

Abstract
Spleen tyrosine kinase (SYK) is the best known for its involvement in immune receptor signalling, mediated by binding of SYK tandem Src-homology 2 domains to tandem phosphotyrosine in immunoreceptor tyrosine-based activation motifs (ITAMs). ITAM adaptors or ITAM-containing receptor tails mediate signalling from B- and T-cell receptors, Fc receptors and many C-type lectins, including dectin-1. Recent data point to constitutive binding of SYK to the cytoplasmic domain of toll-like receptor-4 (TLR4). This SYK-TLR4 binding increases upon TLR4 dimerization and phosphorylation, and SYK plays a prominent role in TLR4 signalling in response to LPS in neutrophils and monocytes. SYK also plays an important role in TLR4-mediated macrophage responses to minimally oxidized low-density lipoprotein (mmLDL), which is a form of oxidized LDL relevant to development of human atherosclerosis. Interestingly, mmLDL-induced effects in macrophages, which occur via TLR4, are predominantly MyD88 independent. This unmasks the role of the SYK branch of TLR4 signalling, which mediates modest cytokine release via activation of AP-1 transcription and robust reactive oxygen species generation and cytoskeletal rearrangements. The latter results in extensive membrane ruffling and macropinocytosis, leading to lipoprotein uptake and foam cell formation, a hallmark of atherosclerotic lesions. Because inhibitors of SYK activity, such as fostamatinib, are in advanced clinical trials for rheumatoid arthritis and other autoimmune diseases, understanding the role of SYK in signalling via TLR4 is of immediate importance. This signalling pathway seems to be particularly important in TLR4 activation by host-derived, damage-associated molecular pattern ligands, such as mmLDL, relevant to development of atherosclerosis and other chronic inflammatory diseases.
Keywords: spleen tyrosine kinase, toll-like receptor-4, macrophage, atherosclerosis, oxidized low-density lipoprotein, inflammation, innate immunity, signalling, reactive oxygen species, lipid uptake
Introduction
The Nobel Prize of 2011 in Physiology or Medicine was awarded to Jules Hoffmann and Bruce Beutler for their discovery of the role of toll-like receptors (TLRs) in innate immunity and to Ralph Steinman for his discovery of dendritic cells and their role in adaptive immunity. Hoffmann discovered that Toll helped Drosophila fend off fungal infection (Lemaitre et al., 1996), and Beutler found that toll-like receptor-4 (TLR4) was the signalling receptor that recognized bacterial LPS and initiated strong antibacterial responses in mammals (Poltorak et al., 1998). TLR4 belongs to the eclectic family of pattern-recognition receptors, the receptors of innate immunity that recognize structurally common pattern motifs on microbial products. Indeed, TLR4 is activated by many different LPS molecules expressed on various Gram-negative bacteria.
To add to the diversity of ligands that activate TLR4, many host-derived molecules, ranging from lipids to proteins, exert cellular responses via TLR4. The common feature of host-derived TLR4 ligands is that they are modified, but not native, host molecules, often associated with the tissue damage or a pathologic process. The modification could be due to oxidation, glycosylation, methylation, cleavage or other post-translational (proteins) or post-synthetic (lipids) alterations. To list just a few of the host-derived ligands, TLR4 recognizes tenascin-C, an extracellular matrix glycoprotein specifically expressed in inflamed rheumatoid joints (Midwood et al., 2009); serum amyloid A3 secreted by pre-metastatic tumours (Hiratsuka et al., 2008); and minimally oxidized low-density lipoprotein (mmLDL), but not native LDL (Miller et al., 2003b).
The downstream signalling from TLR4 is no less complex than the nature of ligands that initiate it. For the reasons not completely understood, different stimuli induce TLR4 signalling via recruitment of different adaptor/signalling molecules to the cytoplasmic domain of the receptor, thus resulting in different cellular responses. This may be due to the structure of a TLR4 agonist (Bowen et al., 2012). In addition, the cell surface or endosomal localization of TLR4 drives recruitment of MyD88 or toll/interleukin-1 receptor (TIR) domain-containing adapter inducing interferon-β (TRIF) respectively (Kenny and O'Neill, 2008). Another possible explanation is that TLR4 is never a stand-alone receptor responding to bacterial or endogenous challenge, but rather part of a receptor cluster – each component of the cluster influences the ultimate signalling pattern (Schmitz and Orso, 2002; Stewart et al., 2010; Triantafilou and Triantafilou, 2010). For example, TLR4 responds to mmLDL (Miller et al., 2009), a lipoprotein composed of a 512 kDa protein and hundreds of lipid and sterol molecules, many of which are modified; thus, in addition to TLR4, mmLDL likely engages several other receptors leading to their clustering with TLR4.
The next level of regulation involves multiple non-receptor protein and lipid kinases (Lee and Kim, 2007). This article will review data on spleen tyrosine kinase (SYK), a signalling kinase directly transducing TLR4 signals, and will highlight mmLDL and its component oxidized cholesterol ester as host-derived TLR4 agonists, which induce primarily SYK-mediated responses.
Classic model of TLR4 signalling
TLR4 is the signalling receptor that mediates a robust inflammatory response to LPS, but it requires several co-receptors for the optimal LPS presentation as well as adaptor molecules for the signal transduction (Kawai and Akira, 2007). In blood, LPS circulates as an oligomer, carried by LPS-binding protein (LBP). It is then transferred in a monomeric form to CD14, the LPS receptor present in both soluble and membrane-bound forms. The membrane-bound CD14 is anchored to the plasma membrane via glycosylphosphatidylinositol and it lacks an intracellular signalling domain. Thus, CD14 further presents a LPS monomer to myeloid differentiation-2 (MD-2). Like CD14, MD-2 has neither transmembrane nor signalling domain; the difference is that MD-2 is constitutively bound to TLR4. In fact, the TLR4/MD-2 complex assembles in the endoplasmic reticulum (Shibata et al., 2011) and MD-2 is required for TLR4 trafficking to the plasma membrane. MD-2 knockout mice do not express TLR4 on the cell surface (Nagai et al., 2002).
According to a crystal structure model (Park et al., 2009), the hydrophobic pocket of MD-2 accommodates five of the six acyl chains of LPS lipid A, while the sixth acyl chain and the oligosaccharide Kdo2 of LPS provide an interface for binding with a second TLR4* from a different TLR4*/MD-2* pair. Accordingly, a second molecule of LPS bound to MD-2* connects it with TLR4 of the first TLR4/MD-2 pair. These interactions result in dimerization of two TLR4/MD-2 pairs on the extracellular surface of the plasma membrane. Intracellular TIR domains of TLR4 also dimerize, change conformation, and these events induce binding of adaptor molecules to TLR4 and thereby initiate signalling cascades.
If the TIR dimerization event occurs at the plasma membrane, it leads to recruitment of Mal, followed by MyD88 (McGettrick and O'Neill, 2010). Downstream from MyD88, there are several more levels of signalling intermediates, ultimately leading to activation of two sets of kinases, IKK and MAPK. Activation of IKK results in translocation of NF-κB transcription factors to the nucleus and initiation of the transcriptional program leading to expression of inflammatory cytokines and other targets. The MAPKs ERK, JNK and p38 activate the AP-1 transcription program as well as mediate cytoskeletal reorganization and regulate other cellular processes. The detailed description of the signalling intermediates in the TLR4/MyD88 pathway can be found in recent reviews (Newton and Dixit, 2012). In the endocytic compartment, following internalization of the LPS-TLR4/MD-2 complex, the TLR4 dimer recruits TRIF-related adaptor molecule (TRAM) and TRIF (McGettrick and O'Neill, 2010). The preference for TRAM and TRIF over Mal and MyD88 may be due to a different mode of TLR4 dimerization, which occurs upon endosome acidification and protonation of key histidines involved in TLR4 dimerization (Gangloff, 2012). Downstream from TRIF, two transcriptional events occur: delayed (compared with MyD88-mediated) activation of NF-κB and initiation of the IRF3 transcription program, the latter resulting in expression of type I interferons.
Spleen tyrosine kinase
SYK is an ubiquitous non-receptor tyrosine kinase that has diverse biological functions in various cell types. SYK is a 72 kDa protein that consists of two tandem Src-homology 2 (SH2) domains and a C-terminal tyrosine kinase domain. SYK is expressed in haematopoietic, endothelial, neuronal and epithelial cells and in fibroblasts (Zarbock and Ley, 2011). The total SYK knockout mice die perinatally because of a failure of the blood and lymph vasculature to separate, but mice with myeloid-specific SYK knockdown (Sykfl/fl LysM-Cre) are viable and fertile (Choi et al., 2012).
SYK plays a central role in immunoreceptor signalling. The classic concept of the B-cell receptor (BCR) and T-cell receptor (TCR) function requires recruitment of signalling proteins with immunoreceptor tyrosine-based activation motifs (ITAMs). Phosphorylation of tandem tyrosines in an ITAM by Src-family kinases leads to the ITAM binding with SYK via SYK's tandem SH2 domains, with subsequent SYK activation. Activated SYK directly binds to Vav, PLCγ, PI3K and SLP76/SLP65, which in turn engage a plethora of signalling intermediates, including MAPKs, Rho family GTPases, components of NLRP3 inflammasome and transcription factors. Resulting cellular responses range from cytoskeletal reorganization and reactive oxygen species (ROS) production to cell differentiation, proliferation and survival (Mocsai et al., 2010; Lowell, 2011).
In addition to ITAM adaptors to BCR, TCR and a number of other immune receptors, SYK also binds to ITAM in receptor tails, for example, in FcγRIIA, and to half-ITAM motifs in dimerized C-type lectins (Kerrigan and Brown, 2011). The role of SYK in function of CLEC7a, also known as dectin-1, received a special attention due to the dectin-1 importance in anti-fungal and anti-mycobacterium immune responses. Binding of β-glucan carbohydrates to dectin-1 leads to SYK recruitment, which is PKCδ dependent (Elsori et al., 2011), and activation of MAPKs, NF-κB and nuclear factor of activated T cells (NFAT), as well as the NALP3 inflammasome assembly and IL-1β maturation (Kerrigan and Brown, 2011). Downstream from dectin-1, PKCδ and SYK stimulate ROS generation and phagocytosis of zymosan (a mimic of fungal pathogens) by human monocytes (Elsori et al., 2011).
SYK also directly binds to β-integrins, but this interaction is independent of ITAM adaptors and, in fact, is a constitutive binding via a site distinct from the site of phosphotyrosine binding (Woodside et al., 2002; Jakus et al., 2007). This interaction is proposed to coordinate integrin signalling cascades with co-opted ITAM-containing transmembrane adaptors (Mocsai et al., 2010).
SYK recruitment to TLR4
Recruitment of SYK to a TLR4-associated receptor complex has been reported in several studies. Experiments in which TLR4 and/or SYK specific antibodies were used to pull down the receptor complex from cell lysates have demonstrated that TLR4 and SYK co-immunoprecipitate in neutrophils, monocytes and macrophages, even in the absence of LPS or any other stimuli (Arndt et al., 2004; Chaudhary et al., 2007; Bae et al., 2009). Using a yeast-two-hybrid assay, we have shown that there is a physical interaction between SYK and the TIR domain of TLR4 and that the N-terminal SH2 domain of SYK is responsible for the interaction (Choi et al., 2009). Because SYK binding to TLR4 occurs in non-stimulated cells and in the yeast-two-hybrid system, this interaction seems to be constitutive and independent of TLR4 tyrosine phosphorylation. The phosphotyrosine-independent, constitutive binding of SYK to TLR4 resembles the SYK binding to β-integrins, both interactions are mediated by the N-terminal SH2 domain of SYK (Woodside et al., 2002; Choi et al., 2009).
Upon neutrophil and monocyte stimulation with LPS and macrophage stimulation with mmLDL, SYK binding to TLR4 increases, and this coincides with TLR4 as well as SYK phosphorylation (Arndt et al., 2004; Chaudhary et al., 2007; Bae et al., 2009; Choi et al., 2012; Lu et al., 2012). Both TLR4 and SYK are phosphorylated by Lyn in neutrophils and macrophages (Medvedev et al., 2007; Tiemi Shio et al., 2009; Lu et al., 2012). Pharmacologic inhibition of SYK prevented LPS-induced TLR4 phosphorylation (Chaudhary et al., 2007), suggesting that SYK may contribute to tyrosine phosphorylation of TIR domain in TLR4. However, it is unclear whether the increased TLR4/SYK association in stimulated cells is mediated by the SH2–phosphotyrosine interactions or via a different mechanism.
Role of SYK in the TLR4 signalling in response to bacterial pathogens
SYK has been implicated in LPS-stimulated, CD14-dependent delivery of TLR4 to endosomes and the TRIF pathway activation in macrophages and dendritic cells (Zanoni et al., 2011). The authors did not investigate whether SYK directly binds to TLR4. An interesting distinction between macrophages and neutrophils is that LPS induces a more robust IL-1β secretion by neutrophils than by macrophages, although both cell types express pro-IL-1β in response to LPS (Rowe et al., 2002; Sutterwala et al., 2006). The inflammasome activation of caspase-1 and subsequent processing of pro-IL-1β into mature IL-1β requires a second signal (e.g. ATP) in macrophages but not in neutrophils. A recent paper suggests that in neutrophils LPS-induced recruitment of SYK to TLR4 mediates the inflammasome assembly and caspase-1 activation, explaining why the LPS signal alone is sufficient to stimulate IL-1β secretion by neutrophils (Lu et al., 2012). Importantly, SYK-dependent generation of ROS is involved in the process. The authors also detail a turn-off mechanism in which the neutrophil adhesion molecule CEACAM1 (CD66a) recruits and activates the phosphatase SHP1, which dephosphorylates SYK, leading to the reduced inflammasome activity. Another prominent role of SYK in LPS-stimulated neutrophils is activation of JNK. The TLR4/SYK/JNK pathway regulates MCP-1 and TNFα but not IL-8 expression in human neutrophils (Arndt et al., 2004).
The involvement of SYK in TLR4 signalling is different in different human monocyte subpopulations. In humans, CD14highCD16-, CD14+CD16+ and CD14dimCD16high monocytes have been described, although some authors combine the last two CD16+ populations into one (Ley et al., 2011). Human CD14highCD16- monocytes express CCR2, L-selectin, and the Fc receptor CD64, whereas CD14+CD16+ monocytes lack CCR2 but express CD32 and higher levels of MHC-II. The CD16+ monocyte subpopulations expand in patients with sepsis and in patients with the high risk of cardiovascular disease (Heine et al., 2012; Shalova et al., 2012). In CD16+ monocytes, CD16 has been reported to regulate the TRIF-dependent TLR4 pathway by activating SYK (Shalova et al., 2012). As a result, CD14+CD16+ monocytes express IFNβ, CCL5 and CXCL10. Interestingly, the authors found that CD16 induced the expression of negative regulators of the MyD88-dependent pathway, IRAKM and IL-1RA, resulting in down-regulation of IL-6, CXCL1 and CCL3 expression. These findings corroborate the earlier reports that in macrophages the integrin CD11b and the adaptor molecule DAP12 induce SYK-dependent phosphorylation and degradation of MyD88 (Hamerman et al., 2005; Han et al., 2010). These mechanisms may explain the dissociation between SYK- and MyD88-mediated TLR4 responses, as it will be described below in the case of mmLDL-induced macrophage activation.
mmLDL, 12/15-lipoxygenase and TLR4 in atherosclerosis
Our interest in TLR4/SYK signalling stems from the finding that mmLDL induces production of ROS and robust cytoskeletal rearrangements in macrophages via a TLR4-dependent mechanism (Miller et al., 2003b; Bae et al., 2009). mmLDL is a form of oxidized LDL that is highly relevant to the pathogenesis of atherosclerosis. It is oxidized by 12/15-lipoxygenase (12/15LO), the enzyme expressed in inflamed endothelial cells, in vascular smooth muscle cells and in macrophages in atherosclerotic lesions (Ylä-Herttuala et al., 1990; 1991; Ezaki et al., 1995; Patricia et al., 2001; Miller et al., 2003b; Natarajan and Nadler, 2003). Mouse model studies have convincingly demonstrated that 12/15LO (Alox15) oxidizes LDL in vivo and that Alox15-/- mice on the Ldlr-/- or Apoe-/- background fed with a high-fat diet develop less atherosclerosis than Alox15 wild-type mice (Cyrus et al., 1999; 2001; George et al., 2001; Huo et al., 2004; Zhao et al., 2005; Poeckel et al., 2009; Rong et al., 2012). Cholesterol ester hydroperoxides, produced by 12/15LO in LDL, are responsible for the majority of TLR4-dependent effects of mmLDL (Harkewicz et al., 2008; Choi et al., 2009; Miller et al., 2009). The oxidized cholesterol esters found in mmLDL have also been identified in mouse and human atherosclerotic lesions, in human plasma and even in cholesterol-fed zebrafish (Upston et al., 2002; Leitinger, 2003; Harkewicz et al., 2008; Fang et al., 2010; Hutchins et al., 2011).
Human and animal studies suggest that TLR4, the receptor for mmLDL, is proatherogenic. TLR4 is strongly expressed in human and mouse atherosclerotic lesions (Xu et al., 2001; Edfeldt et al., 2002). A deficiency in TLR4 or MyD88 attenuates the development of atherosclerosis in hyperlipidemic Apoe-/- mice, likely owing to the reduction in macrophage recruitment to atherosclerotic lesions (Bjorkbacka et al., 2004; Michelsen et al., 2004). Transplantation of bone marrow from Tlr4-/- mice into Ldlr-/- recipients, followed by feeding a high-cholesterol diet, results in reduced atherosclerosis compared with mice transplanted with wild-type bone marrow (Coenen et al., 2009), although similar bone marrow transplantation studies in which angiotensin II induced aortic aneurysm found no role for TLR4 (Owens et al., 2011). Lipid accumulation and foam cell formation in early lesions of Tlr4-/-Apoe-/- mice are reduced by 70–80% compared with Apoe-/- controls (Higashimori et al., 2011). These results strongly imply that one or more endogenous ligands are activating TLR4, leading to proatherogenic effects.
There is evidence for a role of TLR4 in the development of human atherosclerosis as well. Kiechl et al. first reported that the common D299G TLR4 loss-of-function polymorphism was associated with a decreased risk of carotid and femoral artery atherosclerosis and cardiovascular cause of death (Kiechl et al., 2002). In other studies, the D299G TLR4 polymorphism was also associated with a reduced risk of acute coronary events, independent of other coronary risk factors (Ameziane et al., 2003; Boekholdt et al., 2003). However, the D299G polymorphism was not associated with coronary artery stenosis, cerebral ischaemia or progression of atherosclerosis in patients with familial hypercholesterolemia (Yang et al., 2003; Netea et al., 2004; Reismann et al., 2004). Because these studies evaluated different clinical manifestations of atherosclerosis, the observed discrepancies are not necessarily contradictory. Other studies showing increased TLR4 expression in macrophages in symptomatic carotid atherosclerotic plaques (Katsargyris et al., 2011) and increased TLR4 expression in circulating monocytes of patients with coronary atherosclerosis (Geng et al., 2006) and patients with acute coronary syndrome compared with stable angina (Methe et al., 2005; Xie et al., 2010) also suggest that TLR4 is involved in vascular inflammation in humans.
Role of TLR4 and SYK in macrophage responses to mmLDL
The robust and rapid ROS generation in macrophages induced by mmLDL is clearly TLR4 dependent (Bae et al., 2009), and mmLDL-induced TLR4 dimerization is evident from the immunoprecipitation studies with the cells expressing Flag-TLR4 and GFP-TLR4 (Choi et al., 2012). However, the mmLDL-induced ROS generation was absolutely MyD88 independent (Bae et al., 2009). Searching for the kinase, meditating the mmLDL/TLR4 effect, we have found that inhibition of SYK with pharmacologic inhibitors and the SYK knockdown with shRNA abrogate ROS production. Because of our finding of direct SYK binding to the intracellular domain of TLR4 (Choi et al., 2009) and many reports of SYK co-immunoprecipitation with TLR4 (Arndt et al., 2004; Chaudhary et al., 2007; Bae et al., 2009), we suggest that SYK should be considered as a TLR4 adaptor molecule (refer to Figure 1). The difference from the other TLR4 adaptors, Mal and TRAM, is that SYK possesses a kinase domain. Thus, it directly phosphorylates, among other targets, PLCγ and Vav1 (Mocsai et al., 2010). Indeed, mmLDL induces phosphorylation of PLCγ and Vav1 in macrophages. In turn, PLCγ activates PKC, which contributes to activation of the NADPH oxidase Nox2 (Bae et al., 2009). In addition, mmLDL activation of Vav1 initiates a Ras-Raf-MEK1-ERK1/2 signalling cascade, resulting in GTP binding to Rac (Miller et al., 2005; Choi et al., 2009). Binding of GTP-Rac completes the assembly of Nox2-activating proteins and allows for ROS generation.
Figure 1.
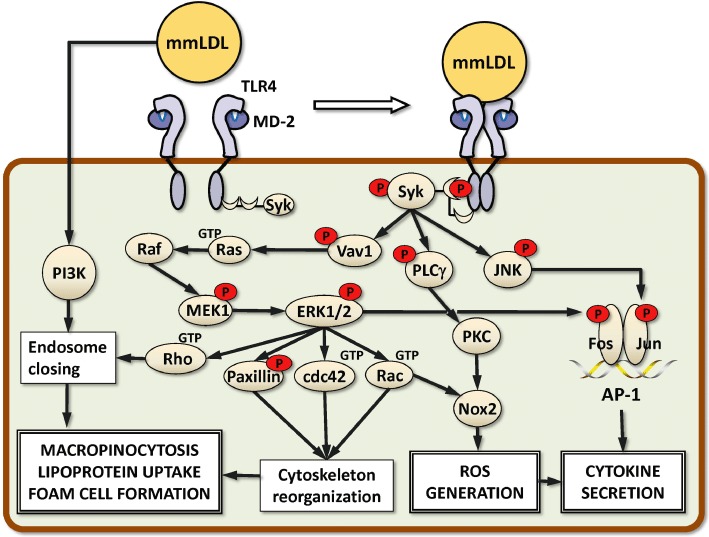
Signalling mechanisms of TLR4/SYK-dependent macrophage activation. SYK is constitutively bound to the cytoplasmic domain of TLR4. Macrophage activation with mmLDL results in TLR4 dimerization and phosphorylation and increased SYK binding to TLR4 (note: SYK SH2 binding to phosphotyrosine in TLR4 is hypothetical; there are yet no specific data to support the SH2-type binding). Phosphorylated (activated) SYK activates Vav1, PLCγ and JNK. In turn, Vav1 activates a Ras-Raf-MEK1-ERK1/2 signalling cascade. ERK1/2-dependent phosphorylation of paxillin and GTP binding (activation) to Rac and Cdc42 lead to robust cytoskeletal rearrangements in macrophages, in part resulting in extensive membrane ruffling. PI3K- and Rho-assisted ruffle closing into macropinosomes captures the surrounding fluid. If this occurs in a LDL-rich medium, macrophages accumulate large quantities of lipid and become the so-called foam cells, a hallmark of atherosclerotic lesions. Downstream from PLCγ, activation of PKC, together with ERK1/2-mediated activation of Rac, contributes to Nox2-dependent generation of ROS. Phosphorylation of the transcription factors Fos and Jun by ERK1/2 and JNK, respectively, results in the assembly of an AP-1 transcription complex and stimulates expression of pro-inflammatory cytokines. Nox2-produced ROS contributes to cytokine expression.
In addition to Rac, phosphorylation of ERK1/2 in mmLDL-activated macrophages results in GTP binding (activation) of Cdc42 and Rho and phosphorylation of paxillin (Choi et al., 2009). These are central components regulating actin polymerization and cytoskeletal reorganization. Within minutes, mmLDL-stimulated macrophages develop extensive membrane ruffling, lamellipodia and filopodia protrusions (Miller et al., 2003a,b; Harkewicz et al., 2008). Membrane ruffles close into macropinosomes and capture the surrounding fluid, the process regulated in part by PI3K and Rho. This mmLDL-induced and TLR4/SYK-dependent macropinocytosis is suggested to constitute an important mechanism of excessive lipid accumulation in macrophages (Choi et al., 2009; Miller et al., 2009; 2011; Stoletov et al., 2009). Although this macropinocytosis process is initiated by a specific lipid modification in mmLDL and is activated via a TLR4/SYK-dependent mechanism illustrated in Figure 1, the fluid phase uptake is indiscriminate towards the cargo and as a result, native LDL and various types of oxidized LDL are taken up by macrophages. These mechanisms of lipid uptake are important because formation of lipid-laden macrophage foam cells in the vascular wall is a hallmark of atherosclerotic lesions (Glass and Witztum, 2001). An interesting and unexpected confirmation of the role of TLR4 and SYK in atherogenesis has recently been reported by the Boren group (Levin et al., 2011). The authors found that the deficiency in Rip2, a serine/threonine kinase that activates NF-κB, in myeloid cells, reduced systemic inflammation but, contrary to their hypothesis, increased the size of atherosclerotic lesions in Ldlr-/-APOB100 mice. This was due to a twofold increased expression of TLR4 in macrophages, constitutive TLR4-dependent macropinocytosis and foam cell formation. Furthermore, inhibition of SYK abrogated macropinocytosis in Rip2-/- macrophages.
Activation of SYK, downstream from mmLDL and TLR4, is also essential for phosphorylation of JNK (Choi et al., 2012). JNK phosphorylates the transcription factor Jun, leading to removal of nuclear co-repressor NCoR (Wiesner et al., 2010) and allowing for the assembly of Jun and Fos into an AP-1 transcription complex. ERK1/2 phosphorylates Fos, and IKKε, together with JNK, phosphorylates Jun. The SYK regulation of AP-1 transcription is essential for expression and secretion of MIP-2 (CXCL2) and significantly contributes to expression of IL-6 in primary macrophages, as shown with the macrophages derived from Sykfl/fl LysM-Cre mice (Choi et al., 2012). Other genes induced by mmLDL in macrophages and dependent on Nox2 are IL-1β and RANTES (CCL5) (Bae et al., 2009). RANTES plays an important role in recruitment of vascular smooth muscle cells into the intima of the vascular wall, forming a fibrous cup of atherosclerotic lesions.
Importantly, the mmLDL-induced activation of AP-1 synergizes with activation of NF-κB induced by low doses of LPS to produce higher levels of pro-inflammatory cytokines, compared with the levels induced separately by either mmLDL or LPS (Wiesner et al., 2010). These findings are important because a subclinical but persistent endotoxemia characterizes many chronic infections that contribute to development and complications of atherosclerosis (Kalayoglu et al., 2002; Scannapieco et al., 2003; Pussinen et al., 2007). Furthermore, many obese and diabetic patients have the so-called metabolic endotoxemia (Amar et al., 2008; Erridge, 2008; Cani and Delzenne, 2009; Laugerette et al., 2011). Similarly to our in vitro and animal model findings, the synergistic pro-inflammatory effects of subclinical levels of LPS and oxidized lipoproteins abundant during atherogenesis may result in accelerated atherogenesis and a higher risk of cardiovascular events in specific cohorts of patients with chronic endotoxemia.
While this article's focus is on TLR4 signalling induced by mmLDL and oxidized cholesterol esters, in comparison to the LPS-induced signalling, this is only one example of pattern-recognition receptor interaction with oxidized lipoproteins and their components. TLR2, TLR6, CD36, SR-A, SR-BI, MARCO, LOX-1 and PSOX contribute to recognition, binding, internalization and inflammatory signalling in response to oxidized LDL and high-density lipoprotein. Oxidized phospholipids and the apolipoproteins modified by lipid-derived hydroperoxides, aldehydes and ketones, as well as by free radicals and peroxidase enzymes, have been described as ligands to pattern-recognition receptors. The reader can find a more detailed summary and discussion of this topic in our recent review article (Miller et al., 2011). It is also important to note that specific oxidized phospholipids, when used at high concentrations, inhibit LPS-induced, TLR4-dependent inflammatory responses, both in vitro and in vivo (Bochkov et al., 2002; Walton et al., 2003; von Schlieffen et al., 2009). This is in contrast to the synergy between mmLDL and low-dose LPS described above, underscoring the complexity of biological responses to host-derived ligands and the importance of in vivo experimentation to assess the quantitative significance of each particular pathway for specific pathology.
SYK as a therapeutic target
Understanding cellular mechanisms of SYK regulation has immediate importance because of the emergence of SYK inhibitors as promising therapeutic agents. The oral SYK inhibitor fostamatinib, being developed by Rigel and Astra-Zeneca, is currently in a phase III clinical trial in patients with rheumatoid arthritis. The Oral SYK Inhibition in Rheumatoid Arthritis (OSKIRA) is a 12 month clinical trial of approximately 900 patients studying two dosing regimens of fostamatinib compared with placebo in patients who are not achieving an adequate response with methotrexate alone (from Rigel's web site). A new drug application for fostamatinib with the Federal Drug Administration is expected in 2013. Several other pharmaceutical companies develop different SYK inhibitors for various clinical application (Mocsai et al., 2010), some of which are entering phase II clinical trials. Although targeting SYK is currently designed to benefit conditions with excessively up-regulated ITAM-mediated responses, such as in rheumatoid arthritis, allergies, asthma, lupus and other autoimmune diseases, our findings and the work of others point to the importance of SYK in regulation of TLR4 responses to either bacterial pathogens or host-derived agonists, such as mmLDL, the latter important in development of atherosclerosis.
Indeed, a recent report has demonstrated that administering fostamatinib attenuates atherosclerosis in Ldlr-/- mice fed with a high-cholesterol diet (Hilgendorf et al., 2011). Fostamatinib impaired macrophage survival and migration into atherosclerotic lesions and reduced expression of pro-inflammatory cytokines. Increased numbers of vascular smooth muscle cells and levels of collagen improved stability of the lesions. These results suggest that inhibition of SYK may be a viable therapeutic strategy to treat atherosclerosis and cardiovascular disease.
To put the prospects of SYK inhibition into perspective, there are numerous therapeutic targets downstream of TLR4, which is not surprising given the central role of TLR4 in inflammation. The relevance of these targets to the treatment of atherosclerosis depends on the significance of specific signalling pathways involved in the disease development and its clinical manifestations. As to the direct inhibitors of TLR4/MD-2 as drug candidates, several molecules have been tested for treatment of sepsis. For example, eritoran (E5564), a lipid A analogue, binds to MD-2 and inhibits LPS activation of TLR4 (Rossignol and Lynn, 2005), and resatorvid (TAK-242) targets Cys747 in the intracellular domain of TLR4 and inhibits its signalling (Ii et al., 2006; Takashima et al., 2009). For the variety of reasons, sepsis clinical trials for both of them were discontinued, and their effects on atherosclerosis have not yet been tested. A detailed review of these and other TLR4 inhibitors can be found elsewhere (Wittebole et al., 2010).
Conclusions
The TLR4 ligand recognition and signalling pathways are well studied in the LPS-stimulated macrophages. Certainly, LPS recognition by TLR4 produces the strongest pro-inflammatory responses in macrophages, and MyD88 is the most important adaptor molecule responsible for the immediate downstream signal transduction. One has to acknowledge, however, the existence of other, MyD88-independent TLR4 responses, which become centrally important under certain conditions. The better known example is the TRIF-dependent TLR4 signalling occurring upon endocytosis of TLR4/MD-2 with its microbial ligand and characterized by the delayed inflammatory responses. The less known pathway is the one mediated by the direct binding of SYK to TLR4, with ensuing activation of SYK targets and resulting in such, often overlooked, TLR4 responses as cytoskeletal rearrangements, macropinocytosis, ROS generation, inflammasome processing of IL-1β and others, discussed in this paper. Several studies suggest that the SYK pathway becomes important in LPS-stimulated neutrophils, where MyD88 signalling is inhibited and SYK mediates NLRP3 inflammasome activation and IL-1β maturation.
The realization that TLR4 as a pattern-recognition receptor is no less important in recognition of host-derived danger (or damage)-associated molecular patterns than microbial pathogens instructs the importance of the careful examination of signalling pathways involved. Muted inflammatory and robust tissue remodelling processes that characterize TLR4-mediated chronic inflammation in response to host-derived ligands also point to a signalling network that is different from that involved in acute and often excessively strong responses to LPS. In this regard, we discussed in this paper TLR4-mediated responses to mmLDL, an oxidized form of LDL apparently involved in a lifelong, asymptomatic inflammation in the vascular wall. The inflammatory processes in vascular atherosclerotic lesions may result, nevertheless, in myocardial infarction and stroke, the leading causes of mortality and morbidity in developed countries. The reasons for the lack of involvement of MyD88 in the majority of mmLDL-induced TLR4 responses in macrophages are yet unclear, but in the absence of a strong MyD88-mediated signalling component, the SYK-mediated pathways become centrally important. They regulate lipid accumulation in macrophages, ROS production and AP-1-dependent cytokine expression (Figure 1), the processes involved in development of atherosclerosis.
Future studies will reveal the detailed mechanisms and crosstalk between different branches of TLR4 signalling when activated by mmLDL and other host-derived ligands and help understand how this intertwined signalling shapes chronic inflammatory processes. Specifically in the case of atherosclerosis, one study has suggested that inhibition of SYK reduces atherosclerosis burden in mice (Hilgendorf et al., 2011). It is yet unknown if this effect is related to the SYK involvement in TLR4 signalling or any other SYK-dependent immune receptor-mediated pathways, which are numerous. It is also unclear what is the cell type(s) in which inhibition of SYK delivers an atheroprotective effect. In vivo studies with cell-specific SYK knockout mice will be able to address these questions.
Acknowledgments
The studies performed in authors' laboratories were supported by NIH grants HL081862, HL088093, HL093767 and GM069338, a grant from the Leducq Fondation, World Class University program (R31-2008-000-10010-0) of the MOEST/KOSEF and the Ewha Global Top 5 grant 2011 from Ewha Womans University.
Glossary
- LDL
low-density lipoprotein
- MD-2
myeloid differentiation-2 (synonym: lymphocyte antigen 96, LY96)
- mmLDL
minimally oxidized LDL
- SYK
spleen tyrosine kinase
- TLR4
toll-like receptor-4 (synonym: CD284)
Conflict of interest
Authors declare no conflicts of interest.
References
- Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr. 2008;87:1219–1223. doi: 10.1093/ajcn/87.5.1219. [DOI] [PubMed] [Google Scholar]
- Ameziane N, Beillat T, Verpillat P, Chollet-Martin S, Aumont MC, Seknadji P, et al. Association of the Toll-like receptor 4 gene Asp299Gly polymorphism with acute coronary events. Arterioscler Thromb Vasc Biol. 2003;23:e61–e64. doi: 10.1161/01.ATV.0000101191.92392.1D. [DOI] [PubMed] [Google Scholar]
- Arndt PG, Suzuki N, Avdi NJ, Malcolm KC, Worthen GS. Lipopolysaccharide-induced c-Jun NH2-terminal kinase activation in human neutrophils: role of phosphatidylinositol 3-kinase and Syk-mediated pathways. J Biol Chem. 2004;279:10883–10891. doi: 10.1074/jbc.M309901200. [DOI] [PubMed] [Google Scholar]
- Bae YS, Lee JH, Choi SH, Kim S, Almazan F, Witztum JL, et al. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ Res. 2009;104:210–218. doi: 10.1161/CIRCRESAHA.108.181040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkbacka H, Kunjathoor VV, Moore KJ, Koehn S, Ordija CM, Lee MA, et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med. 2004;10:416–421. doi: 10.1038/nm1008. [DOI] [PubMed] [Google Scholar]
- Bochkov VN, Kadl A, Huber J, Gruber F, Binder BR, Leitinger N. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature. 2002;419:77–81. doi: 10.1038/nature01023. [DOI] [PubMed] [Google Scholar]
- Boekholdt SM, Agema WR, Peters RJ, Zwinderman AH, Van Der Wall EE, Reitsma PH, et al. Variants of toll-like receptor 4 modify the efficacy of statin therapy and the risk of cardiovascular events. Circulation. 2003;107:2416–2421. doi: 10.1161/01.CIR.0000068311.40161.28. [DOI] [PubMed] [Google Scholar]
- Bowen WS, Minns LA, Johnson DA, Mitchell TC, Hutton MM, Evans JT. Selective TRIF-dependent signaling by a synthetic toll-like receptor 4 agonist. Sci Signal. 2012;5:ra13. doi: 10.1126/scisignal.2001963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Delzenne NM. Interplay between obesity and associated metabolic disorders: new insights into the gut microbiota. Curr Opin Pharmacol. 2009;9:737–743. doi: 10.1016/j.coph.2009.06.016. [DOI] [PubMed] [Google Scholar]
- Chaudhary A, Fresquez TM, Naranjo MJ. Tyrosine kinase Syk associates with toll-like receptor 4 and regulates signaling in human monocytic cells. Immunol Cell Biol. 2007;85:249–256. doi: 10.1038/sj.icb7100030. [DOI] [PubMed] [Google Scholar]
- Choi S-H, Harkewicz R, Lee JH, Boullier A, Almazan F, Li AC, et al. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ Res. 2009;104:1355–1363. doi: 10.1161/CIRCRESAHA.108.192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Wiesner P, Almazan F, Kim J, Miller YI. Spleen tyrosine kinase regulates AP-1 dependent transcriptional response to minimally oxidized LDL. PLoS ONE. 2012;7:e32378. doi: 10.1371/journal.pone.0032378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenen K, Gruen M, Lee-Young R, Puglisi M, Wasserman D, Hasty A. Impact of macrophage toll-like receptor 4 deficiency on macrophage infiltration into adipose tissue and the artery wall in mice. Diabetologia. 2009;52:318–328. doi: 10.1007/s00125-008-1221-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF, et al. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J Clin Invest. 1999;103:1597–1604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyrus T, Pratico D, Zhao L, Witztum JL, Rader DJ, Rokach J, et al. Absence of 12/15-lipoxygenase expression decreases lipid peroxidation and atherogenesis in apolipoprotein e-deficient mice. Circulation. 2001;103:2277–2282. doi: 10.1161/01.cir.103.18.2277. [DOI] [PubMed] [Google Scholar]
- Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002;105:1158–1161. [PubMed] [Google Scholar]
- Elsori DH, Yakubenko VP, Roome T, Thiagarajan PS, Bhattacharjee A, Yadav SP, et al. Protein kinase C delta is a critical component of Dectin-1 signaling in primary human monocytes. J Leukoc Biol. 2011;90:599–611. doi: 10.1189/jlb.0610376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C. The roles of pathogen-associated molecular patterns in atherosclerosis. Trends Cardiovasc Med. 2008;18:52–56. doi: 10.1016/j.tcm.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Ezaki M, Witztum JL, Steinberg D. Lipoperoxides in LDL incubated with fibroblasts that overexpress 15-lipoxygenase. J Lipid Res. 1995;36:1996–2004. [PubMed] [Google Scholar]
- Fang L, Harkewicz R, Hartvigsen K, Wiesner P, Choi SH, Almazan F, et al. Oxidized cholesteryl esters and phospholipids in zebrafish larvae fed a high-cholesterol diet: macrophage binding and activation. J Biol Chem. 2010;285:32343–32351. doi: 10.1074/jbc.M110.137257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff M. Different dimerisation mode for TLR4 upon endosomal acidification? Trends Biochem Sci. 2012;37:92–98. doi: 10.1016/j.tibs.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng HL, Lu HQ, Zhang LZ, Zhang H, Zhou L, Wang H, et al. Increased expression of Toll like receptor 4 on peripheral-blood mononuclear cells in patients with coronary arteriosclerosis disease. Clin Exp Immunol. 2006;143:269–273. doi: 10.1111/j.1365-2249.2005.02982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George J, Afek A, Shaish A, Levkovitz H, Bloom N, Cyrus T, et al. 12/15-Lipoxygenase gene disruption attenuates atherogenesis in LDL receptor-deficient mice. Circulation. 2001;104:1646–1650. doi: 10.1161/hc3901.095772. [DOI] [PubMed] [Google Scholar]
- Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Hamerman JA, Tchao NK, Lowell CA, Lanier LL. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol. 2005;6:579–586. doi: 10.1038/ni1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol. 2010;11:734–742. doi: 10.1038/ni.1908. [DOI] [PubMed] [Google Scholar]
- Harkewicz R, Hartvigsen K, Almazan F, Dennis EA, Witztum JL, Miller YI. Cholesteryl ester hydroperoxides are biologically active components of minimally oxidized LDL. J Biol Chem. 2008;283:10241–10251. doi: 10.1074/jbc.M709006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine GH, Ortiz A, Massy ZA, Lindholm B, Wiecek A, Martinez-Castelao A, et al. Monocyte subpopulations and cardiovascular risk in chronic kidney disease. Nat Rev Nephrol. 2012;8:362–369. doi: 10.1038/nrneph.2012.41. [DOI] [PubMed] [Google Scholar]
- Higashimori M, Tatro JB, Moore KJ, Mendelsohn ME, Galper JB, Beasley D. Role of Toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:50–57. doi: 10.1161/ATVBAHA.110.210971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgendorf I, Eisele S, Remer I, Schmitz J, Zeschky K, Colberg C, et al. The oral spleen tyrosine kinase inhibitor fostamatinib attenuates inflammation and atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:1991–1999. doi: 10.1161/ATVBAHA.111.230847. [DOI] [PubMed] [Google Scholar]
- Hiratsuka S, Watanabe A, Sakurai Y, Kashi-Takamura S, Ishibashi S, Miyake K, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10:1349–1355. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, Burcin T, et al. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;110:2024–2031. doi: 10.1161/01.CIR.0000143628.37680.F6. [DOI] [PubMed] [Google Scholar]
- Hutchins PM, Moore EE, Murphy RC. Electrospray tandem mass spectrometry reveals extensive and non-specific oxidation of cholesterol esters in human peripheral vascular lesions. J Lipid Res. 2011;52:2070–2083. doi: 10.1194/jlr.M019174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ii M, Matsunaga N, Hazeki K, Nakamura K, Takashima K, Seya T, et al. A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-Chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol Pharmacol. 2006;69:1288–1295. doi: 10.1124/mol.105.019695. [DOI] [PubMed] [Google Scholar]
- Jakus Z, Fodor S, Abram CL, Lowell CA, Mocsai A. Immunoreceptor-like signaling by [beta]2 and [beta]3 integrins. Trends Cell Biol. 2007;17:493–501. doi: 10.1016/j.tcb.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Kalayoglu MV, Libby P, Byrne GI. Chlamydia pneumoniae as an emerging risk factor in cardiovascular disease. JAMA. 2002;288:2724–2731. doi: 10.1001/jama.288.21.2724. [DOI] [PubMed] [Google Scholar]
- Katsargyris A, Tsiodras S, Theocharis S, Giaginis K, Vasileiou I, Bakoyiannis C, et al. Toll-like receptor 4 immunohistochemical expression is enhanced in macrophages of symptomatic carotid atherosclerotic plaques. Cerebrovasc Dis. 2011;31:29–36. doi: 10.1159/000320259. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Kenny EF, O'Neill LAJ. Signalling adaptors used by Toll-like receptors: an update. Cytokine. 2008;43:342–349. doi: 10.1016/j.cyto.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Kerrigan AM, Brown GD. Syk-coupled C-type lectins in immunity. Trends Immunol. 2011;32:151–156. doi: 10.1016/j.it.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, et al. Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med. 2002;347:185–192. doi: 10.1056/NEJMoa012673. [DOI] [PubMed] [Google Scholar]
- Laugerette F, Vors C, Peretti N, Michalski MC. Complex links between dietary lipids, endogenous endotoxins and metabolic inflammation. Biochimie. 2011;93:39–45. doi: 10.1016/j.biochi.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem. 2007;76:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- Leitinger N. Cholesteryl ester oxidation products in atherosclerosis. Mol Aspects Med. 2003;24:239–250. doi: 10.1016/s0098-2997(03)00019-0. [DOI] [PubMed] [Google Scholar]
- Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- Levin MC, Jirholt P, Wramstedt A, Johansson ME, Lundberg AM, Trajkovska MG, et al. Rip2 deficiency leads to increased atherosclerosis despite decreased inflammation. Circ Res. 2011;109:1210–1218. doi: 10.1161/CIRCRESAHA.111.246702. [DOI] [PubMed] [Google Scholar]
- Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1506–1516. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell CA. Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harb Persp Biol. 2011;3:a002352. doi: 10.1101/cshperspect.a002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Pan H, Shively JE. CEACAM1 negatively regulates IL-1beta production in LPS activated neutrophils by recruiting SHP-1 to a SYK-TLR4-CEACAM1 complex. PLoS Pathog. 2012;8:e1002597. doi: 10.1371/journal.ppat.1002597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGettrick AF, O'Neill LA. Localisation and trafficking of Toll-like receptors: an important mode of regulation. Curr Opin Immunol. 2010;22:20–27. doi: 10.1016/j.coi.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Medvedev AE, Piao W, Shoenfelt J, Rhee SH, Chen H, Basu S, et al. Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J Biol Chem. 2007;282:16042–16053. doi: 10.1074/jbc.M606781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methe H, Kim JO, Kofler S, Weis M, Nabauer M, Koglin J. Expansion of circulating toll-like receptor 4-positive monocytes in patients with acute coronary syndrome. Circulation. 2005;111:2654–2661. doi: 10.1161/CIRCULATIONAHA.104.498865. [DOI] [PubMed] [Google Scholar]
- Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. PNAS. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–780. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- Miller YI, Worrall DS, Funk CD, Feramisco JR, Witztum JL. Actin polymerization in macrophages in response to oxidized LDL and apoptotic cells: role of 12/15-lipoxygenase and phosphoinositide 3-kinase. Mol Biol Cell. 2003a;14:4196–4206. doi: 10.1091/mbc.E03-02-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller YI, Viriyakosol S, Binder CJ, Feramisco JR, Kirkland TN, Witztum JL. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J Biol Chem. 2003b;278:1561–1568. doi: 10.1074/jbc.M209634200. [DOI] [PubMed] [Google Scholar]
- Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. 2005;25:1213–1219. doi: 10.1161/01.ATV.0000159891.73193.31. [DOI] [PubMed] [Google Scholar]
- Miller YI, Choi SH, Fang L, Harkewicz R. Toll-like receptor-4 and lipoprotein accumulation in macrophages. Trends Cardiovasc Med. 2009;7:227–232. doi: 10.1016/j.tcm.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller YI, Choi S-H, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, et al. Oxidation-specific epitopes are danger associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–248. doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocsai A, Ruland J, Tybulewicz VLJ. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10:387–402. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- Natarajan R, Nadler JL. Lipoxygenases and lipid signaling in vascular cells in diabetes. Front Biosci. 2003;8:s783–s795. doi: 10.2741/1144. [DOI] [PubMed] [Google Scholar]
- Netea MG, Hijmans A, van Wissen S, Smilde TJ, Trip MD, Kullberg BJ, et al. Toll-like receptor-4 Asp299Gly polymorphism does not influence progression of atherosclerosis in patients with familial hypercholesterolaemia. Eur J Clin Invest. 2004;34:94–99. doi: 10.1111/j.1365-2362.2004.01303.x. [DOI] [PubMed] [Google Scholar]
- Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harbor Persp Biol. 2012;4:a006049. doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens AP, III, Rateri DL, Howatt DA, Moore KJ, Tobias PS, Curtiss LK, et al. MyD88 deficiency attenuates angiotensin II-induced abdominal aortic aneurysm formation independent of signaling through Toll-like receptors 2 and 4. Arterioscler Thromb Vasc Biol. 2011;31:2813–2819. doi: 10.1161/ATVBAHA.111.238642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- Patricia MK, Natarajan R, Dooley AN, Hernandez F, Gu JL, Berliner JA, et al. Adenoviral delivery of a leukocyte-type 12 lipoxygenase ribozyme inhibits effects of glucose and platelet-derived growth factor in vascular endothelial and smooth muscle cells. Circ Res. 2001;88:659–665. doi: 10.1161/hh0701.088838. [DOI] [PubMed] [Google Scholar]
- Poeckel D, Zemski Berry KA, Murphy RC, Funk CD. Dual 12/15- and 5-lipoxygenase deficiency in macrophages alters arachidonic acid metabolism and attenuates peritonitis and atherosclerosis in APOE knockout mice. J Biol Chem. 2009;284:21077–21089. doi: 10.1074/jbc.M109.000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler Thromb Vasc Biol. 2007;27:1433–1439. doi: 10.1161/ATVBAHA.106.138743. [DOI] [PubMed] [Google Scholar]
- Reismann P, Lichy C, Rudofsky G, Humpert PM, Genius J, Si TD, et al. Lack of association between polymorphisms of the toll-like receptor 4 gene and cerebral ischemia. J Neurol. 2004;251:853–858. doi: 10.1007/s00415-004-0447-7. [DOI] [PubMed] [Google Scholar]
- Rong S, Cao Q, Liu M, Seo J, Jia L, Boudyguina E, et al. Macrophage 12/15 lipoxygenase expression increases plasma and hepatic lipid levels and exacerbates atherosclerosis. J Lipid Res. 2012;53:686–695. doi: 10.1194/jlr.M022723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol DP, Lynn M. TLR4 antagonists for endotoxemia and beyond. Curr Opin Investig Drugs. 2005;6:496–502. [PubMed] [Google Scholar]
- Rowe SJ, Allen L, Ridger VC, Hellewell PG, Whyte MKB. Caspase-1-deficient mice have delayed neutrophil apoptosis and a prolonged inflammatory response to lipopolysaccharide-induced acute lung injury. J Immunol. 2002;169:6401–6407. doi: 10.4049/jimmunol.169.11.6401. [DOI] [PubMed] [Google Scholar]
- Scannapieco FA, Bush RB, Paju S. Associations between periodontal disease and risk for atherosclerosis, cardiovascular disease, and stroke. A systematic review. Ann Periodontol. 2003;8:38–53. doi: 10.1902/annals.2003.8.1.38. [DOI] [PubMed] [Google Scholar]
- Schmitz G, Orso E. CD14 signalling in lipid rafts: new ligands and co-receptors. Curr Opin Lipidol. 2002;13:513–521. doi: 10.1097/00041433-200210000-00007. [DOI] [PubMed] [Google Scholar]
- Shalova IN, Kajiji T, Lim JY, Gomez-Pina V, Fernandez-Ruiz I, Arnalich F, et al. CD16 regulates TRIF-dependent TLR4 response in human monocytes and their subsets. J Immunol. 2012;188:3584–3593. doi: 10.4049/jimmunol.1100244. [DOI] [PubMed] [Google Scholar]
- Shibata T, Motoi Y, Tanimura N, Yamakawa N, Kashi-Takamura S, Miyake K. Intracellular TLR4/MD-2 in macrophages senses Gram-negative bacteria and induces a unique set of LPS-dependent genes. Int Immunol. 2011;23:503–510. doi: 10.1093/intimm/dxr044. [DOI] [PubMed] [Google Scholar]
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletov K, Fang L, Choi SH, Hartvigsen K, Hansen LF, Hall C, et al. Vascular lipid accumulation, lipoprotein oxidation, and macrophage lipid uptake in hypercholesterolemic zebrafish. Circ Res. 2009;104:952–960. doi: 10.1161/CIRCRESAHA.108.189803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, et al. Critical role for NALP3/CIAS1/cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Takashima K, Matsunaga N, Yoshimatsu M, Hazeki K, Kaisho T, Uekata M, et al. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br J Pharmacol. 2009;157:1250–1262. doi: 10.1111/j.1476-5381.2009.00297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiemi Shio M, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJE, Harder KW, et al. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. 2009;5:e1000559. doi: 10.1371/journal.ppat.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou M, Triantafilou K. Membrane partitioning: is location everything when it comes to endotoxin recognition? Subcell Biochem. 2010;53:173–184. doi: 10.1007/978-90-481-9078-2_8. [DOI] [PubMed] [Google Scholar]
- Upston JM, Niu X, Brown AJ, Mashima R, Wang H, Senthilmohan R, et al. Disease stage-dependent accumulation of lipid and protein oxidation products in human atherosclerosis. Am J Pathol. 2002;160:701–710. doi: 10.1016/S0002-9440(10)64890-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schlieffen E, Oskolkova OV, Schabbauer G, Gruber F, Bluml S, Genest M, et al. Multi-hit inhibition of circulating and cell-associated components of the toll-like receptor 4 pathway by oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2009;29:356–362. doi: 10.1161/ATVBAHA.108.173799. [DOI] [PubMed] [Google Scholar]
- Walton KA, Cole AL, Yeh M, Subbanagounder G, Krutzik SR, Modlin RL, et al. Specific phospholipid oxidation products inhibit ligand activation of toll-like receptors 4 and 2. Arterioscler Thromb Vasc Biol. 2003;23:1197–1203. doi: 10.1161/01.ATV.0000079340.80744.B8. [DOI] [PubMed] [Google Scholar]
- Wiesner P, Choi SH, Almazan F, Benner C, Huang W, Diehl CJ, et al. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor {kappa}B and activator protein-1. Possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ Res. 2010;107:56–65. doi: 10.1161/CIRCRESAHA.110.218420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittebole X, Castanares-Zapatero D, Laterre PF. Toll-like receptor 4 modulation as a strategy to treat sepsis. Mediators Inflamm. 2010;2010:568396. doi: 10.1155/2010/568396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodside DG, Obergfell A, Talapatra A, Calderwood DA, Shattil SJ, Ginsberg MH. The N-terminal SH2 domains of Syk and ZAP-70 mediate phosphotyrosine-independent binding to integrin cytoplasmic domains. J Biol Chem. 2002;277:39401–39408. doi: 10.1074/jbc.M207657200. [DOI] [PubMed] [Google Scholar]
- Xie P, Cao YS, Su P, Li YH, Gao ZL, Borst MM. Expression of toll-like receptor 4, tumor necrosis factor-alpha, matrix metalloproteinase-9 and effects of benazepril in patients with acute coronary syndromes. Clin Med Insights Cardiol. 2010;4:89–93. doi: 10.4137/CMC.S5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, et al. Toll-Like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–3108. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- Yang IA, Holloway JW, Ye S. TLR4 Asp299Gly polymorphism is not associated with coronary artery stenosis. Atherosclerosis. 2003;170:187–190. doi: 10.1016/s0021-9150(03)00286-7. [DOI] [PubMed] [Google Scholar]
- Ylä-Herttuala S, Rosenfeld ME, Parthasarathy S, Glass CK, Sigal E, Witztum JL, et al. Colocalization of 15-lipoxygenase mRNA and protein with epitopes of oxidized low density lipoprotein in macrophage-rich areas of atherosclerotic lesions. Proc Natl Acad Sci USA. 1990;87:6959–6963. doi: 10.1073/pnas.87.18.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylä-Herttuala S, Rosenfeld ME, Parthasarathy S, Sigal E, Sarkioja T, Witztum JL, et al. Gene expression in macrophage-rich human atherosclerotic lesions. 15-lipoxygenase and acetyl low density lipoprotein receptor messenger RNA colocalize with oxidation specific lipid-protein adducts. J Clin Invest. 1991;87:1146–1152. doi: 10.1172/JCI115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Ostuni R, Marek L, Barresi S, Barbalat R, Barton G, et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147:868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbock A, Ley K. Protein tyrosine kinases in neutrophil activation and recruitment. Arch Biochem Biophys. 2011;510:112–119. doi: 10.1016/j.abb.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Zhao L, Pratico D, Rader DJ, Funk CD. 12/15-Lipoxygenase gene disruption and vitamin E administration diminish atherosclerosis and oxidative stress in apolipoprotein E deficient mice through a final common pathway. Prostaglandins Other Lipid Mediat. 2005;78:185–193. doi: 10.1016/j.prostaglandins.2005.07.003. [DOI] [PubMed] [Google Scholar]