

Abstract
Macrophages are the dominant leukocyte population found in the tumor microenvironment. Accumulating evidence suggests that these tumor-associated macrophages (TAMs) actively promote all aspects of tumor initiation, growth, and development. However, TAMs are not a single uniform population; instead, they are composed of multiple distinct pro- and anti-tumoral subpopulations with overlapping features depending on a variety of external factors. Defining and differentiating these subsets remains a challenging work-in-progress. These difficulties are apparent in prognostic studies in lung cancer that initially demonstrated conflicting evidence regarding the significance of TAMs but which have more recently clarified and confirmed the clinical importance of these subsets through improved phenotypic capabilities. Thus, these cells represent potential targets for cancer therapeutic initiatives through translational approaches. In this review, we summarize the current understanding of how the tumor microenvironment takes advantage of macrophage plasticity to mold an immunosuppressive population, the phenotypic heterogeneity of TAMs, and their link to prognosis in human lung cancer.
Keywords: Myeloid cells, tumor-associated macrophages, tumor microenvironment, lung cancer, phenotype, prognosis
Introduction
The tumor microenvironment encompasses a wide variety of cells including malignant and non-malignant populations [1]. Non-malignant populations include stromal cells, an expanding vasculature, and a leukocyte infiltrate [2]. Macrophages comprise the dominant portion of the leukocyte population [3]. These “tumor-associated macrophages (TAMs)” have served as an example of the “smoldering” cancer-related inflammation [4], suggested recently to represent the 7th hallmark of cancer [5]. Not only is this inflammation present in the microenvironment of most neoplastic tissues, but there is accumulating evidence suggesting that this inflammation, with myeloid cells as key mediators, actively promotes all aspects of tumor growth and development [6-9].
While the role of myeloid cells in innate and adaptive immunity has been known for over 100 years, their involvement in cancer biology has only been recently recognized [8]. Most of the experimental work until now has focused on the malignant cells that make up tumors, further expounding on the framework proposed by Weinberg and colleagues that neoplastic cells must acquire six biological capabilities, or “hallmarks”, via mutational events in the multistep development of tumors [5,8,10]. Furthermore, and perhaps consequentially, the translation of our improved understanding of the development of cancer to therapeutics has been slower than expected [11-14]. For example, active immunotherapy using cancer vaccines has largely demonstrated poor results to date most likely as a result of the failure to address the local immunosuppression present in the tumor microenvironment that acts as a metaphorical “brick wall” to anti-tumor leukocytes. While cancer antigen-specific T cells can be generated by direct immunization, these cells most often are ineffective in destroying tumors because of their inability to implement their cytotoxic effector functions in the tumor microenvironment [13]. Along with evidence from experimental tumor models that demonstrate the requirement of an inflammatory response for full neoplastic transformation [6,15-19] and from pathologic studies linking macrophage presence in the tumor microenvironment with poor prognosis [20], these clinical experiences point to the reality that the tumor microenvironment, of which macrophages are key mediators, must be addressed for immunotherapy to succeed.
It is unlikely that the multitude of pro-tumoral functions belong to one population of TAMs. For clinical approaches to succeed, these various populations of TAMs in the tumor microenvironment first must be properly identified and characterized. Accurate identification alone is no small task when considering the plasticity of macrophages [21]. Furthermore, the lack of unique cell surface markers between lineages has made it difficult to pinpoint which myeloid cell populations have been studied [8]. To date, research groups have focused on individual myeloid cell populations in isolation, contributing to a field composed of “fragmented information” [8] in which the bigger picture remains muddled. Making matters more complicated is the fact that most of our knowledge about myeloid cell phenotypes in tumors is derived from animal studies; thus, trans-species differences in the expression of certain markers have made the direct translation to human cancer difficult [22]. Once the populations crucial to the natural history of tumorigenesis have been identified, their functionality must be characterized such that we better understand how they differ from non tumor-associated macrophages and how we can target them therapeutically.
Lastly, prognostic studies in numerous types of human cancers have been central to the realization that not all TAM subsets are protumorigenic. The field of lung cancer research is not immune to this debate, as evidence exists supporting both pro and anti-tumor effector functions of macrophages. As translational research efforts push forward in the realm of lung cancer, it will be important that therapeutic interventions specifically target the pro-tumoral subsets demonstrated to be clinically significant through prognostic studies. The ability to accurately phenotype and characterize these various TAM subsets will be critical for success in these undertakings.
Thus, the purpose of this review is three-fold. First, it will summarize our current understanding of how the tumor microenvironment takes advantage of macrophage plasticity to mold an immunosuppressive population. Second, it will describe the phenotypic heterogeneity of macrophages in the tumor microenvironment. Lastly, it will outline the evidence that links TAMs with prognosis in human lung cancer.
Part I Macrophages: adaptation of normal functions in the tumor microenvironment
Macrophages are a heterogenous population of tissue-resident, terminally differentiated, innate myeloid cells that originate from circulating bone marrow-derived monocytic precursors [23]. They demonstrate a high degree of plasticity in response to local cues from the microenvironment and can assume a spectrum of roles required for tissue homeostasis. These numerous roles, ranging from host defense against infectious agents, to tissue development, wound healing, and immune system regulation, are reflected in the wide spectrum of possible phenotypes [21,23-25]. Broadly speaking, the two extremes of possible differentiation states include the classically-activated type 1 macrophages (M1) and the alternatively-activated type 2 macrophages (M2) (Figure 1) [3]. Bacterial moieties such as lipopolysaccharide (LPS), certain Toll-like receptor (TLR) agonism, and the Th1 cytokine interferon-gamma (IFNγ) polarize macrophages along the M1 pathway. Activated M1 macrophages phagocytose and destroy microbes, eliminate tumor cells, present antigen to T cells for an adaptive immune response, and produce high levels of pro-inflammatory cytokines [3,4]. Characterized by their expression of inducible nitric oxide synthase (iNOS), reactive oxygen species (ROS), and production of the Th1-associated cytokine IL-12, M1 macrophages are well-adapted to promote a strong immune response [15]. On the other hand, exposure to Th2 and tumor-derived cytokines such as IL-4, IL-10, IL-13, transforming growth factorbeta (TGF-beta), or prostaglandin E2 (PGE2) promulgates M2 polarization [2]. In general, this population participates in polarized Th2 responses, suppresses Th1 mediatedinflammation through IL-10 and IL-1b production, and promotes all aspects of tissue remodeling and wound healing i.e. digestion of extracellular matrix with matrix metalloproteinases (MMPs), promotion of angiogenesis via vascular endothelial growth factor (VEGF) production, and debris scavenging [22,25,26]. Thus, in contrast to the M1 subtype, M2 macrophages are well-suited to promote tumor development. While the M1/M2 theory provides a useful framework for thinking about macrophage plasticity, it is important to remember that it is overly simplified and neglects the continuum of intermediate phenotypes that macrophages can adopt [27]. However, this apparent dual nature of macrophages with regards to tumor development is increasingly appreciated and has recently been termed the “macrophage balance hypothesis” [3].
Figure 1.
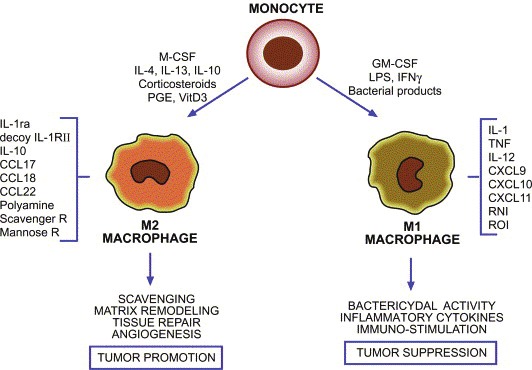
The differentiation pathways of classically-activated M1 macrophages and alternatively-activated M2 macrophages [9].
In line with the “macrophage balance hypothesis” [3], macrophages have been described as a “double-edged sword”, capable of both promoting and opposing tumor development [4]. Macrophages activated by with TLR agonism and/or IFNγ possess the ability to reject tumors cells [28,29]. However, most established tumors lack any substantial immune-mediated limitation on tumor growth. One possible explanation is that something alters or prevents macrophages from killing tumor cells, such as an immunosuppressive microenvironment [15]. Analogous to areas of wound-healing or inflammation that sculpt the phenotypes of local macrophages so that these cells may optimally perform the functions required of them in that context, the tumor microenvironment takes advantage of the normal physiologic functions of macrophages to best serve its interests. Tumor cells coax macrophages to a M2-like phenotype via chemokines and polarizing cytokines, aiding their own escape from destruction, and promoting their development [21,30]. The end result is monocyte/macrophage-mediated modification of every aspect of a tumor’s natural history, from cancer cell proliferation, to cancer cell motility, invasiveness, angiogenesis, immunosuppression, and extraceullular matrix reorganization (Figure 2) [3,4,15,21].
Figure 2.
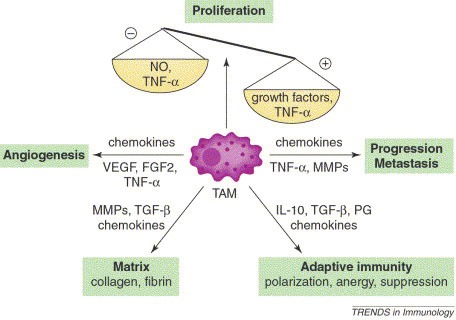
The various pro-tumoral effector functions of TAMs [3].
While literature in the past has equated TAMs with a M2-like phenotype, it has become increasingly clear that TAMs are composed of multiple distinct populations with overlapping features that depend on a variety of factors including location in the microenvironment, stage of the tumor, and type of cancer [3,23]. Pollard et al. have even gone so far as to suggest the existence of unique TAM populations that are educated by microenvironmental cues to adopt a particular phenotype to carry out each of the pro-tumor functions discussed above [15]. Currently, it is still largely unknown whether TAM diversity results from the maturation of unique monocytic precursors or from differences in microanatomical factors [31]. In general however, TAMs isolated from established tumors have a “trophic” immunomodulatory M2-like phenotype similar to those involved in development processes and consistent with the smoldering nature of cancer-related inflammation [21,32]. In fact, it is known that molecular pathways driving TAM polarization can differ considerably in tumors arising at different sites but M2-like skewing reappears as the “recurrent common denominator” [31]. This is in line with the macrophage balance hypothesis that postulates different net effects at different stages of tumor progression: in early stages of carcinogenesis, innate responses are beneficial to the host and involve the activation of effective surveillance by adaptive immunity to eliminate tumor cells, while in established malignancy, TAMs orchestrate “smoldering inflammation” that promotes tumor progression [33]. Typically, this M2-like population lacks expression of reactive nitrogen intermediates, less efficiently presents antigen, displays little tumoricidal activity, and produces angiogenic factors, metalloproteases, and cathepsins [21]. Furthermore, these TAMs promote tumor development via immune and non-immune mechanisms [8].
Non-immune mechanisms directly promote many of the hallmarks ultimately required for tumor development and distal seeding; prominent among these hallmarks are angiogenesis and tumor cell invasion/metastasis [34,35]. Angiogenesis is essential to tumor survival; tumors do not grow beyond 2-3 cubic mm and cannot metastasize unless vascularized [36]. Macrophages are required for the angiogenic switch indicative of a benign-to-malignant transition [34,37,38]. Inhibition of tumor-derived TAM chemoattractants, ablation of TAMs by DNA vaccination, or pharmacological neutralization of TAM-produced proangiogenic molecules have demonstrated impaired tumor angiogenesis in various tumor models [38]. In human breast cancer, TAMs have been shown to cluster in “hot spots” of angiogenesis, primarily in avascular areas [39]. Hypoxia, or cytokines produced secondary to hypoxia, attract macrophages which subsequently up-regulate hypoxia inducible factor 2-alpha (HIF-2α) [40]. HIF-2α then turns on VEGF production, thus promoting new blood vessel formation [41]. In addition, TAMs produce other pro-angiogenic cytokines, including TNFα, MMP-9 (which releases bioactive VEGF from its extracellular matrix-bound latent form), urokinase-type plasminogen activator (uPA), and IL-1 (which up-regulates HIF-2α) [1]. The production of many of these molecules is controlled by the ETS2 transcription factor [37].
Many of the same proteases that are involved in the promotion of angiogenesis are also circumstantially implicated in tumor invasion. In order for invasion to occur, these proteases must destroy the extracellular matrix and stroma. TAMs are implicated as the main producers of proteases, as focal areas of basement membrane penetration at the time of malignant transition have been shown to include high quantities of TAMs [42]. Based on in vivo evidence [43,44], tumor cells are then thought to migrate through these disruptions under the influence of TAM-derived epidermal growth factor (EGF), which is itself produced in response to paracrine tumor-derived colony-stimulating factor (CSF)-1 in a positive feedback cycle. Thus, macrophages are thought to be both the “key that unlocks the gate” [1] to allow tumor cells to escape, as well as the fuel reserves that allow migration to proceed [44].
Immune mechanisms of tumor promotion include elimination of M1 macrophage-mediated innate immune responses, promotion of T regulatory cell (Treg) activity, impairment of T cell activation via direct and indirect mechanisms, and loss of antigen presenting capabilities [8]. M1 macrophages are known to be tumoricidal largely because of the IL-12 dominant cytokine milieu they produce. When TAMs adopt an M2-like phenotype, they are unable to produce the IL-12 required to activate an anti-tumor response mediated by NK cells, Th1 cells, and CTLs [8]. Instead, M2 TAMs produce IL-10 which induces Th2 cell polarization [8]. Th2 cells then produce IL-4 which in turn promotes M2 TAMs in a positive-feedback cycle [45]. Concurrently, M2-derived CCL22 attracts Tregs whose activity is maintained by these high local levels of IL-10 [45-47]. The well-known soluble immunosuppressive cytokines TGF-β and PGE2 [48-50] have also been linked to M2 macrophages. Not only can TAMs produce TGF-β but they can process latent TGF-β to produce its active form [51]. These two cytokines play important roles in tempering T cell activation and tumoricidal capacity. Furthermore, it has recently been found that TAM expression of PD1-ligand1 (PDL1) can directly induce T cell apoptosis after binding its receptor [52]. In an indirect but functionally important manner, TAMs are also thought to inhibit T cell growth by depleting the local concentration of arginine, an essential amino acid for T cell growth, via expression of the enzyme arginase [53]. Lastly, by adopting an M2-like phenotype, macrophages down-regulate their MHC class II molecules and lose most of their ability to link tumor antigen with its respective Agspecific T cell, i.e. they become ineffective antigen presenting cells [26,49].
Part II TAM phenotypes
As evidence accumulates supporting the existence of a complex bi-directional interplay between malignant and non-malignant cells, our conception of tumor structure and organization has changed in appreciation of these dynamics. Namely, tumors are now perceived as complex organ-like structures with regional differences in microenvironments [24]. This complexity is reflected in the wide spectrum of infiltrating macrophages found in tumors that differ functionally and molecularly according to location within the tumor and local cues they encounter there [54,55]. Making matters more complicated, myeloid cell types in tumors are highly related, can express similar markers, and in some instances perform similar functions [38]. Past studies in humans using CD68 as the sole marker of macrophages were not able to distinguish these subsets and it is now common to phenotype based on 2-3 different markers. While these combinations of markers enhance our ability to identify specific subsets, research groups have utilized different combinations of markers to examine individual populations independently, producing “fragmented information” that is difficult to fit into a broader context [8,22].
Macrophages can be phenotyped with four independent but complementary approaches: expression of cell surface markers, expression of transcription factors, production of cytokines, and production of specific enzymes related to function. Common cell-surface targets identified in mouse models of cancer to separate TAM subsets include LY6C, MHC class II molecules, chemokine receptors CX3CR1 and CCR2, CD62L (L-selectin), and TIE2 (angiopoietin receptor) [8,54]. In humans, frequent cell-surface targets include LPS co-receptor (CD14), HLA-DR (MHC class II), CD312, CD115, and most recently, the Fcγ-receptor FcγRIII (CD16) [15]. Subset-specific markers include CD163 and CD204, both scavenger receptors expressed by M2 macrophages, CD301, a galactose-type C-type lectin expressed by M2 macrophages [56,57], and CD206, a scavenger receptor expressed by both tolerogenic macrophages and dendritic cells [26,58]. No unique cell-surface receptor has yet been identified for M1 macrophages [26,58].
Unfortunately, cell surface markers are not static; rather, surface marker profiles are dynamic, often reflecting changes in activation status [59]. Chemokines, cytokines, and enzymes linked to subset-specific functions have helped overcome this challenge. Common targets such as the MMP family of proteases or IL-10 for M2 macrophages and iNOS (inducible nitric oxide synthase), TNF-alpha, or IL-12 for M1 macrophages can be combined with the aforementioned cell-surface markers for more accurate identification. Levels of NO and ROS are controversial markers of M1 macrophages secondary to short half-lives in situ and variable expression in vitro [60]. M2 macrophages also release CCL2 which binds CCR2 on T cells, promoting Th2 responses incapable of tumoricidal activity [61]. Other chemokines include CXCL-10, a Th1 T cell attractant which has been used to identify M1 macrophages, CCL-22, and CCL-18, both which have helped identify M2 macrophages [62,63]. Transcription factors such as IRF-5 and the STAT family have also been used as viable targets given their role in cellular programming and differentiation [64]. IRF-5 has shown promise secondary to its role in up-regulating IFN and IL-10 production in M1 macrophages [65]. Specific members of the STAT protein family have been found to be discriminative markers because of their restricted expression in macrophage subsets: STAT1 in M1 macrophages and STAT3 in M2 macrophages [66].
Much of the recent progress in identifying specific TAM subsets has occurred as a result these evolving complementary approaches to differentiate populations that had previously been lumped together. For example, using advanced microscopy on mouse mammary tumors, two populations of TAMs were identified that differ in their migratory behavior at the tumor-stroma border [24,67]. MCSF-R+CD68+ CD206- dextran -ingesting- TAMs but not M2-type M-CSFR+CD68+ CD206+ dextran-ingesting+ TAMs were associated with migration at the border and invasion into the tumor mass [24,67]. Another study of mouse mammary tumors used a different marker combination in their description of two TAM populations which differed in terms of MHCII expression and intratumoral localization. MHCIIhighCD206- TAMs were more M1-oriented and less able to penetrate hypoxic areas while MHCIIlowCD206+ TAMs were more M2-oriented, able to penetrate hypoxic areas, promote angiogenesis, and even produce IL-10 in human hepatocellular cancer [54,68]. Differences in TAM populations have also been apparent on the level of gene expression, with an invasive MCSF-R+dextran- TAM population that co-migrates with cancer cells showing marked divergence compared to that of MCSF-R+ dextran+ TAMs [32,69]. These differences were not appreciated before assortment along dextran-ingestion but once this filter was applied, all genes of a consensus signature for type II cytokine-associated M2-type macrophages were expressed at a lower level in dextran- TAMs compared to dextran+ TAMs [24]. It has been suggested that each of the aforementioned studies used different marker combinations to describe the same TAM populations [24]. If true, this proposal would involve an invasive M1-like M-CSFR+dextran-CD206-MHCIIhigh TAMs that are hijacked by cancer cells to pave the way for migration and immobile M2-like MCSF-R+ dextran+CD206+MHCIIlow TAMs which are co-opted to promote angiogenesis in hypoxic areas and T cell suppression [24].
The division of tumor-promoting labor by specific TAM populations is gaining momentum, with some groups classifying separate TAM populations based on functional role in the tumor microenvironment (Figure 3) [15]. These functionally distinct TAM subpopulations were defined in animal models of cancer. While they share expression of common macrophage markers including CD11b, F4/80, and CSF-1R, they each express additional markers according to the local cues they encounter in the tumor microenvironment [15]. This classification scheme is still in its infancy; it still remains largely unknown if these subgroups are truly distinct populations and if human correlates exist. Nevertheless, while it remains to be seen how these populations fit into a broader context, this model reinforces the idea of macrophage plasticity and heterogeneity according to local cues. While this knowledge will help focus future investigation on specific subsets, it also has the potential to help clarify past confusion. For instance, the aforementioned findings might help explain why earlier gene expression profiles of total TAM populations showed mixed M1 and M2 characteristics [70,71]. Thus, with better identification of TAM phenotypes, subsets with distinct biological activities can be described.
Figure 3.
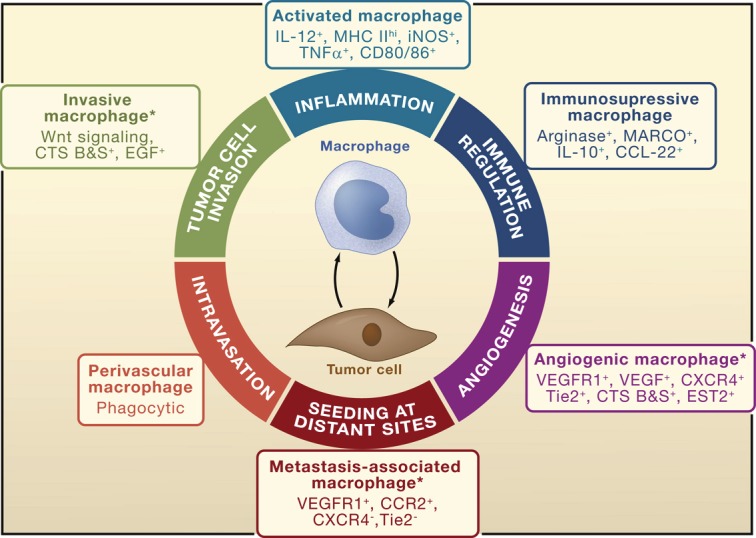
TAM phenotypes according to pro-tumoral function [15].
Ideally, these markers would be unique to macrophage subsets. Unfortunately, this is not the case as many of the macrophage lineage markers are normally expressed by other leukocyte populations [72]. Furthermore such markers as STAT3, CD163, CD204, and MMPs are known to be expressed by tumor cells [22]. As a result, the importance of using three or more concurrent methods of detection to reliably phenotype macrophage subsets cannot be understated. However, until a consensus is reached on accurate, reproducible combinations of markers for specific TAM subsets, separate research groups will continue to describe populations independently (exemplified in the example above) which will contribute to fragmented information that is difficult to fit into a broader context.
Part III TAMs and prognosis in human lung cancer
While it has been known since the mid-19th century that most solid tumors are abundantly populated with leukocytes, their association with clinical outcomes has only recently been appreciated [1]. The presence of extensive TAM infiltration has been shown to correlate with poor prognosis in humans in most studies, lending credence to the idea that TAMs are beneficial for tumor growth and disease progression [20,73]. Furthermore, other parameters of TAM presence such as pro-angiogenic molecules, chemokines, and cytokines have correlated with poor clinical outcome as well [4,15]. However, the literature on this subject is not a consensus; conflicting evidence exists in certain types of cancers. The field of lung cancer research is not immune to this debate, as evidence exists supporting both pro and anti-tumoral functions of macrophages.
A significant portion of the available evidence points to a positive correlation between TAM infiltration and good prognosis. In a study of patients with surgically resected non-small cell lung cancer (NSCLC), increasing tumor islet CD68+ macrophage density and tumor islet/stromal macrophage ratio were significant independent predictors of increased survival. In contrast, increasing stromal macrophage density was an independent predictor of reduced survival [74]. Furthermore, patients with a high tumor islet macrophage density but incomplete resection survived significantly longer than patients with a low tumor islet macrophage density but complete resection. These findings were later confirmed in NSCLC by two independent groups [75,76]. In addition, Dai et al. compared CD68+ macrophages to mature dendritic cells or cytotoxic T cells and found that counting macrophages in the tumor islets and stroma had a higher predictive capacity for patient survival than counting either of the other two lineages [76]. Ohri et al. were the first group to apply additional phenotypic markers to determine the association of specific macrophage subsets with prognosis. In their first study [77], they compared NSCLC patients with extended survival to those with poor survival and found that CD68+ M1 (defined by either HLA-DR+, iNOS+, TNF-alpha+, MRP8/14+) tumor islet density was significantly increased in the extended survival group compared to those with poor survival. Furthermore, the tumor islet:stromal ratio of M1 macrophages was significantly increased compared to CD68+ M2 (defined by CD163+ or VEGF+) in the extended survival group but not the poor survival group. Ma et al. confirmed these findings of significantly higher tumor islet densities of M1 macrophages (defined as CD68+HLA-DR+) in NSCLC patients with extended survival. However, they also demonstrated significantly higher stromal M1 densities in the extended survival group [78].
In addition to TAM density itself, the overexpression of macrophage growth factors, chemokines, or other factors related to TAM-function have also been linked with prognosis. Ohri et al. built upon their analysis of NSCLC patients with extended and poor survival by comparing the levels of cytokines, chemokines, and their respective receptors between the two groups. In one study they found that TNF-α was increased in the tumor islets of patients with extended survival compared to those with poor survival and that increasing tumor islet TNF-α density was a favorable prognostic indicator independent of other predictors such as stage [79]. Interestingly, 100% of the TNF-α+ cells in tumor islets and stroma of patients with extended survival were macrophages or mast cells whereas this figure was less than 50% in patients with poor survival, suggesting that TNF-α levels should be interpreted in the context of the cell types expressing it. In another study, they found that CXCR2-3 and CCR1 were increased in tumor islets of patients with extended survival compared to those with poor survival [80]. Furthermore, there was a positive correlation between macrophage density and CXCR3 and CCR1 expression in tumor islets, suggesting that macrophages were a significant source of these chemokines. Other macrophage -associated chemokine research in NSCLC has been conducted by Nakanishi et al. who found that higher macrophage-derived chemokine (e.g. CCL22) gene expression was correlated with longer disease-free survival and lower risk of recurrence after tumor resection [81]. Another group attempted to discern the molecular factors that influence macrophage distribution but were unable to show that molecular changes of the epidermal growth factor receptor (EGFR) were related to macrophage infiltration, as the degree of infiltration did not significantly correlate with EGFR mutations, gene copy number, or protein expression [75].
Not all evidence supports the correlation between TAMs and good prognosis in human lung cancer. Multiple studies have shown no correlation with prognosis [82-86]. In one of these studies on patients who underwent surgery for NSCLC, CD68+ macrophage content was not associated with stage, nodal status, or survival at 3-years [82]. In another series comparing patients with early versus late-stage NSCLC, CD68+ macrophages were present in similar numbers in tumor islets and stroma regardless of stage and the number of CD68+ cells had no significant relation to survival [83]. Another group investigating tumor angiogenesis as a significant prognostic factor in NSCLC found no statistically significant correlation between tumor stage and CD68+ macrophage count. In a study examining response to chemotherapy in stage IV NSCLC patients, no correlation was found between the number of CD68+ macrophages in either tumor islets or stroma and chemotherapy response [84]. While patients with more TAMs in tumor islets than stroma had significantly better survival than patients who had more TAMs in stroma than islets, no relationship was found between the overall number of TAMs in islets and patient survival. Interestingly, patients with higher overall numbers of stromal TAMs had worse survival than those with lower numbers of stromal TAMs [84]. The last study using CD68+ as the sole macrophage marker found that CD68+ TAMs, M-CSF (the major regulator of the mononuclear phagocytic lineage), or CSFR1 (receptor for M-CSF) in the stroma or islets did not show significant correlation with disease-specific survival, while increasing stromal CD1a+ dendritic cells and CD56+ NK cells correlated with prolonged survival [85]. Studies utilizing other subset-specific TAM markers have also found no correlation with prognosis. One group found that CD68+CD163+ M2 TAM densities were not significantly different in islets or stroma and were not associated with survival time when patients of extended survival were compared to those of poor survival [75]. Lastly, in a study that examined VEGF in patients with NSCLC, no difference was found in survival according to VEGF-C status in stromal macrophages while [87].
Substantial evidence also exists correlating TAMs with poor prognosis in human lung cancer [88-96]. One of the first groups to report such a correlation was Takanami et al. who found significantly lower 5-year survival rates in adenocarcinoma patients with high CD68+ TAM densities compared to low TAM densities [91]. This work was furthered by Chen et al. who found that CD68+ TAM density correlated positively with tumor IL-8 mRNA expression (a potent angiogenic factor) and intratumor microvessel counts but negatively with prognosis in patients with NSCLC [88]. Recently, groups using multiple phenotypic markers to define macrophage subpopulations have also demonstrated poor prognosis with increased TAMs. One study investigated whether TAMs in advanced NSCLC correlated with treatment response to EGFRtyrosine kinase inhibitors and whether these cells predicted survival [90]. They found that greater than 95% CD68+ TAMs were located in tumor stroma and positively co-stained with CD163 suggesting an M2-like phenotype. CD68+CD163+TAM counts were significantly higher in patients with progressive disease, a trend that remained in those with known EGFR mutation status and those with wild type EGFR. High TAM counts were also significantly related to poor progression-free survival and overall survival [90]. Similarly, Ohtaki et al. recently found that expression of the M2 marker CD204 in TAMs was significantly correlated with poor outcome in patients with adenocarcinoma and by a much larger magnitude than TAMs defined only by CD68 [92]. Lastly, in another study of patients with adenocarcinoma, the infiltration of CD68+CD206+ M2 TAMs was significantly associated with p-TNM staging and lymph node metastasis (more strongly than TAMs defined only by CD68+) [93]. Furthermore, higher CD68+CD206+ M2 TAM density was associated with poor prognosis when compared to low M2 TAM density.
TAM-associated factors have also been associated with negative prognostic implications. One group has characterized CD68+ TAM-associated factors in sequential studies of NSCLC patients and linked these factors to stage of disease. In the first study, they demonstrate that CD68+TAMs express high levels of IL-10 and that high levels of IL-10 expression by TAMs are significantly correlated with late stage of disease [97]. In the second study they show that CD68+ IL-10hiTAMs express high levels of MMP9 and VEGF mRNA and that expression of these factors also correlated with late stage of disease [94]. Other groups have also looked at IL-10 expression by TAMs in the context of prognosis. Zeni et al. demonstrate that high IL-10 expression by CD68+ TAMs is a significant independent predictor of advanced tumor stage and is associated with poor overall survival. Furthermore, they suggest that IL-10 expression by TAMs, but not by tumor cells, may play a role in the progression and prognosis of NSCLC because IL-10 expression by tumor cells did not differ between stages [95]. Ohtaki et al. have found that the expression of IL-10 and monocyte chemoattractant protein-1 (MCP-1), both of which are involved in differentiation, accumulation, and migration of M2 macrophages, was significantly correlated with the numbers of CD204+ TAMs within the stroma of lung adenocarcinomas [92]. As discussed above [92], CD204+TAMs were significantly correlated with poor outcome, thus indirectly linking IL-10 and MCP-1 with poor prognosis. Lastly, Ho et al. have demonstrated that TREM-1 expression, an important macrophage-specific molecule for the amplification of the inflammatory response and sepsis biomarker, is increased in CD68+ TAMs isolated from patients with NSCLC. Furthermore, increased TREM-1+ TAMs in tumor tissue of patients with NSCLC were associated with reduced disease-free and overall survival [96].
Conclusion
The importance of the macrophage infiltrate for tumor progression is highlighted by the aforementioned clinical evidence. Furthermore, it has been estimated that 80% of studies that have tried to relate TAM density to prognosis in any type of cancer have found a negative correlation while less than 10% have found a positive correlation [20,73]. Thus, while increased TAM density is usually associated with advanced tumor progression, the question remains as to why the remaining studies do not arrive at the same conclusion. One possible explanation references the “macrophage balance” hypothesis, i.e., the tumors processed for each study were obtained at variable stages thus reflecting the natural variation in function between TAM subsets. Another explanation suggests that many of the macrophage populations used for analysis were not phenotyped with enough precision to distinguish pro- from anti-tumoral TAM subsets. For example, one study on lung cancer initially found that high CD68+ TAM numbers correlated with improved survival. However, when more extensive phenotyping was performed using CD163 and VEGF for M2 and HLA-DR, iNOS, MRP 8/14 and TNF-α for M1, it was determined that better survival was actually associated with a high M1/M2 ratio [22,77]. In fact, most of the earlier studies before the early 2000’s used only one marker of TAMs-CD68. The detection of macrophages on the basis of CD68 does not allow identification of these distinct subsets which is increasingly being recognized [22]. Accordingly, recent studies have started to utilize 2-3 different markers in combination [22].
As the field moves forward with this knowledge and a more thorough toolkit to distinguish TAM subsets, the problem will not be imprecise phenotyping of TAM populations. Instead, the problem will be the opposite, in which overly specific identification with various combinations of 3-4 markers by individual groups will prevent comparison of equivalent TAM populations across the spectrum of human cancers. This concern may be warranted even today, as it is already challenging to integrate the information from different studies on specific populations into a broader context. While no consensus solution has been reached given the early stages of investigation, it is worth revisiting as more progress is made in the near future.
Acknowledgements
JQ and EE developed the idea for the review. JQ wrote the paper. JQ and EE reviewed and helped revise the initial drafts. The project described was supported by the National Center for Research Resources, Grant TL1RR024133, and is now at the National Center for Advancing Translational Sciences, Grant TL1R000138. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- 1.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 2.Siveen KS, Kuttan G. Role of macrophages in tumour progression. Immunol Lett. 2009;123:97–102. doi: 10.1016/j.imlet.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 3.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 4.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg R. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 8.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 11.Waldmann TA. Immunotherapy: past, present and future. Nat Med. 2003;9:269–277. doi: 10.1038/nm0303-269. [DOI] [PubMed] [Google Scholar]
- 12.Waldmann TA. Effective cancer therapy through immunomodulation. Annu Rev Med. 2006;57:65–81. doi: 10.1146/annurev.med.56.082103.104549. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gattinoni L, Powell DJ Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Oshima H, Hioki K, Popivanova BK, Oguma K, Van Rooijen N, Ishikawa TO, Oshima M. Prostaglandin E(2) signaling and bacterial infection recruit tumor-promoting macrophages to mouse gastric tumors. Gastroenterology. 2011;140:596–607. doi: 10.1053/j.gastro.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Popivanova BK, Kostadinova FI, Furuichi K, Shamekh MM, Kondo T, Wada T, Egashira K, Mukaida N. Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res. 2009;69:7884–7892. doi: 10.1158/0008-5472.CAN-09-1451. [DOI] [PubMed] [Google Scholar]
- 19.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 21.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 22.Heusinkveld M, van der Burg SH. Identification and manipulation of tumor associated macrophages in human cancers. J transl med. 2011;9:216. doi: 10.1186/1479-5876-9-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laoui D, Van Overmeire E, Movahedi K, Van den Bossche J, Schouppe E, Mommer C, Nikolaou A, Morias Y, De Baetselier P, Van Ginderachter JA. Mononuclear phagocyte heterogeneity in cancer: different subsets and activation states reaching out at the tumor site. Immunobiology. 2011;216:1192–1202. doi: 10.1016/j.imbio.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 25.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 26.Heusinkveld M, de Vos vS, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJ. M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J Immunol. 2011;187:1157–1165. doi: 10.4049/jimmunol.1100889. [DOI] [PubMed] [Google Scholar]
- 27.Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol. 2009;70:325–330. doi: 10.1016/j.humimm.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Fidler IJ, Schroit AJ. Recognition and destruction of neoplastic cells by activated macrophages: discrimination of altered self. Biochim Biophys Acta. 1988;948:151–173. doi: 10.1016/0304-419x(88)90009-1. [DOI] [PubMed] [Google Scholar]
- 29.Eubank TD, Roberts RD, Khan M, Curry JM, Nuovo GJ, Kuppusamy P, Marsh CB. Granulocyte Macrophage Colony-Stimulating Factor Inhibits Breast Cancer Growth and Metastasis by Invoking an Anti-Angiogenic Program in Tumor-Educated Macrophages. Cancer Research. 2009;69:2133–2140. doi: 10.1158/0008-5472.CAN-08-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, Traver D, van Rooijen N, Weissman IL. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22:33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Ojalvo LS, Whittaker CA, Condeelis JS, Pollard JW. Gene expression analysis of macrophages that facilitate tumor invasion supports a role for Wnt-signaling in mediating their activity in primary mammary tumors. J Immunol. 2010;184:702–712. doi: 10.4049/jimmunol.0902360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222:155–161. doi: 10.1111/j.1600-065X.2008.00607.x. [DOI] [PubMed] [Google Scholar]
- 34.Lin EY. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 35.Qian B. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE. 2009;4:e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Folkman J. Tumor angiogenesis. Adv Cancer Res. 1985;43:175–203. doi: 10.1016/s0065-230x(08)60946-x. [DOI] [PubMed] [Google Scholar]
- 37.Zabuawala T, Taffany DA, Sharma SM, Merchant A, Adair B, Srinivasan R, Rosol TJ, Fernandez S, Huang K, Leone G, Ostrowski MC. An Ets2-Driven Transcriptional Program in Tumor-Associated Macrophages Promotes Tumor Metastasis. Cancer Research. 2010;70:1323–1333. doi: 10.1158/0008-5472.CAN-09-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coffelt SB, Lewis CE, Naldini L, Brown JM, Ferrara N, De Palma M. Elusive identities and overlapping phenotypes of proangiogenic myeloid cells in tumors. Am J Pathol. 2010;176:1564–1576. doi: 10.2353/ajpath.2010.090786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leek RD. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996;56:4625–4629. [PubMed] [Google Scholar]
- 40.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 41.Lewis CE, Hughes R. Inflammation and breast cancer. Microenvironmental factors regulating macrophage function in breast tumours: hypoxia and angiopoietin-2. Breast Cancer Res. 2007;9:209. doi: 10.1186/bcr1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growth factor, CSF-1, in mammary gland development and cancer. J Mammary Gland Biol Neoplasia. 2002;7:147–162. doi: 10.1023/a:1020399802795. [DOI] [PubMed] [Google Scholar]
- 43.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J. A Paracrine Loop between Tumor Cells and Macrophages Is Required for Tumor Cell Migration in Mammary Tumors. Cancer Research. 2004;64:7022–7029. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 44.Wyckoff JB, Wang Y, Lin EY, Li J, Goswami S, Stanley ER, Segall JE, Pollard JW, Condeelis J. Direct Visualization of Macrophage-Assisted Tumor Cell Intravasation in Mammary Tumors. Cancer Research. 2007;67:2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 45.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murai M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nature Immunol. 2009;10:1178–1184. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 48.Quatromoni JG, Wang Y, Vo DD, Morris LF, Jazirehi AR, McBride W, Chatila T, Koya RC, Economou JS. T cell receptor (TCR)-transgenic CD8 lymphocytes rendered insensitive to transforming growth factor beta (TGFbeta) signaling mediate superior tumor regression in an animal model of adoptive cell therapy. J Transl Med. 2012;10:127. doi: 10.1186/1479-5876-10-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eruslanov E, Daurkin I, Ortiz J, Vieweg J, Kusmartsev S. Pivotal Advance: Tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE(2) catabolism in myeloid cells. J Leukoc Biol. 2010;88:839–848. doi: 10.1189/jlb.1209821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eruslanov E, Daurkin I, Vieweg J, Daaka Y, Kusmartsev S. Aberrant PGE2 metabolism in bladder tumor microenvironment promotes immunosuppressive phenotype of tumor-infiltrating myeloid cells. Int Immunopharmacol. 2011;11:848–855. doi: 10.1016/j.intimp.2011.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chong H, Vodovotz Y, Cox GW, Barcellos-Hoff M. Immunocytochemical localization of latent transforming growth factor-[beta] activation by stimulated macrophages. J Cell Physiol. 1999;178:275–283. doi: 10.1002/(SICI)1097-4652(199903)178:3<275::AID-JCP1>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 52.Kuang DM. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206:1327–1337. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodriguez PC. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigenspecific T-cell responses. Cancer Res. 2004;64:5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 54.Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, Mack M, Pipeleers D, In't Veld P, De Baetselier P, Van Ginderachter JA. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 55.Pettersen JS, Fuentes-Duculan J, Suarez-Farinas M, Pierson KC, Pitts-Kiefer A, Fan L. Tumor-associated macrophages in the cutaneous SCC microenvironment are heterogeneously activated. J Invest Dermatol. 2011;131:1322–1330. doi: 10.103/jid.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prokop S, Heppner FL, Goebel HH, Stenzel W. M2 Polarized Macrophages and Giant Cells Contribute to Myofibrosis in Neuromuscular Sarcoidosis. The American Journal of Pathology. 2011;178:1279–1286. doi: 10.1016/j.ajpath.2010.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raes G, Brys L, Dahal BK, Brandt J, Grooten J, Brombacher F, Vanham G, Noël W, Bogaert P, Boonefaes T, Kindt A, Van den Bergh R, Leenen PJM, De Baetselier P, Ghassabeh GH. Macrophage galactose-type C-type lectins as novel markers for alternatively activated macrophages elicited by parasitic infections and allergic airway inflammation. Journal of Leukocyte Biology. 2005;77:321–327. doi: 10.1189/jlb.0304212. [DOI] [PubMed] [Google Scholar]
- 58.Verreck FA, de BT, Langenberg DM, van dZ, Ottenhoff TH. Phenotypic and functional profiling of human proinflammatory type-1 and anti-inflammatory type-2 macrophages in response to microbial antigens and IFN-gamma- and CD40L-mediated costimulation. J Leukoc Biol. 2006;79:285–293. doi: 10.1189/jlb.0105015. [DOI] [PubMed] [Google Scholar]
- 59.Pander J, Heusinkveld M, van dS, Jordanova ES, Baak-Pablo R, Gelderblom H. Activation of Tumor-Promoting Type 2 Macrophages by EGFR-Targeting Antibody Cetuximab. Clin Cancer Res. 2011;17:5668–5673. doi: 10.1158/1078-0432.CCR-11-0239. [DOI] [PubMed] [Google Scholar]
- 60.Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol. 2011;89:557–563. doi: 10.1189/jlb.0710409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 62.Chen J, Yao Y, Gong C, Yu F, Su S, Chen J. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell. 2011;19:541–555. doi: 10.1016/j.ccr.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110:4319–4330. doi: 10.1182/blood-2007-02-072587. [DOI] [PubMed] [Google Scholar]
- 64.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 65.Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 66.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nature Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Egeblad M, Ewald AJ, Askautrud HA, Truitt ML, Welm BE, Bainbridge E, Peeters G, Krummel MF, Werb Z. Visualizing stromal cell dynamics in different tumor microenvironments by spinning disk confocal microscopy. Disease Models & Mechanisms. 2008;1:155–167. doi: 10.1242/dmm.000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuang DM, Wu Y, Chen N, Cheng J, Zhuang SM, Zheng L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood. 2007;110:587–595. doi: 10.1182/blood-2007-01-068031. [DOI] [PubMed] [Google Scholar]
- 69.Ghassabeh GH, De Baetselier P, Brys L, Noel W, Van Ginderachter JA, Meerschaut S, Beschin A, Brombacher F, Raes G. Identification of a common gene signature for type II cytokine-associated myeloid cells elicited in vivo in different pathologic conditions. Blood. 2006;108:575–583. doi: 10.1182/blood-2005-04-1485. [DOI] [PubMed] [Google Scholar]
- 70.Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW, Johnson RS. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70:7465–7475. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, Okada M, Balazs M, Adany R, Shibata T, Takami T. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol. 2008;83:1136–1144. doi: 10.1189/jlb.0907611. [DOI] [PubMed] [Google Scholar]
- 72.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 73.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 74.Welsh TJ, Green RH, Richardson D, Waller DA, O'Byrne KJ, Bradding P. Macrophage and mastcell invasion of tumor cell islets confers a marked survival advantage in non-small-cell lung cancer. J. Clin. Oncol. 2005;23:8959–8967. doi: 10.1200/JCO.2005.01.4910. [DOI] [PubMed] [Google Scholar]
- 75.Kim DW, Min HS, Lee KH, Kim YJ, Oh DY, Jeon YK, Lee SH, Im SA, Chung DH, Kim YT, Kim TY, Bang YJ, Sung SW, Kim JH, Heo DS. High tumour islet macrophage infiltration correlates with improved patient survival but not with EGFR mutations, gene copy number or protein expression in resected non-small cell lung cancer. Br J Cancer. 2008;98:1118–1124. doi: 10.1038/sj.bjc.6604256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dai F, Liu L, Che G, Yu N, Pu Q, Zhang S, Ma J, Ma L, You Z. The number and microlocalization of tumor-associated immune cells are associated with patient's survival time in non-small cell lung cancer. BMC Cancer. 2010;10:220. doi: 10.1186/1471-2407-10-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur Respir J. 2009;33:118–126. doi: 10.1183/09031936.00065708. [DOI] [PubMed] [Google Scholar]
- 78.Ma J, Liu L, Che G, Yu N, Dai F, You Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer. 2010;10:112. doi: 10.1186/1471-2407-10-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. Tumour necrosis factor-alpha expression in tumour islets confers a survival advantage in non-small cell lung cancer. BMC Cancer. 2010;10:323. doi: 10.1186/1471-2407-10-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. Chemokine receptor expression in tumour islets and stroma in non-small cell lung cancer. BMC Cancer. 2010;10:172. doi: 10.1186/1471-2407-10-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nakanishi T, Imaizumi K, Hasegawa Y, Kawabe T, Hashimoto N, Okamoto M, Shimokata K. Expression of macrophage-derived chemokine (MDC)/CCL22 in human lung cancer. Cancer Immunol Immunother. 2006;55:1320–1329. doi: 10.1007/s00262-006-0133-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Toomey D, Smyth G, Condron C, Kelly J, Byrne AM, Kay E, Conroy RM, Broe P, Bouchier-Hayes D. Infiltrating immune cells, but not tumour cells, express FasL in non-small cell lung cancer: No association with prognosis identified in 3-year follow-up. Int J Cancer. 2003;103:408–412. doi: 10.1002/ijc.10836. [DOI] [PubMed] [Google Scholar]
- 83.Miotto D, Lo Cascio N, Stendardo M, Querzoli P, Pedriali M, De Rosa E, Fabbri LM, Mapp CE, Boschetto P. CD8+ T cells expressing IL-10 are associated with a favourable prognosis in lung cancer. Lung Cancer. 2010;69:355–360. doi: 10.1016/j.lungcan.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 84.Kawai O, Ishii G, Kubota K, Murata Y, Naito Y, Mizuno T, Aokage K, Saijo N, Nishiwaki Y, Gemma A, Kudoh S, Ochiai A. Predominant infiltration of macrophages and CD8(+) T Cells in cancer nests is a significant predictor of survival in stage IV nonsmall cell lung cancer. Cancer. 2008;113:1387–1395. doi: 10.1002/cncr.23712. [DOI] [PubMed] [Google Scholar]
- 85.Al-Shibli K, Al-Saad S, Donnem T, Persson M, Bremnes RM, Busund LT. The prognostic value of intraepithelial and stromal innate immune system cells in non-small cell lung carcinoma. Histopathology. 2009;55:301–312. doi: 10.1111/j.1365-2559.2009.03379.x. [DOI] [PubMed] [Google Scholar]
- 86.Tataroglu C, Kargi A, Ozkal S, Esrefoglu N, Akkoclu A. Association of macrophages, mast cells and eosinophil leukocytes with angiogenesis and tumor stage in non-small cell lung carcinomas (NSCLC) Lung Cancer. 2004;43:47–54. doi: 10.1016/j.lungcan.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 87.Ogawa E, Takenaka K, Yanagihara K, Kurozumi M, Manabe T, Wada H, Tanaka F. Clinical significance of VEGF-C status in tumour cells and stromal macrophages in non-small cell lung cancer patients. Br J Cancer. 2004;91:498–503. doi: 10.1038/sj.bjc.6601992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen JJ, Yao PL, Yuan A, Hong TM, Shun CT, Kuo ML, Lee YC, Yang PC. Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer. Clin Cancer Res. 2003;9:729–737. [PubMed] [Google Scholar]
- 89.Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, Tsai MF, Chen CH, Yang PC. Tumorassociated macrophages: the double-edged sword in cancer progression. J. Clin. Oncol. 2005;23:953–964. doi: 10.1200/JCO.2005.12.172. [DOI] [PubMed] [Google Scholar]
- 90.Chung FT, Lee KY, Wang CW, Heh CC, Chan YF, Chen HW, Kuo CH, Feng PH, Lin TY, Wang CH, Chou CL, Chen HC, Lin SM, Kuo HP. Tumor-associated macrophages correlate with response to epidermal growth factor receptor-tyrosine kinase inhibitors in advanced non-small cell lung cancer. Int J Cancer. 2012;131:E227–35. doi: 10.1002/ijc.27403. [DOI] [PubMed] [Google Scholar]
- 91.Takanami I, Takeuchi K, Kodaira S. Tumor-associated macrophage infiltration in pulmonary adenocarcinoma: association with angiogenesis and poor prognosis. Oncology. 1999;57:138–142. doi: 10.1159/000012021. [DOI] [PubMed] [Google Scholar]
- 92.Ohtaki Y, Ishii G, Nagai K, Ashimine S, Kuwata T, Hishida T, Nishimura M, Yoshida J, Takeyoshi I, Ochiai A. Stromal macrophage expressing CD204 is associated with tumor aggressiveness in lung adenocarcinoma. J Thorac Oncol. 2010;5:1507–1515. doi: 10.1097/JTO.0b013e3181eba692. [DOI] [PubMed] [Google Scholar]
- 93.Zhang B, Yao G, Zhang Y, Gao J, Yang B, Rao Z, Gao J. M2-polarized tumor-associated macrophages are associated with poor prognoses resulting from accelerated lymphangiogenesis in lung adenocarcinoma. Clinics. 2011;66:1879–1886. doi: 10.1590/S1807-59322011001100006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang R, Zhang J, Chen S, Lu M, Luo X, Yao S, Liu S, Qin Y, Chen H. Tumor-associated macrophages provide a suitable microenvironment for non-small lung cancer invasion and progression. Lung Cancer. 2011;74:188–196. doi: 10.1016/j.lungcan.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 95.Zeni E, Mazzetti L, Miotto D, Lo Cascio N, Maestrelli P, Querzoli P, Pedriali M, De Rosa E, Fabbri LM, Mapp CE, Boschetto P. Macrophage expression of interleukin-10 is a prognostic factor in nonsmall cell lung cancer. Eur Respir J. 2007;30:627–632. doi: 10.1183/09031936.00129306. [DOI] [PubMed] [Google Scholar]
- 96.Ho CC, Liao WY, Wang CY, Lu YH, Huang HY, Chen HY, Chan WK, Chen HW, Yang PC. TREM-1 expression in tumor-associated macrophages and clinical outcome in lung cancer. Am J Respir Crit Care Med. 2008;177:763–770. doi: 10.1164/rccm.200704-641OC. [DOI] [PubMed] [Google Scholar]
- 97.Wang R, Lu M, Zhang J, Chen S, Luo X, Qin Y, Chen H. Increased IL-10 mRNA expression in tumor-associated macrophage correlated with late stage of lung cancer. J Exp Clin Cancer Res. 2011;30:62. doi: 10.1186/1756-9966-30-62. [DOI] [PMC free article] [PubMed] [Google Scholar]