

Abstract
BACKGROUND AND PURPOSE
The use of tubulin-binding compounds, which act in part by inhibiting tumour angiogenesis, has become an integral strategy of tumour therapy. Recently, tubulysins were identified as a novel class of natural compounds of myxobacterial origin, which inhibit tubulin polymerization. As these compounds are structurally highly complex, the search for simplified precursors [e.g. pretubulysin (Prt)] and their derivatives is mandatory to overcome supply problems hampering clinical development. We tested the anti-angiogenic efficacy of Prt and seven of its derivatives in comparison to tubulysin A (TubA).
EXPERIMENTAL APPROACH
The compounds were tested in cellular angiogenesis assays (proliferation, cytotoxicity, cell cycle, migration, chemotaxis, tube formation) and in vitro (tubulin polymerization). The efficacy of Prt was also tested in vivo in a murine subcutaneous tumour model induced with HUH7 cells; tumour size and vascularization were measured.
KEY RESULTS
The anti-angiogenic potency of all the compounds tested ran parallel to their inhibition of tubulin polymerization in vitro. Prt showed nearly the same efficacy as TubA (EC50 in low nanomolar range in all cellular assays). Some modifications in the Prt molecule caused only a moderate drop in potency, while others resulted in a dramatic loss of action, providing initial insight into structure–activity relations. In vivo, Prt completely prevented tumour growth and reduced vascular density to 30%.
CONCLUSIONS AND IMPLICATIONS
Prt, a chemically accessible precursor of some tubulysins is a highly attractive anti-angiogenic compound both in vitro and in vivo. Even more simplified derivatives of this compound still retain high anti-angiogenic efficacy.
Keywords: angiogenesis, endothelial cells, tubulysin, pretubulysin, microtubule, tubulin, migration
Introduction
Natural compounds have turned out to be a valuable source for the development of drugs, especially in the treatment of tumours (Kingston, 2009), and some of the most broadly used therapeutics against cancer are of natural origin or have been inspired by natural compounds (e.g. etoposide, doxorubicin). The main issue with natural compounds that hampers their clinical use is their supply. Often, they cannot be isolated from their source in adequate quantities, or they are chemically so complicated that their total synthesis is not feasible on an industrial scale. This problem has, for example, retarded the advent of taxanes in the clinical setting for many years (Dumontet and Jordan, 2010). Therefore, a constant need exists for simplified and chemically accessible, yet still highly potent derivatives or precursors of natural compounds, which have been shown to have promising properties.
Besides DNA, the microtubule cytoskeleton is a main target for anti-cancer drugs. This is due to the central role of the microtubule cytoskeleton not only during mitosis, but also in organelle positioning and other transport phenomena (Lomakin and Nadezhdina, 2010). A part of the relative selectivity of microtubule-targeting drugs towards tumour cells as compared to normal cells stems from the highly dynamic nature of cancer cells. In recent years, not only the tumour cells as such but also the newly formed tumour vessels have turned out to be a valid clinical target. Anti-angiogenic approaches range from therapeutic antibodies (e.g. bevacizumab) to kinase inhibitors (e.g. sunitinib), and also comprise microtubule-targeting drugs (Schwartz, 2009). Thus, it is of clinical importance to test novel microtubule-targeting compounds also in the context of angiogenesis.
A novel group of microtubule-depolymerizing natural products of myxobacterial origin, the tubulysins, has recently been described (Sasse et al., 2000), and has turned out to be highly potent against some tumour cell lines (Sasse et al., 2000; Kaur et al., 2006). Due to the supply issue mentioned earlier, several attempts have been made to synthesize simplified tubulysin derivatives (Raghavan et al., 2008; Ullrich et al., 2009a). The most promising approach to date is the synthesis of pretubulysin (Prt), a putative precursor of some tubulysins, which has only a slight loss of potency as compared to tubulysin itself (Ullrich et al., 2009a). With Prt as a starting point, several biologically active derivatives have been established (Ullrich et al., 2009b; Burkhart et al., 2011). Tubulysin A (TubA) has previously been described as a microtubule-binding drug that has anti-angiogenic properties (Kaur et al., 2006). However, no detailed information on cellular effects or anti-angiogenic actions of Prt or its derivatives is available yet. Therefore, in the present study, we characterized the effects of Prt and seven of its derivatives on various parameters of angiogenesis in vitro, and also the most potent compound (Prt) in vivo.
Methods
Compounds
Prt and its derivatives (Figure 1) were synthesized as described previously (Ullrich et al., 2009b; Burkhart et al., 2011). Tub A was purchased from Merck Calbiochem (Darmstadt, Germany). The compounds were prepared as 10 mM stock solutions in dimethylsulfoxide (DMSO). The final DMSO concentrations did not exceed 0.3%, a concentration verified not to affect respective experimental parameters.
Figure 1.
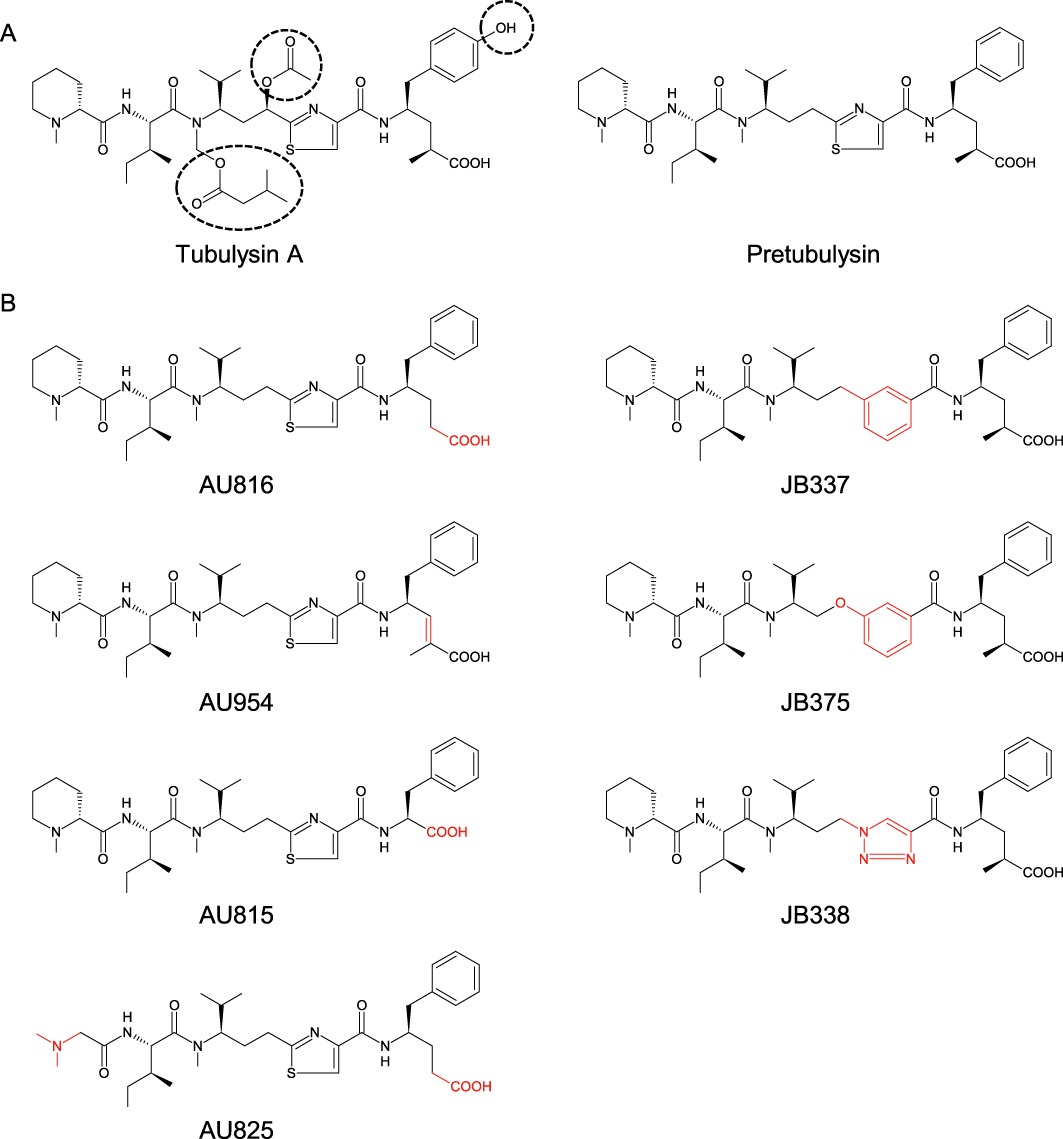
(A) Chemical structures of tubulysin A and pretubulysin. (B) Derivatives of pretubulysin modified either at an N/C terminal position (AU816, AU954, AU825, AU815) or at a central position (JB337, JB375, JB338). Structural alterations from pretubulysin are marked by dotted lines (A) or in red (B).
Cell culture
Human microvascular endothelial cells [HMEC-1, (Ades et al., 1992)] were obtained from CDC (Atlanta, GA, USA) and used until the 12th passage maximum. Primary human umbilical vein endothelial cells (HUVECs) were isolated by collagenase treatment of umbilical cords as previously described (Liebl et al., 2010), and used at the third passage.
Endothelial cell growth medium (PromoCell, Heidelberg, Germany), containing 10% heat inactivated fetal calf serum (FCS) and a supplemental mix of vitamins and growth factors (PromoCell), was used as culture medium,. HUH7 cells were obtained from the Japan Health Science Research Resources Bank (JCRB0403) and cultivated in Dulbecco's modified Eagle's medium (DMEM) containing 10% FCS at 37°C and 5% CO2. They were used between passage 10 and 14.
Immunocytochemistry
HMEC-1 were seeded on 0.001% collagen G-coated 8-well slides (IBIDI, Martinsried, Germany) and incubated with TubA, Prt or the Prt derivatives for 16 h. HMEC-1 were washed with pre-warmed PBS. Cell extraction buffer (80 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) pH 6.8; 1 mM MgCl2, 5 mM EGTA-K and 0.5% Triton X-100) was added to remove mono and dimeric tubulin subunits. After 30 s of extraction, cells were fixed for 10 min by adding glutaraldehyde to a final level of 0.5%. Excess glutaraldehyde was removed and quenched with 0.1% NaBH4 in PBS for 7 min. Cells were washed thoroughly with PBS again, blocked with PBS containing 0.2% BSA and stained for α-tubulin and nuclei.
The anti α-tubulin antibody (ab18251) and the AlexaFluor 488 secondary antibody (A 11008) were purchased from Abcam (Cambridge, UK) and Invitrogen (Darmstadt, Germany), respectively. Hoechst 33342 (bisbenzimide), purchased from Sigma-Aldrich (Taufkirchen, Germany), was used for nuclei staining.
Proliferation assay
The proliferation assay was performed according to National Cancer Institute (NCI) protocols for angiogenesis.
Briefly, 1500 HMEC-1 per well were seeded into 96-well plates in 100 µL of media. After 24 h, one plate of control cells was fixed and stained with crystal violet (see later) to determine the initial cell number. The other plates were incubated with increasing concentrations of the compounds to be tested for 72 h. After this time, cells were stained with crystal violet solution (0.5% crystal violet in 20% methanol) for 10 min. Unbound crystal violet was removed by rinsing with distilled water and cells were subsequently air-dried. Crystal violet, which mainly binds to DNA, was eluted from cells with 0.1 M sodium citrate in 50% ethanol. The absorbance of crystal violet is proportional to the cell number and was determined at 540 nm with a Magellan 6 plate reader (TECAN, Männedorf, Switzerland). For each compound, three independent experiments were performed, each of them included six replicates. The same protocol was used to investigate the effects of tubulysin and Prt on the proliferation of the HUH7 tumour cells in the in vivo assay (see later).
Detection and quantification of nuclear fragmentation, membrane integrity and cell cycle
In order to determine whether cytotoxicity plays a role in the effects of the compounds on functional parameters of angiogenesis (migration, tube formation), HMEC-1 or HUVECs were incubated with the compounds and seeded on 24-well plates at the same density, drug : cell relationship, and for the appropriate time as in the respective functional assay (24 h or 48 h). Quantification of nuclear fragmentation as one indicator of cell death was performed according to Nicoletti (Nicoletti et al., 1991). In brief, after treatment, cells were harvested on ice and incubated in a hypotonic buffer [0.1% sodium citrate, 0.1% Triton X-100 and 50 µg·mL−1 propidium iodide (PI)] overnight at 4°C, and then analysed by flow cytometry on a FACSCalibur (Becton Dickinson, Heidelberg, Germany) using Cell Quest Pro Software (Becton Dickinson). Nuclei to the left of the G1 peak containing hypodiploid DNA were considered as fragmented. In the same set of experiments, the percentage of cells in the G2/M phase was evaluated using the FlowJo software (Tree Star Inc., Ashland, OR, USA). All experiments were performed in triplicate and each experiment was repeated at least three times.
As an independent marker for cytotoxicity, membrane integrity was tested by examining the uptake of PI in non-permeabilized cells. Here, again, the assay conditions were adapted to the respective functional assay. Cells were incubated with 10 µg·mL−1 PI not containing any detergent for 30 min. Under these conditions, PI only enters cells with a damaged cell membrane (i.e. dead cells). Cells were harvested and analysed by flow cytometry for PI fluorescence. PI-positive HMEC-1 were gated and analysed using Cell Quest Pro Software. Data are expressed as means ± SEM of three independent experiments (each of which was performed in triplicate). Alternatively, tubes generated on Matrigel were incubated with PI to detect dead cells in situ by fluorescence microscopy.
Migration (scratch assay)
For assays involving migration, primary endothelial cells (HUVECs) were used instead of HMEC-1, since they yielded more stable effects. HUVECs were seeded onto 24-well plates and grown to confluency. A wound of approximately 1 mm was inflicted to the monolayers by scratching them with a pipette tip. Detached cells were removed by washing with PBS, the remaining cells then were incubated for 16 h in either serum-free medium (free of growth factors and serum, 0% migration), culture medium (100% migration) or culture medium containing increasing concentrations of Tub A, Prt or one of the Prt derivatives. Cells were washed with PBS after the incubation and then fixed with 4% formaldehyde. One image was taken of each well (centre position) on an inverted light microscope (Axiovert 200; Zeiss, Jena, Germany) with a 5× lens using an Imago-QE camera system and the appending software (Till Photonics, Gräfelfing, Germany). For quantification, these images were analysed with the WimScratch Wound Healing Module (WIMASIS, Munich, Germany). This online software tool is able to distinguish the cell-covered from the wounded area by using an algorithm based on brightness and contrast values. The cell-free area correlates with the ability of the HUVECs to migrate into the wounded area. Relative migration was calculated relative to the control and the serum-deprived control. In previous experiments, we demonstrated that cell proliferation does not contribute to wound closure in this setting (Liebl et al., 2010). Data are presented as means ± SEM of at least three independent experiments, each performed in triplicate.
Chemotaxis assay
HUVECs were seeded into ‘µ-slides chemotaxis’ chambers coated with collagen IV (IBIDI) and allowed to attach for 2 h. Then a FCS gradient from 0% FCS to 10% FCS was generated according to the manufacturer's instructions. Time-lapse microscopy was performed with the setup described above for ‘Scratch assay’. An incubation chamber from IBIDI was used to keep cells at 37°C, 5% CO2 and 80% relative humidity. Images of cells were obtained every 5 min for 20 h. The Image J plugins ‘Manual Tracking’ and ‘Chemotaxis Analysis’ (National Institutes of Health, USA) were used for image analysis. In each image sequence, 100 cells were tracked. The tracking module allows the assessment of both the overall way the cell has covered (accumulative distance) and the ‘air-line distance’ (Euclidean distance, as a measure of directionality). Data are expressed as means ± SEM of each of the three independent experiments.
Tube formation assay
Growth factor-reduced Matrigel™ (BD Discovery Labware, Bedford, MA, USA) was placed into the lower chambers of µ-slide angiogenesis wells (IBIDI) and polymerized for 30 min at 37°C. Test compounds (500 µL of 2× the final concentration) were each mixed with 500 µL of HMEC-1 (4 × 105 mL−1), and 50 µL of this suspension was placed into µ-slides angiogenesis wells containing Matrigel. Cells were incubated for 16 h. One image per well was taken on the Axiovert 200 microscope as described earlier. The images were analysed with the tube formation module of WIMASIS Image Analysis. This online software module identifies cellular tubes on a multiparametric basis (depending on brightness and contrast differences, length and width of the structure) and interprets tubes and non-tube complexes using an automated mathematic algorithm. Drug effects were assessed by analysing the total tube length and number of tube connecting nodes. Data are presented as means ± SEM of at least three independent experiments, each performed in triplicate.
Tubulin polymerization assay
Microtubule-associated protein-rich tubulin from porcine brain was obtained from Cytoskeleton Inc. (Cat. # ML116, Denver, CO, USA). Tubulin polymerization was monitored by use of turbidimetry. Samples (200 µL, 10 µM tubulin) in polymerization buffer (0.1 M PIPES, pH 6.6, 1 mM EGTA, 1 mM MgSO4 and 1 mM GTP) were rapidly warmed to 37°C in a water-jacketed cuvette holder of a diode array photometer (Spectrophotometer DU 7500, Beckmann Coulter, Krefeld, Germany). Absorbance at 350 nm was monitored in the absence and presence of the compounds at the indicated concentrations.
Subcutaneous murine tumour xenograft model
HUH7 liver tumour cells (5 million per mouse) were injected s.c. into 6 to 8-week-old female severe combined immunodeficiency mice (CB17/lcr-PrkdcSCID/lcrlcocrl from Charles River; Wilmington, MA, USA). Six days after tumour cell application, when tumours had reached a detectable volume of approximately 10–15 mm3, mice were treated i.v. every second day either with 0.1 mg kg−1 Prt dissolved in PBS or with PBS alone for a total of five treatments. Tumour volume was determined continuously with a caliper for the duration of treatment. Mouse weight and general health status were observed throughout the experiment to exclude severe side effects caused by the treatment. At day 16, after tumour cell application, mice were killed, tumours were excised and tumour mass was determined. Tumours were fixed with formalin, embedded in paraffin and cut into 5 µm slices. Immunostaining for microvessels and nuclei was performed, according to the manufacturer's protocol, using VECTASTAIN Elite ABC kit (Vector Laboratories Inc., Burlingame, CA, USA), a CD31 antibody (BD Biosciences 553370) and haematoxylin (Sigma-Aldrich), respectively. Four images were obtained from every tumour using a 10× lens. Within each of the fields of view, four high power (40×) images were obtained (image area 0.0355 mm2). These 40× pictures were used to count vessels. Data for tumour volume, tumour weight, microvessel density and mice weight are expressed as means ± SEM. Five and six mice were used in the control and Prt group, respectively. All in vivo experiments were performed according to the legal terms for animal experiments of the local administration (Government of Upper Bavaria). The number of animals used was 12 (the data from 11 mice were used, one died during the experiment). The animals were kept in 12h day/night cycles (200 lux) at a constant temperature of 24°C with free access to water and food (Sniff, Soest, Germany). The results of all studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010).
Statistical analysis
Sigma Plot software with Sigma Stat integration (Systat, Erkrath, Germany) was used for statistical calculations. For comparison of two groups, an unpaired t-test was performed. Three or more groups were compared by one-way anova, followed by multiple comparisons versus control (Bonferroni t-test). All data are expressed as means ± SEM. Graph Pad Prism (Graph Pad Software, La Jolla, CA, USA) was used for curve fitting, and for calculation of the EC50 values for each compound. Nonlinear curve fitting for sigmoidal dose–response curves with a variable slope was used. In Supporting Information Table S1, EC50 values and their respective 95% confidence intervals are presented.
Results
Depolymerization of the microtubule cytoskeleton in HMEC-1
We initially tested whether and at which concentrations Prt and its derivatives act on microtubules in endothelial cells using TubA as a reference compound. To investigate the influence of the compounds on microtubule depolymerization in intact endothelial cells, we performed an immunostaining in HMEC-1 treated with the respective compound for 16 h. The concentrations were chosen in order to achieve complete microtubule depolymerization. Control cells show intact, long and polarized microtubules (Figure 2, left panels), whereas addition of Prt or TubA (middle and right panels of Figure 2) decreased both microtubule polarization and total mass to a similar extent at 10 and 30 nM, whereas 100 nM AU816 were needed to achieve the same level of microtubule breakdown (Supporting Information Figure S1). Doses of either 300 nM or 1 µM of the intermediately potent derivatives, JB337, JB375 and AU954, respectively, had to be administered to induce similar effects (Supporting Information Figure S1A and B). The substances with the lowest potency, AU825, AU815 and JB338, had to be used at concentrations of 30 µM or 10 µM, that is, about three orders of magnitude higher than those of TubA and Prt (Supporting Information Figure S1C), which clearly indicates a loss of activity due to chemical variation of Prt.
Figure 2.
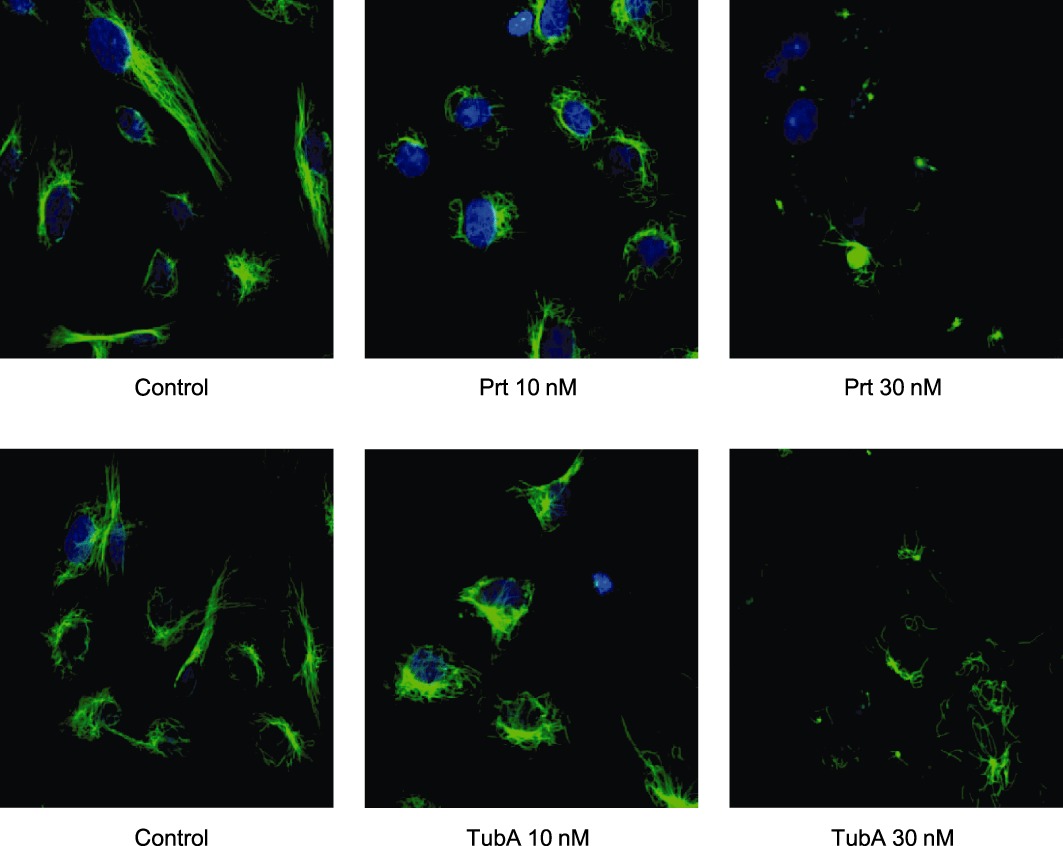
Representative immunofluorescence stainings of microtubules (green) of proliferating HMEC-1 after 16 h treatment with TubA or Prt. Prt and TubA similarly lead to a degradation of the microtubule cytoskeleton in HMEC-1, starting at a concentration of 10 nM. Blue: nuclear staining.
Prt inhibits the proliferation of HMEC-1 to a similar degree as TubA
EC50 values for effects on proliferation of HMEC-1 cells were calculated for each compound including TubA ( Supporting Information Table S1), and a ranking according to potency was established. TubA (EC50 1.2 nM), Prt (EC50 2.3 nM) and AU816 (EC50 4.4 nM) were the most active antiproliferative agents (Figure 3A left panel). The phenyl- and phenoxypretubulysins JB337 and JB375 still exhibited effects at low nanomolar levels (EC50 values: 13.2 nM and 55 nM, respectively), and the 2,3-didehydropretubulysin AU954 was moderately effective with an EC50 of 60 nM (Figure 3A middle panel). The remaining Prt derivatives were three orders of magnitude less potent than TubA and Prt; AU825 and AU815, both Prt derivatives with N- or C-terminal alterations (Figure 1B), had EC50 values of 1.3 µM and 1.6 µM. The central substitution of a triazole ring in JB338 showed a further loss of function (EC50 2.1 µM, Figure 3A right panel).
Figure 3.
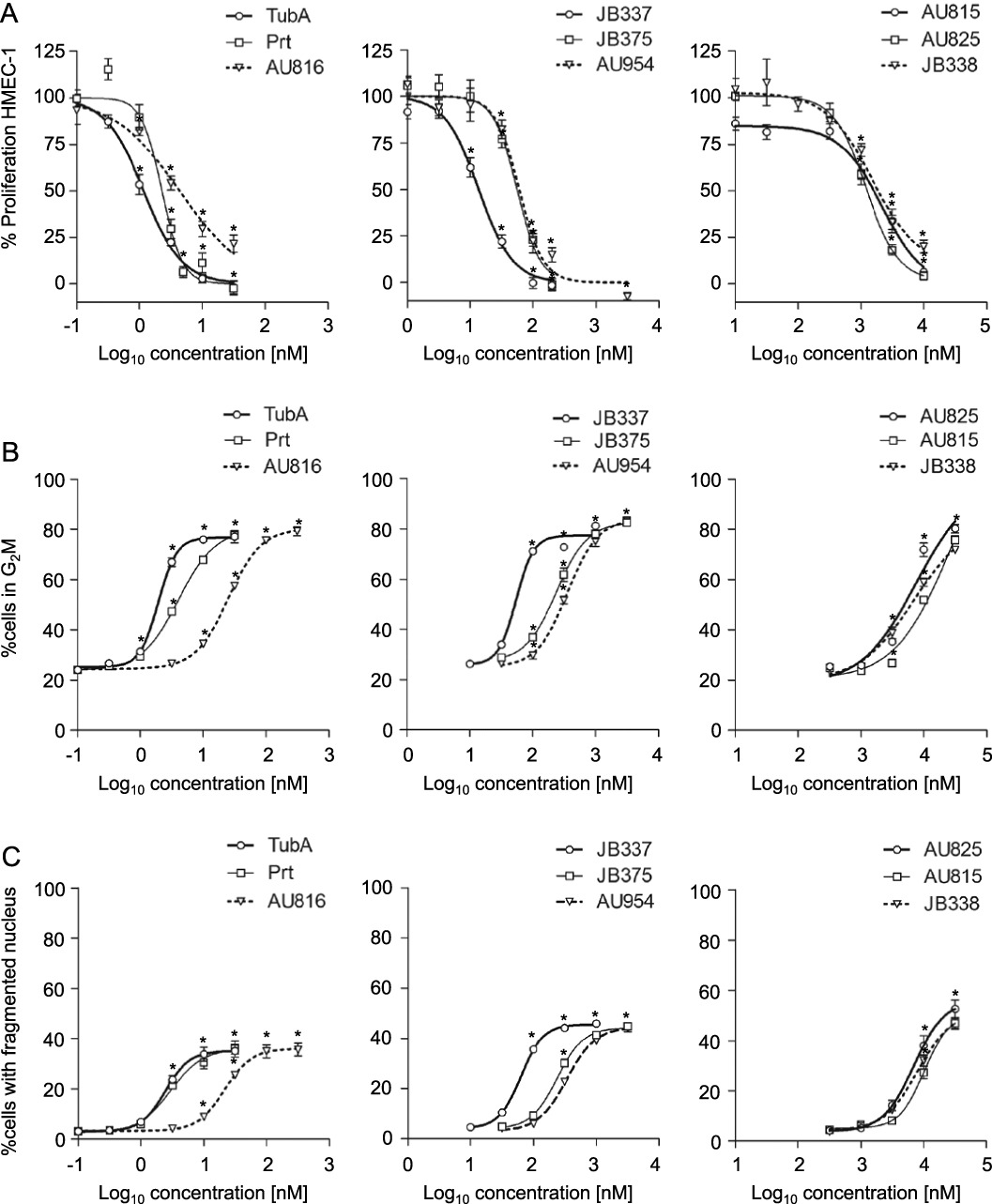
TubA, Prt and Prt derivatives inhibit endothelial cell proliferation (A), cause cell cycle arrest (B) and induce nuclear fragmentation (C). For all three parameters, Prt had a potency comparable to TubA. Compounds were classified into three groups of different potencies (left, middle and right panels). Data are means ± SEM of three independent experiments, *P < 0.05 vs. untreated controls.
Prt and its derivatives cause cell cycle arrest and nuclear fragmentation in HMEC-1 similar to TubA
For investigation of the cell cycle status and induction of nuclear fragmentation as an indicator of cell death in proliferating endothelial cells, HMEC-1 were treated for 48 h with TubA, Prt and its derivatives at the same sets of concentrations as in the proliferation assay. TubA and Prt induced G2/M arrest and nuclear fragmentation at lower concentrations than the other Prt derivatives (Figure 3B and C left panels, Supporting Information Table S1). The rank order of potency of these compounds in this assay totally matched that in the proliferation assay (Figure 3A). As an additional indicator of cell death, membrane integrity was investigated by measuring PI uptake into cells after treatment with the highest concentrations of compounds used. Interestingly, the number of PI positive cells was consistently lower than the number of cells with fragmented nuclei (Supporting Information Figure S2), suggesting that different modes of cell death might be triggered at the same time.
Prt inhibits migration and chemotaxis of endothelial cells at a similar level as TubA
Migration of endothelial cells is a hallmark of angiogenesis. Therefore, the effects of the compounds on both endothelial cell migration into a wounded HUVEC monolayer and on the chemotactic migration towards a serum gradient were evaluated. Prt, TubA and AU816 effectively inhibited the wound closure in a dose-dependent manner with EC50 values of 5.3 nM, 3.4 nM and 11 nM, respectively, whereas JB337, JB375 and AU954 exhibited EC50 values of 26 nM, 260 nM and 200 nM. Chemical variations in the Prt structure as performed for the third group of compounds led to a drastic decrease in anti-migratory potential (Figure 4A). All EC50 values are summarized in Supporting Information Table S1. Figure 4B depicts representative images of scratch assays. Importantly, Prt was only slightly less active at inhibiting wound repair than TubA itself, once more indicating Prt to be a potent alternative to TubA. To determine whether both TubA and Prt inhibit cellular motility per se or only the directional component of the cells, a chemotaxis assay was performed. Figure 4C shows three representative trackings of endothelial cells in a FCS gradient. The directional components of migration (y-forward migration, Euclidean distance) were significantly inhibited by either 3 nM of Prt or TubA (Figure 4D). The non-directional accumulated distance was not significantly affected by the treatment with 3 nM Prt and TubA, indicating that at this concentration, the cells still were able to move. However, as velocity of movement was significantly reduced by treatment (Figure 4D), it can be assumed that overall motility at least begins to be hampered by these concentrations of Prt or TubA, at least in the time frame investigated. Measurement of nuclear fragmentation with TubA or Prt in time-matched experiments showed no dramatic increase in HUVECs at concentrations up to 30 nM (Supporting Information Figure S3), indicating that effects on migration are not primarily due to cytotoxicity.
Figure 4.
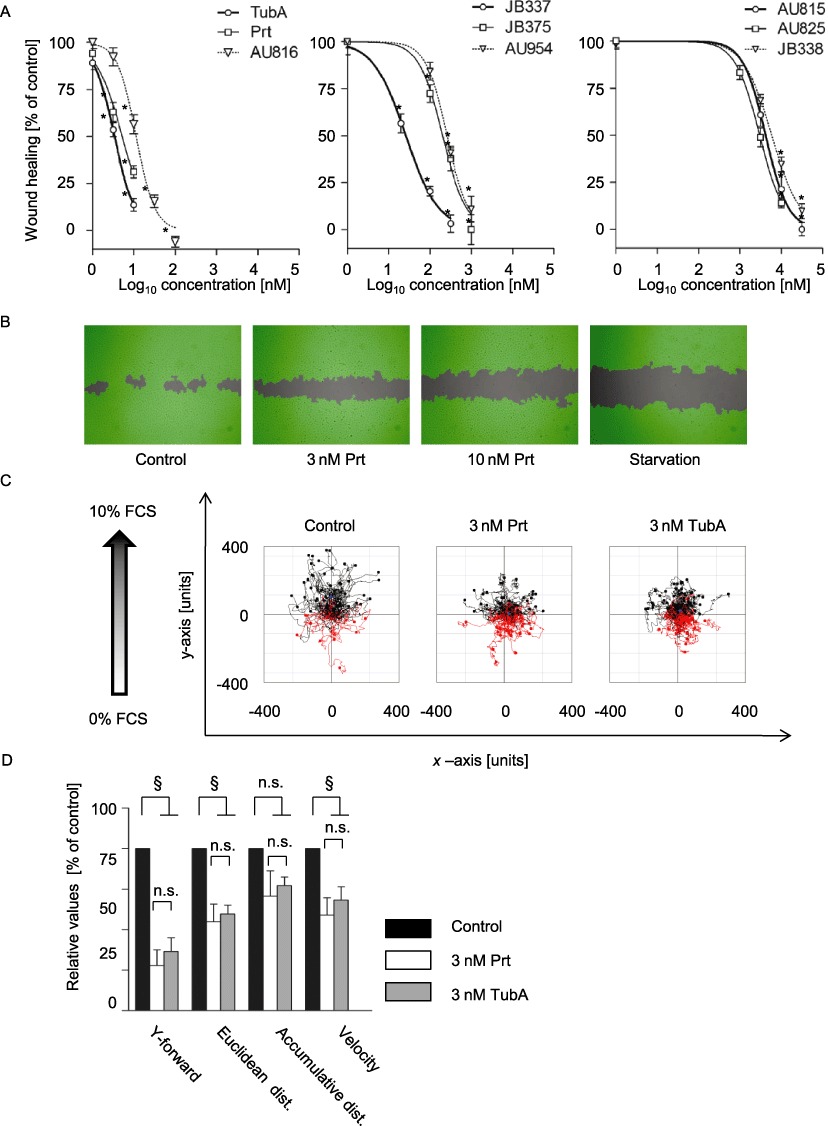
TubA, Prt and Prt derivatives concentration-dependently inhibit endothelial cell migration (A) TubA, Prt and Prt derivatives inhibited wound closure of a scratched HUVEC monolayer with potency similar to the observed effects on proliferation, cell cycle or apoptosis. Data are means ± SEM of three independent experiments, *P < 0.05 vs. untreated controls. (B) Representative images of the wound closure in the absence or presence of pretubulysin or in the absence of growth factors ('starvation’). The cell-free area is grey, the area covered with cells as detected by the imaging software is depicted in green. (C) Representative tracking of the chemotactic movement of endothelial cells in a serum gradient. The starting point of each single cell is placed in the centre of the diagram. Red tracks: cells migrating against the gradient; black tracks: cells migrating along the gradient. In controls, most cells migrated directionally, while cells lost their sense of direction in the presence of Prt or Tub A. (D) Quantitative analysis of the chemotaxis experiments shows reduced parameters of directionality (Y-forward and Euclidean distance), while motility as such (accumulative distance) is not significantly inhibited. Data are means ± SEM of three independent experiments. §Significantly different from controls, P < 0.05; n.s., not significant.
Prt and its derivatives inhibit endothelial tube formation
The tube formation on Matrigel is an in vitro assay that recapitulates many complex features of angiogenesis. By measuring number of node points and total tube length, tube formation was quantitatively determined after 16 h. Again, we could see a totally different inhibitory potential among the derivatives, with Prt (representative images shown in Figure 5A) and TubA being similarly active and the strongest tube formation inhibitors, followed by AU816 (Figure 5B upper left and right panels). TubA and Prt, at about 30 nM, decreased the tube length and node number by 50–70%. The resulting ranking order of potency of the compounds in this assay also accorded with that from all previous experiments. To clarify whether cytotoxic effects could explain the effects of Prt and its derivatives in the tube formation assay, we used similar conditions as in those settings and performed a nuclear fragmentation assay. We could not see any marked cytotoxic effects (Supporting Information Figure S4A). In addition, no substantial amount of dead cells was detected by fluorescence microscopy of PI uptake during tube formation in situ (Supporting Information Figure S4B).
Figure 5.
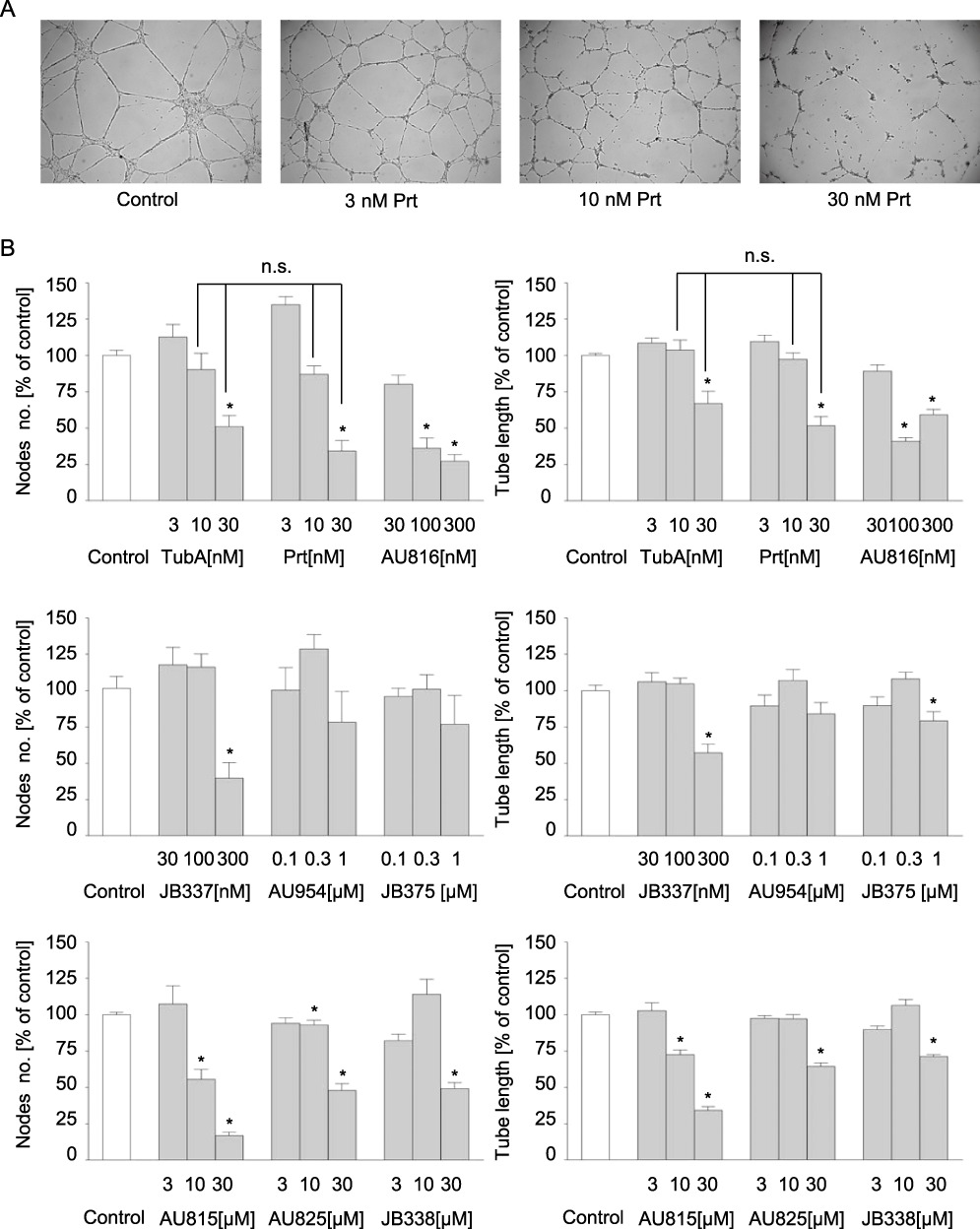
Tub A, Prt and Prt derivatives inhibit endothelial tube formation. (A) Representative pictures of Prt-treated HMEC-1 after 16 h of tube formation, 5× magnification. (B) Quantitative evaluation of branching point number connecting the tubes (left panels) and total tube length (right panels) using WIMASIS software. Data are means ± SEM of three independent experiments. *Significantly different from controls, P≤ 0.05; n.s. not significantly different.
Inhibition of tubulin polymerization in vitro
To get an impression, whether the different efficacies of the compounds might have a cell-based origin (e.g. by different membrane penetration, metabolism or exclusion), all compounds were also tested in a cell-free assay. Among all the compounds tested, TubA, Prt and AU816 (2-desmethylpretubulysin) were the most potent (Figure 6, left panel): 1 µM TubA, as well as 2 µM Prt or AU816 induced comparable residual tubulin polymerization of about 50% or less. To obtain similar effects, a 10-fold higher concentration of all other Prt derivatives had to be used (Figure 6, middle and right panel). JB375 (18%), JB337 (35.7%) and AU954 (37.2%) had an intermediate potency (Figure 6, middle panel), whereas JB338 (40.3%), AU815 (57%) and AU825 (75.15%) had the weakest influence on tubulin polymerization (Figure 6, right panel).
Figure 6.
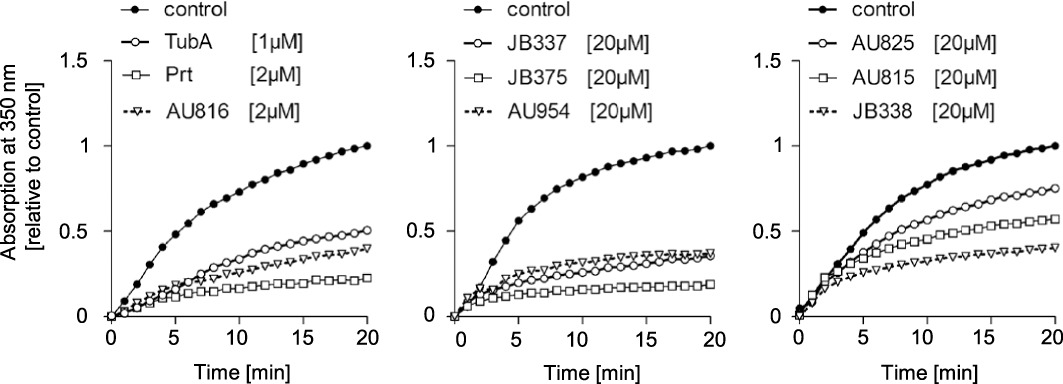
Tubulysin A (TubA), pretubulysin (Prt) and Prt derivatives inhibit in vitro polymerization of tubulin. Purified tubulin was allowed to polymerize in vitro alone or in the presence of selected concentrations of TubA, Prt and Prt derivatives. Changes in relative absorption correlate to tubulin polymerization. Compounds with similar potency are grouped together (left, middle and right panel).
Prt reduces tumour size and vascularization in vivo
In a murine xenograft tumour model with HUH7 cells (hepatocellular carcinoma), Prt, in contrast to solvent treatment, arrested tumour growth (Figure 7A), which finally led to a significant 17-fold reduction in tumour mass 16 days after tumour cell application (Figure 7B) in comparison to the control group. By focusing on the angiogenesis aspects in the immunohistological analysis (Figure 7C), we detected a significantly lower microvessel density in tumour slices of Prt-treated mice in comparison to the control group (Figure 7D). A further indication of reduced tumour vascularization under treatment with Prt is the different phenotype of the tumours: while in controls, the tumours were dark red and filled with blood, the Prt treated tumours were not only smaller, but also strikingly pale (Figure 7E). Importantly, the animal weight did not change significantly during the treatment period either in the Prt or the control group (Figure 7F), which indicates that Prt does not induce acute, severe side effects in mice. In vitro, Prt reduced proliferation of HUH7 cells to a similar degree as TubA with an EC50 of 1.5 nM (Supporting Information Figure S5).
Figure 7.
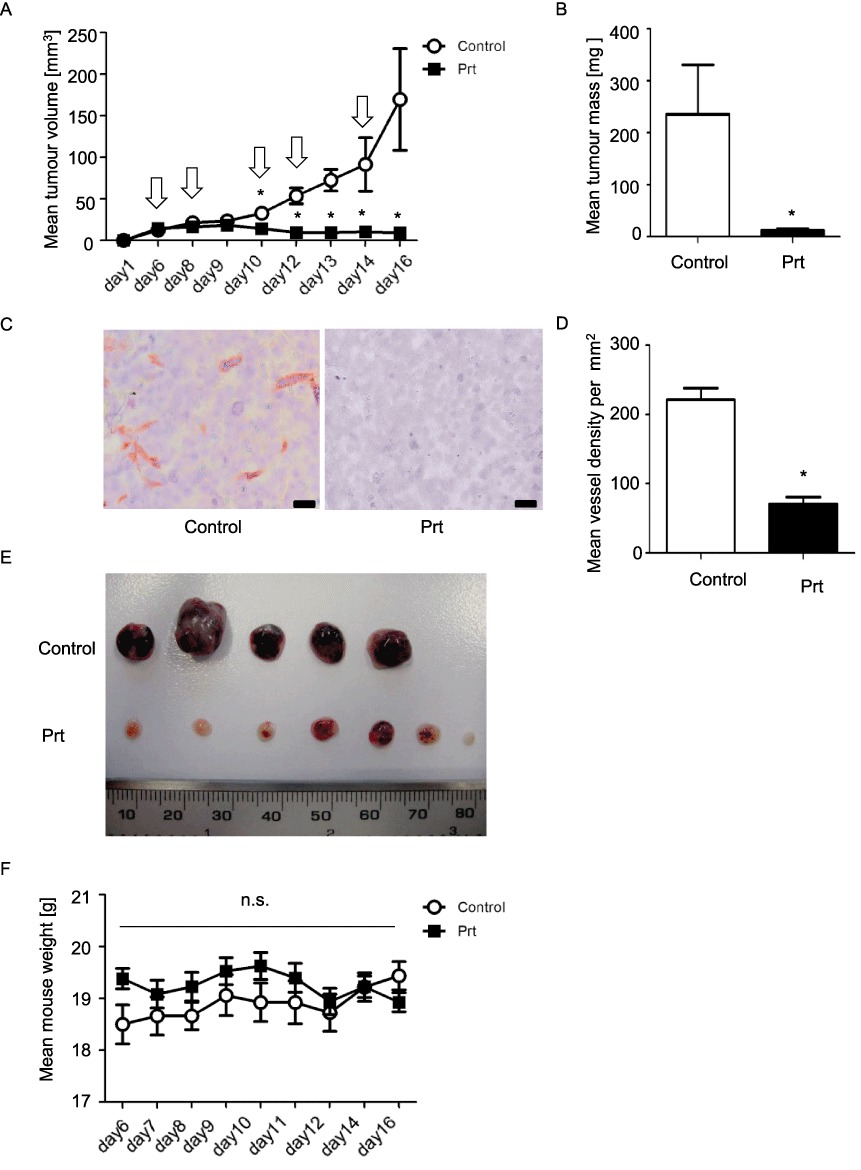
(A) HUH7 tumour growth curves. Starting at day 6 after tumour cell application, mice were treated i.v. either with 0.1 mg·kg−1 Prt (dissolved in 200 µL PBS) or 200 µL PBS alone. Arrows indicate days of treatment. (B) Final HUH7 tumour mass. (C) Microvessel density: slices were stained for CD31 (red) and nuclei (haematoxylin, blue). Scale bars indicate 20 µm. (D) Quantitative evaluation of vessel density. (E) Images of the explanted tumours. Note the dark red, blood filled appearance of the untreated tumours. (F) Animal weight curves. To exclude severe side effects, the weight of the mice was determined routinely throughout the period of treatment. n= 5 mice for Ctrl and six mice for the Prt group, respectively; n= 80 (40× images for both Control and Prt treated groups) for microvessel counting. Data are expressed as mean ± SEM. *P < 0.05; n.s., no significant difference between treatment and control.
Discussion and conclusions
The concept of targeting microtubules with natural compounds or their derivatives has turned out to be extremely attractive, which is mirrored by the plethora of compounds tested in phase I to III clinical trials (Jordan and Wilson, 2004; Kingston, 2009; Dumontet and Jordan, 2010). Established compounds like the taxanes or vinca alkaloids have long since become an integral part of tumour therapy (Pasquier and Kavallaris, 2008). Recently, the spectrum of cells to be targeted by anti-tumour therapy has been extended from tumour cells to endothelial cells in the context of tumour angiogenesis (Bijman et al., 2006; Pasquier et al., 2007). Targeting the endothelium has several advantages as compared to the conventional effort to hit the tumour cells: the genetically relatively stable endothelial cells are, for example, much less susceptible to resistance phenomena than tumour cells, and they can be influenced by a therapeutic regimen of sustained low-dose treatment (‘metronomic chemotherapy’) with the aim of reducing side effects (Kerbel and Kamen, 2004).
In the present study, we used Prt, a chemically accessible precursor of the microtubule-dissociating natural compound tubulysin (Ullrich et al., 2009a), and its derivatives to test for anti-angiogenic effects. TubA itself has already been evaluated as potentially anti-angiogenic agent(Kaur et al., 2006), and a polymeric tubulysin peptide has been shown to reduce tumour growth in colorectal and non-small cell lung carcinoma murine xenograft models (Schluep et al., 2009). The mode of binding of tubulysins to tubulin has not yet been elucidated. Competition studies suggest a binding site close to the peptide site of the vinca domain of β tubulin (Khalil et al., 2006). Due to the limited natural supply of tubulysin and its complicated synthesis, clinical development has, to date, been hampered (Raghavan et al., 2008). By studying the pathway of tubulysin biosynthesis in myxobacteria, Prt was first postulated and then identified as a biosynthetic intermediate (Ullrich et al., 2009a) that is synthetically accessible, and can be easily derivatized (Ullrich et al., 2009b; Burkhart et al., 2011).
A comparison of Prt with TubA showed that they have similar efficacy in all the in vitro and cellular assays employed in this study. This makes Prt an attractive simplified alternative, and an ideal candidate for studying structure-activity relations. To this end, derivatives with alterations in the C- or N-terminus or in the core of the molecule were tested. Surprisingly, the ranking of the efficacy of the compounds was identical in all the test systems: three groups were identified as either highly potent (TubA itself, Prt and AU816), intermediate (JB337, JB375 and AU954) or of low potency (AU825, AU815 and JB338). This ranking was the same in the in vitro tubulin polymerization assay to that in the cellular assays, indicating that the differences in efficacy are not due to uptake kinetics into the cells or to metabolic stability, but to a direct effect on tubulin polymerization. The most likely explanation would be that the derivatives show different affinities towards tubulin. In terms of structure-activity relations, it can be said that an intact piperidine ring at the N-terminus and an intact (2-desmethyl or 2,3-didehydro)-tubuphenylalanine at the C-terminus seem to be inevitable for high pharmacological efficacy, since small alterations at these positions led to a dramatic loss of potency. Replacement of the tubuvalin subunit by simplified spacers (JB337 and JB375) led to a moderate loss of efficacy, while introduction of a triazole deleted its efficacy.
Interestingly, there is growing evidence that the effects of tubulin-targeting drugs in endothelial cells are much more subtle than originally thought. Initially, it was assumed that these drugs inhibited mitosis by damaging the spindle apparatus, whereas now microtubules are considered to be functionally highly relevant for the transport phenomena of vesicles, as well as non-membranous cell components and signalling complexes (Vacca et al., 1999; Hotchkiss et al., 2002; Pasquier et al., 2005; Rothmeier et al., 2009; Lomakin and Nadezhdina, 2010). In addition, they play a critical role in cell polarity (Ueda et al., 1997; McCue et al., 2006), a prerequisite for directional migration. Our group has recently reported that the effects of MT targeting drugs, used at equipotent concentrations, on kinase signalling networks in endothelial cells vary between the different drugs (Rothmeier et al., 2009). In the present study, we found that Prt and its derivatives caused cell cycle arrest in the G2/M phase and cell death after 48 h, which does not come as a surprise. Importantly, at lower concentrations and at earlier time points, before cell death is noticeable, migration and tube formation were reduced, which seems to be primarily due to a loss of directionality. It is interesting to note that the induction of cell death depends on the type of endothelial cells used; while HMEC-1 cells show a three- to fourfold induction of cells with nuclear fragmentation after treatment with 10 or 30 nM Prt or TubA for 48 h, HUVECs are much less sensitive and show only a twofold increase. This latter result accords well with findings from a previous study (Kaur et al., 2006).
As a proof of principle, we applied Prt to an ectopic murine xenograft model with hepatocellular carcinoma cells (HUH7), since hepatocellular carcinomas are the most vascularized solid tumours. TubA itself has already been tested in vivo in colorectal and non-small cell lung carcinoma murine xenograft models (Schluep et al., 2009). The doses used, which were limited by its toxicity, did not induce a significant reduction in tumour growth, and only linking this compound to nanoparticles resulted in smaller tumours and prolonged survival. In our hands, Prt showed no obvious toxic effects at the dose used (0.1 mg·kg−1 every second day), as indicated by the absence of weight loss, but still caused a marked reduction in tumour growth. This might be in part due to direct effects on the proliferation of HUH7 cells, since proliferation of these cells was also inhibited in vitro at the same concentrations as the endothelial cells. However, this compound also has a noticeable, additional inhibitory effect on tumour angiogenesis, as demonstrated by the reduced mean vascular density of the tumours and the macroscopically visible absence of blood in the treated tumours.
It is obvious that a supply of structurally interesting novel microtubule-targeting drugs is not only be desirable from a translational point of view (hopefully presenting better clinical alternatives with lower side effects or better efficacy in resistant tumours), but also from a mechanistic point of view for chemical biology. Novel structures that are ideally chemically accessible and can be derivatized might serve as an important tool to understand the role of microtubules in signalling processes, based on detailed investigations of structure–activity relations.
In conclusion, we demonstrated that Prt and its even more simplified derivatives are highly potent anti-angiogenic compounds that can be reliably obtained by chemical synthesis, and which, therefore, are ideal candidates for clinical development and for chemical biology studies on the function of microtubules.
Acknowledgments
This work was in part supported by the German Research Council (DFG) in the frame of the Research Group FOR1406. The expert technical assistance of Jana Peliskowa, Rita Socher, Bianca Hager and Kerstin Loske is gratefully acknowledged.
Glossary
- HMEC-1
human dermal microvascular endothelial cells
- HUVECs
human umbilical vein endothelial cells
- PI
propidium iodide
- Prt
pretubulysin
- TubA
tubulysin A
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Depolymerization of microtubules by derivatives of pretubulysin in endothelial cells. Blue: nuclear strain, green: microtubules.
Figure S2 Loss of membrane integrity (induction of PI uptake) by tubulysin A, pretubulysin and its derivatives.
Figure S3 Induction of nuclear fragmentation by tubulysin A and pretubulysin in the migration setting.
Figure S4 (A) Induction of nuclear fragmentation by tubulysin A, pretubulysin and its derivatives in a cell density which matches the tube formation setting. (B) Induction of PI uptake (cell death) by pretubulysin in the tube formation setting (upper panel: transmission images, lower panel: PI flourescence).
Figure S5 Effects of tubulysin A and pretubulysin on proliferation of a human hepatocellular tumor cell line (HUH7).
Table S1 Overview over the quantitativefunctional effects (EC50 values) of tubulysin A, pretubulysin and its derivatives. Ninety-five percent confidence intervals in parenthesis.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, et al. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- Bijman MN, van Nieuw Amerongen GP, Laurens N, van Hinsbergh VW, Boven E. Microtubule-targeting agents inhibit angiogenesis at subtoxic concentrations, a process associated with inhibition of Rac1 and Cdc42 activity and changes in the endothelial cytoskeleton. Mol Cancer Ther. 2006;5:2348–2357. doi: 10.1158/1535-7163.MCT-06-0242. [DOI] [PubMed] [Google Scholar]
- Burkhart JL, Müller R, Kazmaier U. Syntheses and evaluation of simplified pretubulysin analogues. Eur J Org Chem. 2011;16:3050–3059. [Google Scholar]
- Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss KA, Ashton AW, Mahmood R, Russell RG, Sparano JA, Schwartz EL. Inhibition of endothelial cell function in vitro and angiogenesis in vivo by docetaxel (Taxotere): association with impaired repositioning of the microtubule organizing center. Mol Cancer Ther. 2002;1:1191–1200. [PubMed] [Google Scholar]
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Kaur G, Hollingshead M, Holbeck S, Schauer-Vukasinovic V, Camalier RF, Domling A, et al. Biological evaluation of tubulysin A: a potential anticancer and antiangiogenic natural product. Biochem J. 2006;396:235–242. doi: 10.1042/BJ20051735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4:423–436. doi: 10.1038/nrc1369. [DOI] [PubMed] [Google Scholar]
- Khalil MW, Sasse F, Lunsdorf H, Elnakady YA, Reichenbach H. Mechanism of action of tubulysin, an antimitotic peptide from myxobacteria. Chembiochem. 2006;7:678–683. doi: 10.1002/cbic.200500421. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston DG. Tubulin-interactive natural products as anticancer agents. J Nat Prod. 2009;72:507–515. doi: 10.1021/np800568j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebl J, Weitensteiner SB, Vereb G, Takacs L, Furst R, Vollmar AM, et al. Cyclin-dependent kinase 5 regulates endothelial cell migration and angiogenesis. J Biol Chem. 2010;285:35932–35943. doi: 10.1074/jbc.M110.126177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin AY, Nadezhdina ES. Dynamics of nonmembranous cell components: role of active transport along microtubules. Biochemistry (Mosc) 2010;75:7–18. doi: 10.1134/s0006297910010025. [DOI] [PubMed] [Google Scholar]
- McCue S, Dajnowiec D, Xu F, Zhang M, Jackson MR, Langille BL. Shear stress regulates forward and reverse planar cell polarity of vascular endothelium in vivo and in vitro. Circ Res. 2006;98:939–946. doi: 10.1161/01.RES.0000216595.15868.55. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- Pasquier E, Kavallaris M. Microtubules: a dynamic target in cancer therapy. IUBMB Life. 2008;60:165–170. doi: 10.1002/iub.25. [DOI] [PubMed] [Google Scholar]
- Pasquier E, Honore S, Pourroy B, Jordan MA, Lehmann M, Briand C, et al. Antiangiogenic concentrations of paclitaxel induce an increase in microtubule dynamics in endothelial cells but not in cancer cells. Cancer Res. 2005;65:2433–2440. doi: 10.1158/0008-5472.CAN-04-2624. [DOI] [PubMed] [Google Scholar]
- Pasquier E, Andre N, Braguer D. Targeting microtubules to inhibit angiogenesis and disrupt tumour vasculature: implications for cancer treatment. Curr Cancer Drug Targets. 2007;7:566–581. doi: 10.2174/156800907781662266. [DOI] [PubMed] [Google Scholar]
- Raghavan B, Balasubramanian R, Steele JC, Sackett DL, Fecik RA. Cytotoxic simplified tubulysin analogues. J Med Chem. 2008;51:1530–1533. doi: 10.1021/jm701321p. [DOI] [PubMed] [Google Scholar]
- Rothmeier AS, Ischenko I, Joore J, Garczarczyk D, Furst R, Bruns CJ, et al. Investigation of the marine compound spongistatin 1 links the inhibition of PKCalpha translocation to nonmitotic effects of tubulin antagonism in angiogenesis. FASEB J. 2009;23:1127–1137. doi: 10.1096/fj.08-117127. [DOI] [PubMed] [Google Scholar]
- Sasse F, Steinmetz H, Heil J, Hofle G, Reichenbach H. Tubulysins, new cytostatic peptides from myxobacteria acting on microtubuli. Production, isolation, physico-chemical and biological properties. J Antibiot (Tokyo) 2000;53:879–885. doi: 10.7164/antibiotics.53.879. [DOI] [PubMed] [Google Scholar]
- Schluep T, Gunawan P, Ma L, Jensen GS, Duringer J, Hinton S, et al. Polymeric tubulysin-peptide nanoparticles with potent antitumor activity. Clin Cancer Res. 2009;15:181–189. doi: 10.1158/1078-0432.CCR-08-1848. [DOI] [PubMed] [Google Scholar]
- Schwartz EL. Antivascular actions of microtubule-binding drugs. Clin Cancer Res. 2009;15:2594–2601. doi: 10.1158/1078-0432.CCR-08-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda M, Graf R, MacWilliams HK, Schliwa M, Euteneuer U. Centrosome positioning and directionality of cell movements. Proc Natl Acad Sci U S A. 1997;94:9674–9678. doi: 10.1073/pnas.94.18.9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich A, Chai Y, Pistorius D, Elnakady YA, Herrmann JE, Weissman KJ, et al. Pretubulysin, a potent and chemically accessible tubulysin precursor from Angiococcus disciformis. Angew Chem Int Ed Engl. 2009a;48:4422–4425. doi: 10.1002/anie.200900406. [DOI] [PubMed] [Google Scholar]
- Ullrich A, Herrmann JE, Müller R, Kazmaier U. Synthesis and biological evaluation of pretubulysin and derivatives. Eur J Org Chem. 2009b;2009:6367–6378. [Google Scholar]
- Vacca A, Iurlaro M, Ribatti D, Minischetti M, Nico B, Ria R, et al. Antiangiogenesis is produced by nontoxic doses of vinblastine. Blood. 1999;94:4143–4155. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.