receptor trafficking, but they also act as scaffolding molecules on which a number of signalling cascades are initiated (for review, see Sorkin and von Zastrow, 2009; Rajagopal et al., 2010). Thus, functional selectivity may be observed by ligands promoting G-protein-dependent or -independent signalling or both. The concept of ligand-directed signalling has important implications from a therapeutic perspective and holds the promise of designing drugs to selectively avoid undesirable biological effects targeted by receptor activation. This review will focus on ligand-directed signalling within the family of opioid receptors as an example of GPCRs with diverse structural and functional ligands and high therapeutic importance.
Opium has been used for many centuries for its medicinal and euphoric properties, and the use and abuse of this plant ultimately led to the discovery of the endogenous opioid system. One of the first breakthroughs in understanding the unique pharmacology of opium occurred in 1806, when Friedrich Wilhelm Serturner isolated the primary active ingredient in opium and called it morphine, after Morpheus, the god of dreams (Scott, 1969). The elucidation of the alkaloid structure of morphine led to the development of the synthetic opioid heroin, which was found to be more potent than morphine and even more problematic for triggering addictive behaviours. Many other opioid agonists have since been characterized, but to date, there are still no commercially available opioid therapeutics that are both effective analgesics and free from abuse liability.
The opioid receptor family includes three members: the µ, δ and κ opioid receptors. The existence of opioid receptors was discovered in 1973 by three separate groups, all using opioid radioligand binding in brain homogenates (Pert and Snyder, 1973; Simon et al., 1973; Terenius, 1973), and genes encoding µ, δ and κ receptors were subsequently cloned in the early 1990s (Evans et al., 1992; Kieffer et al., 1992; Chen et al., 1993; Minami et al., 1993). The µ, δ, and κ opioid receptors are encoded by Oprm1, Oprd1 and Oprk1 genes, respectively. Opioid receptors are activated by a family of naturally occuring endogenous peptides, the first of which was discovered in 1975, and genes identified in the early 1980s. These neuropeptides, which include the enkephalins, endorphins and dynorphins, are processed from larger precursor proteins encoded by Penk, Pdyn and Pomc genes. Opioid receptors are located throughout the body, and regulate a number of important behaviours such as reward, pain, stress, gastrointestinal transport and mood through receptors in both the central and peripheral nervous systems (for recent reviews, see Al Hasani and Bruchas, 2011; Sauriyal et al., 2011).
The three opioid receptors show a high degree of sequence homology, and a common opioid receptor binding pocket within the helical transmembrane core has been postulated based upon modelling and structure activity studies (Metzger and Ferguson, 1995; Paterlini, 2005). The greatest divergence in sequence between the receptors occurs at extracellular domains and in vitro studies using mutant receptors have identified these regions as important for ligand selectivity (Kane et al., 2006). Likewise, these studies have identified helical domain-mediated mechanisms for opioid receptor activation within the membrane core receptor domain (Decaillot et al., 2003), which is highly similar across the three receptors. Very early on, in vitro receptor expression in transfected cells identified the first example of opioid ligand-directed trafficking (Arden et al., 1995; Keith et al., 1996), and site-directed mutagenesis experiments also provided indirect evidence for the existence of multiple active receptor conformations (Befort et al., 1996), preparing the ground for biased agonism at opioid receptors. As may be inferred from the breadth of structural diversity of the peptide and alkaloid agonists that bind to opioid receptors, not all ligands interact with the same components of the receptor protein. Numerous structure–activity studies have identified key amino acids in the opioid receptors that selectively disrupt binding and signalling of some but not all agonists (Kane et al., 2006). The ligand diversity and therapeutic importance of opioid drugs makes the opioid receptors excellent model GPCRs to understand the basis of ligand-directed signalling.
Ligand-directed signalling at the µ opioid receptor
Ligand directed signalling via the µ opioid receptor has important implications given the wide use of µ opioid receptor targeting drugs such as morphine, fentanyl, oxycodone and heroin both as analgesics and drugs of abuse. In mice null for the µ opioid receptor, morphine loses both its analgesic efficacy and rewarding properties (Matthes et al., 1996; Contet et al., 2004), as well as many other well-described biological activities (for review, see Gaveriaux-Ruff and Kieffer, 2002), demonstrating that this receptor mediates multiple effects of the prototypic opiate drug throughout brain circuits. Given their therapeutic importance, agonists that selectively induce discrete µ opioid receptor signalling complexes could be critical in developing pharmacotherapies that dissociate µ agonist-induced pain relief from reward and constipation, or distinguish adaptations to exogenous opioids such as tolerance and hyperalgesia (Kieffer and Evans, 2002; Evans, 2004).
As with most GPCRs, ligand binding to the µ opioid receptor can induce receptor internalization, a complex regulatory process that can lead to diminished receptor activation despite the continued presence of ligand [for review see (Evans, 2004; Kelly et al., 2008)]. µ Receptor internalization is followed by receptor recycling back to the cell surface, leading to restoration of receptor function (Koch et al., 2005). Early evidence for agonist-selective trafficking was revealed by the differential effects of morphine, DAMGO ([D-Ala2, N-MePhe4, Gly-ol]-enkephalin), and fentanyl on receptor trafficking in transfected cells, with morphine inducing poor internalization compared with DAMGO and fentanyl. Further early work indicated differential phosphorylation upon agonist activation, whereby morphine appeared to induce little receptor phosphorylation compared to DAMGO and fentanyl. Subsequent studies revealed differential agonist-dependent signalling and desensitization, suggesting biased responses involving GPCR kinases (GRKs), PKC and β-arrestins (for review, see Evans, 2004; Kelly et al., 2008).
Recent technological developments have started to further characterize ligand-directed signalling at the µ opioid receptor. A new approach, fluorescence recovery after photobleaching (FRAP), has revealed agonist-receptor-specific biophysical events at the level of the plasma membrane (Sauliere-Nzeh et al., 2010). In these studies, morphine triggered diffusion of the fluorescent recombinant receptor in neuroblastoma cells, which was pertussis toxin-sensitive. In contrast, DAMGO induced a sucrose-dependent aggregation to small isolated domains for half the receptors, and free long-range receptor diffusion for the other half. Another recent approach developed to investigate ligand-directed signalling at the µ opioid receptor measured dynamic mass redistribution (DMR) of the receptor upon agonist binding. This promising approach provides real-time optical fingerprints of GPCR signalling in living cells, and a first heat map based on the numerical analysis of DMR parameters for about 50 µ ligands under 13 experimental conditions was recently reported (Morse et al., 2011). This study revealed a number of novel pharmacological properties for several commonly used opioid ligands; including differences in opioid receptor affinity for specific G-proteins and activation of divergent signalling cascades (Morse et al., 2011). Although these studies may not definitively prove the existence of ligand-directed signalling, they do provide the basis for further investigation.
Agonist-directed signalling at the µ opioid receptor is also currently being characterized within native cell systems. In the case of ERK phosphorylation, morphine was shown to activate ERK pathways in the cytosol via PKCε leading to ribosomal S6 kinase stimulation. In contrast, etorphine triggered phospho-ERK translocation into the nucleus through a β-arrestin pathway, modulating Elk-1 and gene expression. First observed in HEK293 cells (Zheng et al., 2008), this differential signalling event was later linked to spine stability and morphology of hippocampal neurons, where morphine decreased dendritic spine volume while etorphine, fentanyl and DAMGO did not (Zheng et al., 2011). In addition, biased agonism has been observed in primary cultures of dorsal root ganglia cells where DAMGO but not morphine-activated P38 MAPK. Pharmacological blockade of P38 MAPK disrupted desensitization of DAMGO but not morphine signalling to calcium channels (Tan et al., 2009), and this supports previous data showing DAMGO-induced P38 activation regulates endocytosis via the early endosomal antigen 1 and Rabenosyn5, both components of the early endosome (Mace et al., 2005). Interestingly, in primary cultures from dorsal root ganglia, DAMGO and clonidine, but not morphine, induced cross-desensitization and co-internalization of both µ receptors and α2A-adrenoceptors. The cross-desensitization of adrenoceptors and opioid receptors was disrupted both by the absence of β-arrestin 2 and the blockade of P38 MAPK, suggesting that biased agonism can also be important in regulating signalling cascades initiated by other GPCRs (Tan et al., 2009). In the case of morphine, desensitization of the µ opioid receptor in dorsal root ganglia was dependent on β-arrestin 2, which was not observed for other higher efficacy agonists (Mittal et al., 2012). Furthermore, whole cell patch clamp experiments in locus coeruleus slices found that desensitization of the µ opioid receptor by morphine was mediated by PKCα, while DAMGO used a GRK2-dependent mechanism (Bailey et al., 2009).
A recent behavioural study demonstrated that JNK signalling is selectively involved in morphine but not fentanyl analgesic tolerance (Melief et al., 2010). Thus, inhibition of JNK signalling either genetically or pharmacologically prevented acute tolerance to morphine but not fentanyl. Conversely, GRK3 knockout mice maintained acute tolerance to morphine but did not show acute tolerance to fentanyl (Terman et al., 2004). Together, these data demonstrate the existence of µ agonist-specific signalling mechanisms in the development of analgesic tolerance, with JNK and GRK3 kinases mediating distinct forms of tolerance. Effector systems associated with µ opioid receptors and the potential distinct signalling complexes that probably operate in vivo are schematized in Figure 1.
Figure 1.
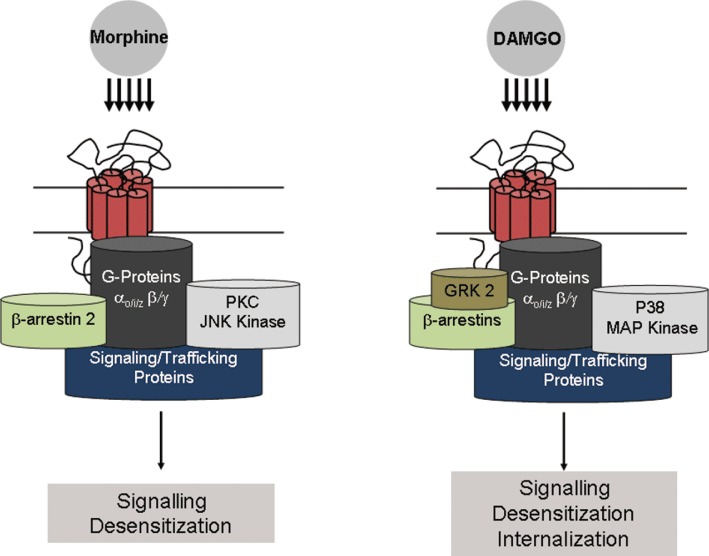
Ligand-specific signalling complexes at the µ opioid receptor. Treatment with morphine or DAMGO elicits differential signalling and trafficking of the µ opioid receptor. Activation of the µ opioid receptor by the low-internalizing agonist morphine is thought to result in receptor desensitization via a β-arrestin and PKC-dependent pathway. In contrast, the high-internalizing agonist DAMGO desensitizes the receptor in a GRK2-dependent manner and recruits P38 MAPK, which appears to be critical for µ opioid receptor desensitization and internalization. Such differences could feasibly be explained by different receptor conformations that allow similar G-protein activation but different kinase recruitment and hence desensitization. Further work is needed to explain how these ligand-specific complexes relate to tolerance.
Finally, knock-in mice expressing a mutant µ opioid receptor that is able to internalize and recycle in response to morphine showed increased analgesia and reward, and reduced tolerance, dependence and addictive behaviour (Kim et al., 2008; Berger and Whistler, 2011). Importantly, morphine but not methadone produced enhanced analgesia and low tolerance with methadone exhibiting similar effects in knock-in and wild-type animals (Kim et al., 2008). These observations show that facilitated receptor internalization improved the drug profile specifically for the low-internalizing agonist (morphine) and suggest that the high-internalizing properties of methadone contribute to optimal analgesic efficacy and duration. µ opioid receptor internalizing agonists and the associated signalling complexes therefore represent valuable molecular targets for more effective analgesics (Berger and Whistler, 2010).
Together, these data demonstrate that agonist-biased signalling has behavioural consequences. These findings also strongly suggest that morphine preferably recruits β-arrestin 2-mediated pathways in vivo, and recent evidence suggests that other µ agonists (methadone, fentanyl) may recruit both β-arrestin 1 and 2 signalling (Groer et al., 2011). It will be interesting to examine in vivo properties of novel compounds such as herkinorin that, in contrast to all the known µ agonists including morphine, do not recruit β-arrestin 2 (Tidgewell et al., 2008). Indeed, very recent work suggests that herkinorin, as compared with morphine, shows attenuated tolerance following chronic use (Lamb et al., 2012).
Ligand-directed signalling at the δ opioid receptor
Compared with the more clinically relevant µ opioid receptor – at least at the time of writing –δ opioid receptors have been relatively understudied. However, recent advances in the pharmacological and genetic tools used to study this receptor have revealed its importance in a number of physiological processes (Pradhan et al., 2011). Stimulation of δ opioid receptors does not result in many of the adverse effects associated with µ agonists, including addictive liability (Stevenson et al., 2005; Codd et al., 2009), respiratory depression (Takita et al., 1997; Codd et al., 2009) and constipation (Petrillo et al., 2003; Codd et al., 2009). Although δ agonists are poor analgesics in acute pain (Gallantine and Meert, 2005), they are highly effective in animal models of chronic inflammatory and neuropathic pain (Fraser et al., 2000; Hurley and Hammond, 2000; Cahill et al., 2003; Nadal et al., 2006; Gaveriaux-Ruff et al., 2008). Interestingly, δ opioid receptors also modulate emotional state. Genetic deletion of either the δ opioid receptor or its endogenous ligand, enkephalin, results in anxiogenic and depressive-like behaviours (Konig et al., 1996; Filliol et al., 2000), and δ opioid receptor agonists produce anxiolytic and anti-depressant effects (Broom et al., 2002a; Saitoh et al., 2004; Perrine et al., 2006).
Agonist activation of the δ opioid receptor can initiate both G-protein-dependent and -independent signalling pathways. Agonist-induced activation of the δ opioid receptor leads to receptor desensitization, which for some agonists is attributed to receptor internalization. δ Opioid receptor internalization has been observed following binding of endogenous opioids (leu and met-enkephalin), peptides ([D-Pen2,5]enkephalin, [D-Pen2,D-Pen5]enkephalin (DPDPE), deltorphin) and small molecules (SNC80) (Bradbury et al., 2009; Pradhan et al., 2009). Unlike internalization of the µ opioid receptor, which results in rapid redistribution of most receptors back to the cell membrane, δ opioid receptors are predominantly targeted for degradation through the endosomal sorting complex required for transport (ESCRT) machinery (Henry et al., 2011).
Convergent evidence from a number of different in vitro studies reveals ligand-directed signalling and trafficking of the δ opioid receptor. In bioluminescence resonance energy transfer (BRET) studies, δ opioid receptor ligands did not equally engage β-arrestin 2. Most ligands were agonists for inducing G-protein coupling to the receptor but showed variable efficacy for arrestin-receptor interactions. In addition, ligands that induced strong physical interactions with G-proteins but weak or no β-arrestin 2 interaction acted as competitive antagonists for arrestin binding (Molinari et al., 2010). This subset of ligands could be explained as partial agonists for β-arrrestin 2 recruitment, and presumably arrestin-mediated signalling; however, they did show full agonist activity in recruiting G-protein subunits. In addition, it was found that δ opioid receptors were in constitutive complexes with G-proteins, and that each δ ligand induced a specific conformation resulting in divergent activation of second messenger transducers (Audet et al., 2008). Evidence from BRET studies may more closely reveal true ligand directed signalling, as this technique directly measures protein–protein interactions that can reflect specific ligand-induced receptor conformations.
Cellular studies examining ligand-specific desensitization and receptor trafficking have also found biased agonism at the δ opioid receptor. Differential desensitization of the δ opioid receptor was observed following stimulation with either peptide (DPDPE and deltorphin II) or alkaloid (etorphine) agonists (Allouche et al., 1999). Furthermore, each class of ligand differentially activated kinases (Marie et al., 2008) and distinctly mediated β-arrestin 1 recruitment (Aguila et al., 2012) in order to initiate receptor desensitization. In addition, agonists differentially regulated sorting of the δ opioid receptor following internalization. In SK-N-BE cells expressing Flag-tagged human δ opioid receptor, the peptides DPDPE and deltorphin, and SNC80 appeared to promote receptor degradation while the endogenous enkephalins and etorphine promoted receptor recycling (Marie et al., 2003b; Lecoq et al., 2004). Taken together, these in vitro studies indicate that the δ opioid receptor exists in multiple active conformation states.
Until recently, very little was known about the in vivo consequences of agonist-induced δ opioid receptor trafficking. The development of a knock-in mouse model expressing fluorescent δ opioid receptor (DOR-eGFP) (Scherrer et al., 2006; 2009) has opened the possibility to correlate ligand-induced receptor trafficking with receptor function in vivo. These animals express functional δ opioid receptors at physiological levels, which are directly visible in vivo. At behaviourally relevant doses, the prototypic agonist SNC80 (Bilsky et al., 1995) produces internalization of DOR-eGFP throughout the peripheral and central nervous systems of these animals (Scherrer et al., 2006; Pradhan et al., 2009; Poole et al., 2011). Acute administration of SNC80 also reversed hyperalgesia in a model of inflammatory pain. However, this initial treatment with SNC80 also produced robust receptor internalization and G-protein uncoupling resulting in acute behavioural desensitization (Pradhan et al., 2009). Subsequently, chronic treatment with SNC80 resulted in extensive receptor degradation, as was predicted from in vitro studies, leading to generalized behavioural tolerance to all agonist effects (Pradhan et al., 2010) and Figure 2). Contrary to SNC80, the δ agonist ARM390 did not produce detectable receptor phosphorylation or internalization following agonist binding, although efficacy and potency for G-protein activation (Marie et al., 2003a; Pradhan et al., 2009), and analgesic properties (Pradhan et al., 2010) were similar. The different receptor trafficking properties of ARM390 had remarkable consequences in vivo. Acute treatment with ARM390 did not produce behavioural desensitization, and δ opioid receptors remained located on the cell surface and coupled to G proteins. However, chronic treatment ultimately led to analgesic tolerance, despite unchanged δ opioid receptor number, G-protein coupling and cell membrane localization. Unlike the generalized tolerance produced by SNC80, ARM390 produced an analgesic tolerance, probably due to changes in second messenger responses in pain-specific pathways (Pradhan et al., 2010 and Figure 2). These data indicate that ligand-specific trafficking of the δ opioid receptor can produce two distinct types of behavioural tolerance. From a therapeutic perspective, these findings imply that non-internalizing δ receptor agonists may be more efficacious for the treatment of diseases unrelated to pain, such as anxiety and depression.
Figure 2.
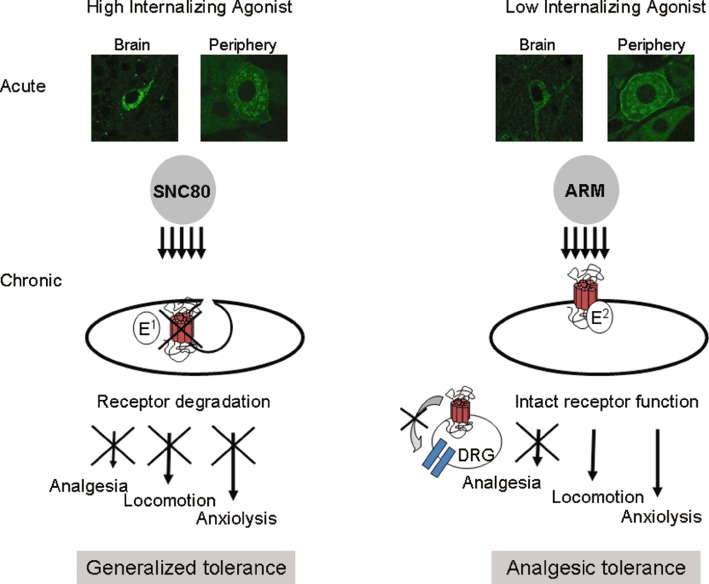
The behavioural consequences of functional selectivity at the δ opioid receptor. SNC80 and ARM390 (ARM) have comparable selectivity and potencies for the δ opioid receptor, but highly distinct internalization properties. Systemic SNC80, but not ARM390, produces clear receptor internalization in vivo as shown in slices from DOR-eGFP knock-in mice (representative images from the hippocampus and dorsal root ganglia) (Pradhan et al., 2009). Chronic administration of either agonist produces two distinct forms of tolerance. Repeated administration of SNC80 produces widespread receptor down-regulation, thus resulting in a generalized tolerance where all δ agonist-induced behaviours are affected. In contrast, chronic administration of the low-internalizing agonist, ARM390, appears to affect δ opioid receptors only at the level of the dorsal root ganglia, thus producing tolerance at the level of pain processing (Pradhan et al., 2010).
In other studies examining tolerance following chronic administration of systemically available δ opioid receptor agonists, analgesic tolerance was not observed in models of chronic pain (Petrillo et al., 2003; Jutkiewicz et al., 2005; Beaudry et al., 2009; Codd et al., 2009). To date, very few δ agonists have been characterized at the level of in vivo receptor trafficking, and in order to truly determine the relationship between δ opioid receptor internalization and tolerance, a thorough study examining a number of different ligands under experimentally controlled conditions needs to be performed.
A major caveat to the development of δ agonists is that some, but not all, δ agonists also produce convulsions, an effect that is specific to the activation of the δ opioid receptor (Broom et al., 2002b; Scherrer et al., 2006). Currently, it is impossible to screen for this ligand-specific behavioural effect in vitro. Understanding which signalling pathways mediate this agonist-selective response would allow for the development of in vitro screening tools for novel agonists, thus saving time, money and the necessary behavioural studies. Importantly, this type of screen would further encourage the development of δ opioid receptor agonists for clinical use.
Ligand-directed signalling at the κ opioid receptor
κ Opioid receptors have been implicated in a number of physiological responses, including nociception, stress, mood, feeding, gut motility and diuresis. Therapeutically, κ opioid receptor agonists are being explored as alternatives to µ analgesics, as they have low abuse potential and produce minimal effects on gastrointestinal transit and respiration. In addition, κ agonists may relieve or prevent hyperalgesia produced by chronic use of µ opioid receptor therapies (for review, see Kivell and Prisinzano, 2010; Vanderah, 2010). However, the clinical relevance of κ agonists is limited as central activation of κ opioid receptors produces dose-dependent dysphoria and some agonists such as salvinorin A produce hallucinations (Gonzalez et al., 2006). The endogenous κ ligand, dynorphin, can be released during stress and produce behavioural correlates of dysphoria, depression and anxiety, effects that have been linked to pro-addictive behaviours and drug relapse (for review, see Bruchas et al., 2010).
A recent study in mice examining dysphoria induced by κ opioid receptor activation has identified the signalling pathway through which this behaviour is regulated. Conditioned place aversion to the κ agonist U50,488 and stress-induced reinstatement of drug seeking (probably a result of dynorphin release) was mediated by the specific activation of P38 MAPK by κ opioid receptors in the dorsal raphe nucleus (Land et al., 2009). In addition, κ-opioid receptor activation of the P38 MAPK pathway in glia also appeared to be important for the development of hyperalgesia following peripheral neuropathy (Xu et al., 2007). κ Opioid receptors activate the P38 MAPK pathway through G-protein-independent signalling via GRK3 and β-arrestins (Bruchas et al., 2006). These results suggest that the development of κ ligands that only activate G-protein-dependent events may produce analgesia and circumvent the dysphoric effects. Although no such ligands presently exist, this is a clear example of how the characterization of the signalling pathways that mediate specific behaviours may ultimately be used to tailor drug development.
A prime illustration of functional selectivity is observed at the level of κ antagonists. Unlike antagonists for the µ and δ opioid receptors, certain κ opioid receptor antagonists (antagonist defined as ability to block κ agonist activity both in vitro and in vivo) have an extremely long duration of action. For example, a single injection of the κ selective antagonists, norbinaltorphimine (norBNI), guanidinonaltrindole or JDTic, maintains continual blockade of κ opioid receptors for up to 3 weeks (Horan et al., 1992; Carroll et al., 2004; Bruchas et al., 2007). This duration of action is in sharp contrast to the pan-opioid receptor antagonist naloxone, which only lasts for several hours. Recent work has shown that this long duration of action is mediated by activation of JNK. Surprisingly, norBNI and other long acting antagonists have been found to activate JNK through the κ opioid receptor, and treatment with these antagonists was found to increase phospho-JNK levels in the brain and spinal cord in wild-type mice but not κ opioid receptor knockout mice (Bruchas et al., 2007; Melief et al., 2011). In addition, the administration of the JNK inhibitor SP600125 blocked the long lasting antagonism induced by norBNI on the analgesic effects of the κ agonist U50, 488 (Bruchas et al., 2007). JNK1 in particular appears to mediate long-term antagonism, as norBNI and other long-lasting antagonists act as short duration competitive antagonists in JNK1 KO mice (Melief et al., 2010; 2011). Interestingly, this is a case where a functional antagonist is not just a classical antagonist in blocking agonist binding but a ‘collateral agonist’ for the JNK signalling cascade, the activation of which produces long duration inactivation of κ receptor signalling via a mechanism yet to be elucidated (Bruchas et al., 2007). Therapeutically, κ opioid receptor antagonists are being developed for the treatment of stress, anxiety and depression, and as an aid to curb drug relapse. Understanding the mechanisms regulating the unique kinetics of κ opioid receptor antagonists has important implications for future drug design.
Perspectives
It should be recognized that beyond ligand-directed signalling, there are multiple mechanisms that could readily mediate differential in vivo activities of drugs targeting opioid receptors. Factors such as intrinsic drug efficacy, pharmacodynamics, drug selectivity and ligand accessibility to selective receptor populations probably explain the differential effects of many drugs. In addition, the ability of a ligand to differentially activate splice variants of receptors or selectively activate homo- or heterodimeric receptor complexes are also possibilities that may masquerade as ligand-directed signalling (reviewed by Evans, 2004). Overall, biased agonism is one of the many mechanisms by which opioid ligands could produce diverse physiological effects.
The concept of biased agonism has profound implications, both in terms of understanding the complexity of GPCR pharmacology and for facilitating drug development (Bosier and Hermans, 2007; Galandrin et al., 2007). The very recent crystal structures of all three opioid receptors with ligands in place promises to visualize different receptor conformations as a result of different ligand interactions (Granier et al., 2012; Manglik et al., 2012; Wu et al., 2012). The ability of ligands to discretely activate particular signalling pathways, which may in turn regulate specific in vivo responses, opens the possibility of separating desirable from adverse drug effects. However, the evidence for this phenomenon is primarily based on in vitro experiments using recombinant cell systems, with only a few studies demonstrating clear agonist-selective activity in vivo. A major challenge in GPCR research will be to demonstrate the physiological relevance of agonist-biased signalling and regulation. There is clearly a need for further studies to bridge the gap between in vitro findings showing differential agonist signalling cascades and the in vivo behavioural consequence of signalling specificity. In the opioid receptor field, the concept of biased agonism in vivo is emerging, and the promise of biased µ agonists that are better analgesics with less abuse liability, or δ and κ agonists with targeted and sustained efficacy in the treatment of pain and mood disorders has now been validated in animal models. Future drug development will be required to determine the clinical relevance of these findings.
Acknowledgments
All authors were supported by NIH-NIDA Grant DA05010 and the Shirley and Stefan Hatos Research Foundation. AAP was additionally supported by NIH-NIDA grant K99DA031243. BK was also supported by the CNRS, INSERM, the Université de Strasbourg and the French ANR grant IMOP. All drug and molecular target (e.g. receptors, ion channels) nomenclature conforms to the British Journal of Pharmacology Guide to Receptors and Channels (Alexander et al., 2011).
Glossary
- ARM390
N,N-diethyl-4-(phenyl-piperidin-4-ylidenemethyl)-benzamide
- BRET
bioluminescence resonance energy transfer
- DAMGO
[D-Ala2, N-MePhe4, Gly-ol]-enkephalin
- DMR
dynamic mass redistribution
- DPDPE
[D-Pen2,5]enkephalin, [D-Pen2,D-Pen5]enkephalin
- ESCRT
endosomal sorting complex required for transport
- FRAP
fluorescence recovery after photobleaching
- GRKs
GPCR kinases
- JDTic
(3R)-7-hydroxy-N-((1S)-1-([(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethylpiperidin-1-yl]methyl)-2-methylpropyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxamide
- norBNI
norbinaltorphimine
- SNC80
(+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethyl benzamide
- U50
488, trans-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl]-benzeneacetamide
Conflict of interest
The authors declare no conflict of interest in the preparation of this manuscript.
References
- Aguila B, Coulbault L, Davis A, Marie N, Hasbi A, Le Bras F, et al. ssarrestin1-biased agonism at human delta-opioid receptor by peptidic and alkaloid ligands. Cell Signal. 2012;24:699–707. doi: 10.1016/j.cellsig.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. 2011;115:1363–1381. doi: 10.1097/ALN.0b013e318238bba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allouche S, Roussel M, Marie N, Jauzac P. Differential desensitization of human delta-opioid receptors by peptide and alkaloid agonists. Eur J Pharmacol. 1999;371:235–240. doi: 10.1016/s0014-2999(99)00180-6. [DOI] [PubMed] [Google Scholar]
- Arden JR, Segredo V, Wang Z, Lameh J, Sadee W. Phosphorylation and agonist-specific intracellular trafficking of an epitope-tagged mu-opioid receptor expressed in HEK 293 cells. J Neurochem. 1995;65:1636–1645. doi: 10.1046/j.1471-4159.1995.65041636.x. [DOI] [PubMed] [Google Scholar]
- Audet N, Gales C, Archer-Lahlou E, Vallieres M, Schiller PW, Bouvier M, et al. Bioluminescence resonance energy transfer assays reveal ligand-specific conformational changes within preformed signaling complexes containing delta-opioid receptors and heterotrimeric G proteins. J Biol Chem. 2008;283:15078–15088. doi: 10.1074/jbc.M707941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Oldfield S, Llorente J, Caunt CJ, Teschemacher AG, Roberts L, et al. Involvement of PKC alpha and G-protein-coupled receptor kinase 2 in agonist-selective desensitization of mu-opioid receptors in mature brain neurons. Br J Pharmacol. 2009;158:157–164. doi: 10.1111/j.1476-5381.2009.00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudry H, Proteau-Gagne A, Li S, Dory Y, Chavkin C, Gendron L. Differential noxious and motor tolerance of chronic delta opioid receptor agonists in rodents. Neuroscience. 2009;161:381–391. doi: 10.1016/j.neuroscience.2009.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Befort K, Tabbara L, Kling D, Maigret B, Kieffer BL. Role of aromatic transmembrane residues of the delta-opioid receptor in ligand recognition. J Biol Chem. 1996;271:10161–10168. doi: 10.1074/jbc.271.17.10161. [DOI] [PubMed] [Google Scholar]
- Berger AC, Whistler JL. How to design an opioid drug that causes reduced tolerance and dependence. Ann Neurol. 2010;67:559–569. doi: 10.1002/ana.22002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AC, Whistler JL. Morphine-induced mu opioid receptor trafficking enhances reward yet prevents compulsive drug use. EMBO Mol Med. 2011;3:385–397. doi: 10.1002/emmm.201100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsky EJ, Calderon SN, Wang T, Bernstein RN, Davis P, Hruby VJ, et al. SNC 80, a selective, nonpeptidic and systemically active opioid delta agonist. J Pharmacol Exp Ther. 1995;273:359–366. [PubMed] [Google Scholar]
- Bosier B, Hermans E. Versatility of GPCR recognition by drugs: from biological implications to therapeutic relevance. Trends Pharmacol Sci. 2007;28:438–446. doi: 10.1016/j.tips.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Bradbury FA, Zelnik JC, Traynor JR. G protein independent phosphorylation and internalization of the delta-opioid receptor. J Neurochem. 2009;109:1526–1535. doi: 10.1111/j.1471-4159.2009.06082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broom DC, Jutkiewicz EM, Folk JE, Traynor JR, Rice KC, Woods JH. Convulsant activity of a non-peptidic delta-opioid receptor agonist is not required for its antidepressant-like effects in Sprague-Dawley rats. Psychopharmacology (Berl) 2002a;164:42–48. doi: 10.1007/s00213-002-1179-y. [DOI] [PubMed] [Google Scholar]
- Broom DC, Nitsche JF, Pintar JE, Rice KC, Woods JH, Traynor JR. Comparison of receptor mechanisms and efficacy requirements for delta-agonist-induced convulsive activity and antinociception in mice. J Pharmacol Exp Ther. 2002b;303:723–729. doi: 10.1124/jpet.102.036525. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. Kappa opioid receptor activation of p38 MAPK is. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S, et al. Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem. 2007;282:29803–29811. doi: 10.1074/jbc.M705540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Hoffert C, O'Donnell D, Beaudet A. Up-regulation and trafficking of delta opioid receptor in a model of chronic inflammation: implications for pain control. Pain. 2003;101:199–208. doi: 10.1016/s0304-3959(02)00333-0. [DOI] [PubMed] [Google Scholar]
- Carroll I, Thomas JB, Dykstra LA, Granger AL, Allen RM, Howard JL, et al. Pharmacological properties of JDTic: a novel kappa-opioid receptor antagonist. Eur J Pharmacol. 2004;501:111–119. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
- Chen Y, Mestek A, Liu J, Hurley JA, Yu L. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol Pharmacol. 1993;44:8–12. [PubMed] [Google Scholar]
- Codd EE, Carson JR, Colburn RW, Stone DJ, Van Besien CR, Zhang SP, et al. JNJ-20788560 [9-(8-azabicyclo[3.2.1]oct-3-ylidene)-9H-xanthene-3-carboxylic acid diethylamide], a selective delta opioid receptor agonist, is a potent and efficacious antihyperalgesic agent that does not produce respiratory depression, pharmacologic tolerance, or physical dependence. J Pharmacol Exp Ther. 2009;329:241–251. doi: 10.1124/jpet.108.146969. [DOI] [PubMed] [Google Scholar]
- Contet C, Kieffer BL, Befort K. Mu opioid receptor: a gateway to drug addiction. Curr Opin Neurobiol. 2004;14:370–378. doi: 10.1016/j.conb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Decaillot FM, Befort K, Filliol D, Yue S, Walker P, Kieffer BL. Opioid receptor random mutagenesis reveals a mechanism for G protein-coupled receptor activation. Nat Struct Biol. 2003;10:629–636. doi: 10.1038/nsb950. [DOI] [PubMed] [Google Scholar]
- Evans CJ. Secrets of the opium poppy revealed. Neuropharmacology. 2004;47(Suppl 1):293–299. doi: 10.1016/j.neuropharm.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Evans CJ, Keith DE, Jr, Morrison H, Magendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, et al. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- Fraser GL, Gaudreau GA, Clarke PB, Menard DP, Perkins MN. Antihyperalgesic effects of delta opioid agonists in a rat model of chronic inflammation. Br J Pharmacol. 2000;129:1668–1672. doi: 10.1038/sj.bjp.0703248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galandrin S, Oligny-Longpre G, Bouvier M. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci. 2007;28:423–430. doi: 10.1016/j.tips.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Gallantine EL, Meert TF. A comparison of the antinociceptive and adverse effects of the mu-opioid agonist morphine and the delta-opioid agonist SNC80. Basic Clin Pharmacol Toxicol. 2005;97:39–51. doi: 10.1111/j.1742-7843.2005.pto_97107.x. [DOI] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Kieffer BL. Opioid receptor genes inactivated in mice: the highlights. Neuropeptides. 2002;36:62–71. doi: 10.1054/npep.2002.0900. [DOI] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Karchewski LA, Hever X, Matifas A, Kieffer BL. Inflammatory pain is enhanced in delta opioid receptor-knockout mice. Eur J Neurosci. 2008;27:2558–2567. doi: 10.1111/j.1460-9568.2008.06223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez D, Riba J, Bouso JC, Gomez-Jarabo G, Barbanoj MJ. Pattern of use and subjective effects of Salvia divinorum among recreational users. Drug Alcohol Depend. 2006;85:157–162. doi: 10.1016/j.drugalcdep.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, et al. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groer CE, Schmid CL, Jaeger AM, Bohn LM. Agonist-directed interactions with specific beta-arrestins determine mu-opioid receptor trafficking, ubiquitination, and dephosphorylation. J Biol Chem. 2011;286:31731–31741. doi: 10.1074/jbc.M111.248310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry AG, White IJ, Marsh M, von Zastrow M, Hislop JN. The role of ubiquitination in lysosomal trafficking of delta-opioid receptors. Traffic. 2011;12:170–184. doi: 10.1111/j.1600-0854.2010.01145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan P, Taylor J, Yamamura HI, Porreca F. Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharmacol Exp Ther. 1992;260:1237–1243. [PubMed] [Google Scholar]
- Hurley RW, Hammond DL. The analgesic effects of supraspinal mu and delta opioid receptor agonists are potentiated during persistent inflammation. J Neurosci. 2000;20:1249–1259. doi: 10.1523/JNEUROSCI.20-03-01249.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutkiewicz EM, Kaminsky ST, Rice KC, Traynor JR, Woods JH. Differential behavioral tolerance to the delta-opioid agonist SNC80 ([(+)-4-[(alphaR)-alpha-[(2S,5R)-2,5-dimethyl-4-(2-propenyl)-1-piperazinyl]-(3-methoxyphenyl)methyl]-N,N-diethylbenzamide) in Sprague-Dawley rats. J Pharmacol Exp Ther. 2005;315:414–422. doi: 10.1124/jpet.105.088831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane BE, Svensson B, Ferguson DM. Molecular recognition of opioid receptor ligands. AAPS J. 2006;8:E126–E137. doi: 10.1208/aapsj080115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, et al. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- Kelly E, Bailey CP, Henderson G. Agonist-selective mechanisms of GPCR desensitization. Br J Pharmacol. 2008;153(Suppl 1):S379–S388. doi: 10.1038/sj.bjp.0707604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Principles: receptor theory in pharmacology. Trends Pharmacol Sci. 2004;25:186–192. doi: 10.1016/j.tips.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Evans CJ. Opioid tolerance-in search of the holy grail. Cell. 2002;108:587–590. doi: 10.1016/s0092-8674(02)00666-9. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Befort K, Gaveriaux-Ruff C, Hirth CG. The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc Natl Acad Sci USA. 1992;89:12048–12052. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Bartlett S, He L, Nielsen CK, Chang AM, Kharazia V, et al. Morphine-induced receptor endocytosis in a novel knockin mouse reduces tolerance and dependence. Curr Biol. 2008;18:129–135. doi: 10.1016/j.cub.2007.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivell B, Prisinzano TE. Kappa opioids and the modulation of pain. Psychopharmacology (Berl) 2010;210:109–119. doi: 10.1007/s00213-010-1819-6. [DOI] [PubMed] [Google Scholar]
- Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg LO, Wundrack N, et al. Receptor endocytosis counteracts the development of opioid tolerance. Mol Pharmacol. 2005;67:280–287. doi: 10.1124/mol.104.004994. [DOI] [PubMed] [Google Scholar]
- Konig M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ, et al. Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature. 1996;383:535–538. doi: 10.1038/383535a0. [DOI] [PubMed] [Google Scholar]
- Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- Lamb K, Tidgewell K, Simpson DS, Bohn LM, Prisinzano TE. Antinociceptive effects of herkinorin, a MOP receptor agonist derived from salvinorin A in the formalin test in rats: new concepts in mu opioid receptor pharmacology: from a symposium on new concepts in mu-opioid pharmacology. Drug Alcohol Depend. 2012;121:181–188. doi: 10.1016/j.drugalcdep.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, et al. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A. 2009;106:19168–19173. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoq I, Marie N, Jauzac P, Allouche S. Different regulation of human delta-opioid receptors by SNC-80 [(+)-4-[(alphaR)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenz yl]-N,N-diethylbenzamide] and endogenous enkephalins. J Pharmacol Exp Ther. 2004;310:666–677. doi: 10.1124/jpet.103.063958. [DOI] [PubMed] [Google Scholar]
- Ma P, Zemmel R. Value of novelty? Nat Rev Drug Discov. 2002;1:571–572. doi: 10.1038/nrd884. [DOI] [PubMed] [Google Scholar]
- Mace G, Miaczynska M, Zerial M, Nebreda AR. Phosphorylation of EEA1 by p38 MAP kinase regulates mu opioid receptor endocytosis. EMBO J. 2005;24:3235–3246. doi: 10.1038/sj.emboj.7600799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes AC, Dunn H, Ferguson SS. Regulation of g protein-coupled receptor activity, trafficking and localization by gpcr-interacting proteins. Br J Pharmacol. 2012;165:1717–1736. doi: 10.1111/j.1476-5381.2011.01552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, et al. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie N, Landemore G, Debout C, Jauzac P, Allouche S. Pharmacological characterization of AR-M1000390 at human delta opioid receptors. Life Sci. 2003a;73:1691–1704. doi: 10.1016/s0024-3205(03)00489-2. [DOI] [PubMed] [Google Scholar]
- Marie N, Lecoq I, Jauzac P, Allouche S. Differential sorting of human delta-opioid receptors after internalization by peptide and alkaloid agonists. J Biol Chem. 2003b;278:22795–22804. doi: 10.1074/jbc.M300084200. [DOI] [PubMed] [Google Scholar]
- Marie N, Aguila B, Hasbi A, Davis A, Jauzac P, Allouche S. Different kinases desensitize the human delta-opioid receptor (hDOP-R) in the neuroblastoma cell line SK-N-BE upon peptidic and alkaloid agonists. Cell Signal. 2008;20:1209–1220. doi: 10.1016/j.cellsig.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Melief EJ, Miyatake M, Bruchas MR, Chavkin C. Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc Natl Acad Sci U S A. 2010;107:11608–11613. doi: 10.1073/pnas.1000751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melief EJ, Miyatake M, Carroll FI, Beguin C, Carlezon WA, Jr, Cohen BM, et al. Duration of action of a broad range of selective kappa-opioid receptor antagonists is positively correlated with c-Jun N-terminal kinase-1 activation. Mol Pharmacol. 2011;80:920–929. doi: 10.1124/mol.111.074195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger TG, Ferguson DM. On the role of extracellular loops of opioid receptors in conferring ligand selectivity. FEBS Lett. 1995;375:1–4. doi: 10.1016/0014-5793(95)01185-h. [DOI] [PubMed] [Google Scholar]
- Minami M, Toya T, Katao Y, Maekawa K, Nakamura S, Onogi T, et al. Cloning and expression of a cDNA for the rat kappa-opioid receptor. FEBS Lett. 1993;329:291–295. doi: 10.1016/0014-5793(93)80240-u. [DOI] [PubMed] [Google Scholar]
- Mittal N, Tan M, Egbuta O, Desai N, Crawford C, Xie CW, et al. Evidence that behavioral phenotypes of morphine in β-arr2-/- mice are due to the unmasking of JNK signalling. J Neuropsychop. 2012;37:1953–1962. doi: 10.1038/npp.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari P, Vezzi V, Sbraccia M, Gro C, Riitano D, Ambrosio C, et al. Morphine-like opiates selectively antagonize receptor-arrestin interactions. J Biol Chem. 2010;285:12522–12535. doi: 10.1074/jbc.M109.059410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse M, Tran E, Sun H, Levenson R, Fang Y. Ligand-directed functional selectivity at the mu opioid receptor revealed by label-free integrative pharmacology on-target. Plos ONE. 2011;6:e25643. doi: 10.1371/journal.pone.0025643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal X, Banos JE, Kieffer BL, Maldonado R. Neuropathic pain is enhanced in delta-opioid receptor knockout mice. Eur J Neurosci. 2006;23:830–834. doi: 10.1111/j.1460-9568.2006.04569.x. [DOI] [PubMed] [Google Scholar]
- Paterlini MG. The function of the extracellular regions in opioid receptor binding: insights from computational biology. Curr Top Med Chem. 2005;5:357–367. doi: 10.2174/1568026053544579. [DOI] [PubMed] [Google Scholar]
- Perrine SA, Hoshaw BA, Unterwald EM. Delta opioid receptor ligands modulate anxiety-like behaviors in the rat. Br J Pharmacol. 2006;147:864–872. doi: 10.1038/sj.bjp.0706686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pert CB, Snyder SH. Opiate receptor: demonstration in nervous tissue. Science. 1973;179:1011–1014. doi: 10.1126/science.179.4077.1011. [DOI] [PubMed] [Google Scholar]
- Petrillo P, Angelici O, Bingham S, Ficalora G, Garnier M, Zaratin PF, et al. Evidence for a selective role of the delta-opioid agonist [8R-(4bS*,8aalpha,8abeta, 12bbeta)]7,10-Dimethyl-1-methoxy-11-(2-methylpropyl)oxycarbonyl 5,6,7,8,12,12b-hexahydro-(9H)-4,8-methanobenzofuro[3,2-e]pyrrolo[2,3-g]iso quinoline hydrochloride (SB-235863) in blocking hyperalgesia associated with inflammatory and neuropathic pain responses. J Pharmacol Exp Ther. 2003;307:1079–1089. doi: 10.1124/jpet.103.055590. [DOI] [PubMed] [Google Scholar]
- Poole DP, Pelayo JC, Scherrer G, Evans CJ, Kieffer BL, Bunnett NW. Localization and regulation of fluorescently labeled delta opioid receptor, expressed in enteric neurons of mice. Gastroenterology. 2011;141:982–991. doi: 10.1053/j.gastro.2011.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Becker JA, Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, et al. In vivo delta opioid receptor internalization controls behavioral effects of agonists. Plos ONE. 2009;4:e5425. doi: 10.1371/journal.pone.0005425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Walwyn W, Nozaki C, Filliol D, Erbs E, Matifas A, et al. Ligand-directed trafficking of the delta-opioid receptor in vivo: two paths toward analgesic tolerance. J Neurosci. 2010;30:16459–16468. doi: 10.1523/JNEUROSCI.3748-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Befort K, Nozaki C, Gaveriaux-Ruff C, Kieffer BL. The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci. 2011;32:581–590. doi: 10.1016/j.tips.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh A, Kimura Y, Suzuki T, Kawai K, Nagase H, Kamei J. Potential anxiolytic and antidepressant-like activities of SNC80, a selective delta-opioid agonist, in behavioral models in rodents. J Pharmacol Sci. 2004;95:374–380. doi: 10.1254/jphs.fpj04014x. [DOI] [PubMed] [Google Scholar]
- Sauliere-Nzeh NA, Millot C, Corbani M, Mazeres S, Lopez A, Salome L. Agonist-selective dynamic compartmentalization of human Mu opioid receptor as revealed by resolutive FRAP analysis. J Biol Chem. 2010;285:14514–14520. doi: 10.1074/jbc.M109.076695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauriyal DS, Jaggi AS, Singh N. Extending pharmacological spectrum of opioids beyond analgesia: multifunctional aspects in different pathophysiological states. Neuropeptides. 2011;45:175–188. doi: 10.1016/j.npep.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, Laustriat D, Cao YQ, et al. Knockin mice expressing fluorescent delta-opioid receptors uncover G protein-coupled receptor dynamics in vivo. Proc Natl Acad Sci USA. 2006;103:9691–9696. doi: 10.1073/pnas.0603359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O'Donnell D, et al. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell. 2009;137:1148–1159. doi: 10.1016/j.cell.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JM. The White Poppy – a History of Opium. New York: Funk & Wagnalls; 1969. [Google Scholar]
- Simon EJ, Hiller JM, Edelman I. Stereospecific binding of the potent narcotic analgesic (3H) Etorphine to rat-brain homogenate. Proc Natl Acad Sci USA. 1973;70:1947–1949. doi: 10.1073/pnas.70.7.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol. 2009;10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson GW, Folk JE, Rice KC, Negus SS. Interactions between delta and mu opioid agonists in assays of schedule-controlled responding, thermal nociception, drug self-administration, and drug versus food choice in rhesus monkeys: studies with SNC80 [(+)-4-[(alphaR)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenz yl]-N,N-diethylbenzamide] and heroin. J Pharmacol Exp Ther. 2005;314:221–231. doi: 10.1124/jpet.104.082685. [DOI] [PubMed] [Google Scholar]
- Takita K, Herlenius EA, Lindahl SG, Yamamoto Y. Actions of opioids on respiratory activity via activation of brainstem mu-, delta- and kappa-receptors; an in vitro study. Brain Res. 1997;778:233–241. doi: 10.1016/s0006-8993(97)01105-0. [DOI] [PubMed] [Google Scholar]
- Tan M, Walwyn WM, Evans CJ, Xie CW. p38 MAPK and beta-arrestin 2 mediate functional interactions between endogenous micro-opioid and alpha2A-adrenergic receptors in neurons. J Biol Chem. 2009;284:6270–6281. doi: 10.1074/jbc.M806742200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terenius L. Characteristics of the ‘receptor’ for narcotic analgesics in synaptic plasma membrane fraction from rat brain. Acta Pharmacol Toxicol (Copenh) 1973;33:377–384. doi: 10.1111/j.1600-0773.1973.tb01539.x. [DOI] [PubMed] [Google Scholar]
- Terman GW, Jin W, Cheong YP, Lowe J, Caron MG, Lefkowitz RJ, et al. G-protein receptor kinase 3 (GRK3) influences opioid analgesic tolerance but not opioid withdrawal. Br J Pharmacol. 2004;141:55–64. doi: 10.1038/sj.bjp.0705595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidgewell K, Groer CE, Harding WW, Lozama A, Schmidt M, Marquam A, et al. Herkinorin analogues with differential beta-arrestin-2 interactions. J Med Chem. 2008;51:2421–2431. doi: 10.1021/jm701162g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidehi N, Kenakin T. The role of conformational ensembles of seven transmembrane receptors in functional selectivity. Curr Opin Pharmacol. 2010;10:775–781. doi: 10.1016/j.coph.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Vanderah TW. Delta and kappa opioid receptors as suitable drug targets for pain. Clin J Pain. 2010;26(Suppl 10):S10–S15. doi: 10.1097/AJP.0b013e3181c49e3a. [DOI] [PubMed] [Google Scholar]
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Bruchas MR, Ippolito DL, Gendron L, Chavkin C. Sciatic nerve ligation-induced proliferation of spinal cord astrocytes is mediated by kappa opioid activation of p38 mitogen-activated protein kinase. J Neurosci. 2007;27:2570–2581. doi: 10.1523/JNEUROSCI.3728-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Loh HH, Law PY. Beta-arrestin-dependent mu-opioid receptor-activated extracellular signal-regulated kinases (ERKs) Translocate to Nucleus in Contrast to G protein-dependent ERK activation. Mol Pharmacol. 2008;73:178–190. doi: 10.1124/mol.107.039842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Chu J, Zhang Y, Loh HH, Law PY. Modulating micro-opioid receptor phosphorylation switches agonist-dependent signaling as reflected in PKCepsilon activation and dendritic spine stability. J Biol Chem. 2011;286:12724–12733. doi: 10.1074/jbc.M110.177089. [DOI] [PMC free article] [PubMed] [Google Scholar]