

Abstract
Oculocutaneous albinism (OCA) is a group of inherited disorders characterized by defective melanin biosynthesis. OCA1, the most common and severe form, is caused by mutations in the tyrosinase (TYR) gene. OCA4, caused by mutations in the SLC45A2 gene, has frequently been reported in the Japanese population. To determine the mutational spectrum in Korean OCA patients, 12 patients were recruited. The samples were first screened for TYR mutations, and negative samples were screened for SLC45A2 mutations. OCA1 was confirmed in 8 of 12 (66.7%) patients, and OCA4 was diagnosed in 1 (8.3%) patient. In the OCA1 patients, a total of 6 distinct TYR mutations were found in 15 of 16 (93.8%) alleles, all of which had been previously reported. Out of the 6 alleles, c.929insC was the most frequently detected (31.3%), and was mainly associated with OCA1A phenotypes. Other TYR mutations identified included c.1037-7T>A/c.1037-10delTT, p.D383N, p.R77Q and p.R299H. These largely overlapped with mutations found in Japanese and Chinese patients. The SLC45A2 gene analysis identified 1 novel mutation, p.D93N, in 1 patient. This study has provided information on the mutation spectrum in Korean OCA patients, and allows us to estimate the relative frequencies of OCA1 and OCA4 in Korea.
Keywords: oculocutaneous albinism, tyrosinase, TYR, SLC45A2, MATP
Introduction
Oculocutaneous albinism (OCA) is a group of inherited disorders characterized by defective melanin biosynthesis, which results in congenital hypopigmentation of the hair, skin and eyes. At least 16 genes have been found to be associated with various forms of albinism. Mutations in these genes, which regulate the processes of melanin synthesis and distribution, mcause various types of OCA (1), which are classified into 2 groups: non-syndromic and syndromic. Non-syndromic OCA is subdivided into 4 forms based on clinical and genetic findings as follows: OCA1, OCA2, OCA3 and OCA4. OCA1, the most severe, is caused by mutations in the tyrosinase (TYR) gene and is further divided into two types; OCA1A and OCA1B. OCA1A is characterized by a life-long absence of melanin pigmentation, whereas patients with OCA1B may develop certain cutaneous and ocular pigmentation with age. The other forms are milder clinical phenotypes revealing some pigment accumulation over time, and include OCA2, OCA3 and OCA4, caused by mutations in the OCA2, TRYP1 and SLC45A2 genes, respectively (1).
The prevalence of the various forms of OCA varies widely in different populations. OCA1 is the most common, with a prevalence of 1 per 40,000 individuals in most populations (2). Although OCA2 and OCA3 are common in African OCA patients, these forms are uncommon in Caucasian and Asian populations (3,4). OCA4 has been reported to be the second most common form in Far East Asian populations, with a prevalence of 24–27% in Japanese patients (5,6) and 12.6% in Chinese patients (7). In addition, it is well known that a certain mutation has been commonly identified in a specific ethnic group due to a founder effect in OCA. Although different forms of OCA are caused by mutations in a variety of genes, clinical phenotypes are not always distinguishable; therefore, molecular diagnosis has become a useful and essential tool for genetic counseling.
Only 5 studies of mutational screenings involving 13 Korean OCA patients exist (8,9). Of these studies, causative mutations in TYR and SLC45A2 were found in 6 (46.2%) patients and 1 (7.7%) patient, respectively. The present study included 11 Korean OCA patients and their parents, who were referred to the Genetics Clinic at Ajou University Hospital. We analyzed the mutations found in TYR and SLC45A2 to determine their mutation spectrums. The clinical features of the mutations were compared and analyzed to determine genotype-phenotype correlations.
Materials and methods
Study patients and clinical evaluations
Korean OCA patients and their parents were recruited for the study at the Genetics Clinic of Ajou University Hospital between December 2004 and May 2011. The study protocol was reviewed and approved by the Institutional Review Board of the Ajou University Hospital, and written informed consent was obtained from all subjects or from their parents. The diagnostic inclusion criteria were based on the presence of the following clinical characteristics: varying degrees of hypopigmentation of the skin and body hair, and abnormal opthalmological findings, including photophobia, nystagmus, reduced visual acuity, strabismus, iris translucency, fundus hypopigmentation or foveal hypoplasia. Patients with OCA forms, including Hermansky-Pudlak, Chediak-Higashi and Griscelli syndromes were excluded based on clinical features. The typical clinical features in the OCA patients were analyzed to predict genotype-phenotype correlations in the cases with detected mutations.
If OCA1 was determined by TYR gene analysis, OCA1 patients were further classified into 2 types: OCA1A and OCA1B. OCA1A was defined as the presence of white hair and skin throughout life, and OCA1B was defined as the presence of white hair at birth, particularly eyelash hair, that subsequently developed pigmentation in the first decade of life.
Mutation identification
Genomic DNA was isolated from the peripheral blood leukocytes of the study subjects using a DNA isolation kit (Qiagen GmbH, Hilden, Germany). The DNA samples were first screened for TYR mutations. All 5 coding exons and the intronic flanking regions of the TYR gene were amplified using a polymerase chain reaction (PCR) using 6 specific primer pairs (Table I). PCR was performed in a reaction volume of 25 μl containing 100 ng of genomic DNA template, 200 nM of each primer, 200 nM of each dNTP, 1X PCR buffer and 2.5 units of LA Taq DNA polymerase (Takara Bio Inc., Shiga, Japan). Amplifications were conducted over 35 cycles; each cycle consisted of denaturation at 95°C for 30 sec, annealing at 51°C for 1 min and extension at 72°C for 1 min, with a final extension at 72°C for 10 min. If no mutation was identified using TYR analysis, the sample was analyzed for SLC45A2 mutations. All 7 coding exons and the intronic flanking regions were PCR-amplified using 10 specific primer pairs (Table I). Amplifications were conducted over 35 cycles; each cycle consisted of denaturation at 95°C for 30 sec, annealing at 58°C for 30 sec and extension at 72°C for 40 sec, with a final extension at 68°C for 10 min.
Table I.
PCR primer pairs used for the amplification of TYR and SLC45A2 coding sequences.
| TYR | ||
|---|---|---|
|
| ||
| Exon | Forward | Reverse |
| 1–1 | CCC ACT GGT GGG ATA CGA | GGT CCC CAA AAG CCA AAC |
| 1–2 | CCC TAG AGC CTG TGT CTC C | CCC TGC CTG AAG AAG TGA T |
| 2 | CCT CAG GAG AAG TCT AAC AAC | ACA ACA CAT ATT CTT GGT C |
| 3 | TGG GTA TCC AGA ATG TAA A | TTT AAA TCC AAT GAG CAC G |
| 4 | TTT TAA TAT ATG CCT TAT TTT AC | GGT AAC ACT AGA TTC AGC AA |
| 5 | CTC CAA AGG ACT GTG AAA GGA | GGT CTT TAC AGA AAA ATA C |
| SLC45A2 | ||
|---|---|---|
|
| ||
| Exon | Forward | Reverse |
| 1-1 | AGG CTC CAC GTC AAA TCC AG | GGT CAC ATA CGC TGC CTC CA |
| 1-2 | CAG ACT CAT CAT GCA CAG CA | ATG CCC ACG AGC ATC ATG AC |
| 1-3 | CAG CAT TGT GTG GTT CCT CA | GGT CAA ACA CAT GAA CAT CCT C |
| 2 | AAC GTG GAT GAT TCT AAA ACA GGA | CTC ATT GTC TGG GGA GCT GA |
| 3-1 | GGG AGT GTC TAT GCA TGA GG | GAT AGA ACC ATA CTC GTA CAT TCC |
| 3-2 | GCC CCA CTT ACA GAG GTT GC | CAA CAA AGA GCA AGA ATA TTT TCC CTT G |
| 4 | AGC TGG CTG AGT TTC TGC AG | CCT CAA CAG GTG TTA ATG GAG G |
| 5 | AGA GGT GGA GAA GCA GAG TG | GAA GAC ATC CTT AGG AGA GAG |
| 6 | ATG AGG CAC TGC CAG CTG TA | CCC AAG GCA GAG GTT CAA TG |
| 7 | GCC CTA AAT GAC AGT TCC TTG | TGT GCT TCA CTG TCT CTG AG |
TYR, tyrosinase.
PCR products were separated on 1.2% agarose gels and bands were visualized with ethidium bromide. Subsequently, DNA sequencing reactions were conducted using the same primer pairs and a BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions. The sequencing reaction mixtures were electrophoresed and analyzed using an ABI3130xl Genetic Analyzer (Applied Biosystems) and Sequencing Analysis v.5.2 software.
To predict the functional impact of novel amino acid changes, we performed additional molecular analyses on DNA from the patients’ parents and 50 healthy controls. Additionally, novel sequence alterations were assessed using in silico prediction algorithms, Polymorphism Phenotyping (PolyPhen) and Sorting Intolerant from Tolerant (SIFT), and the In the PolyPhen program. A position-specific independent count (PSIC) score of >2.0 for an amino acid variant indicates ‘probably damaging to protein function’; a score of 1.5–2.0, ‘possibly damaging’ and a score of <1.5, indicates ‘benign or unknown’ (10,11).
Results
Study population and clinical findings in Korean OCA patients
A total of 12 patients (M:F=5:7) and their parents were enrolled. The ages at enrollment ranged from 0.8 to 29.0 years (median, 3.4 years). OCA subtypes were previously unknown. There were 5 patients with a complete absence of pigmentation who had been clinically diagnosed with OCA1A, and the remaining 7 patients had minimal to moderate pigmentation that had gradually accumulated with age. Patient clinical characteristics are shown in Table II. Ultimately, 24 alleles from 12 OCA patients from nonconsanguineous families were examined.
Table II.
Clinical parameters and mutations in 12 Korean OCA families.
| Patient | Age (years) | Gender | Diagnosis | Causing gene | Mutation | Clinical phenotype | |||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Paternal | Maternal | Hair color | Skin color | Nystagmus | |||||
| 1 | 2.2 | M | OCA1A | TYR | c.929insC | c.929insC | White | White | + |
| 2 | 0.9 | F | OCA1A | TYR | p.R299H | c.929insC | White | White | + |
| 3 | 2.1 | M | OCA1A | TYR | c.929insC | p.D383N | White | White | + |
| 4 | 14.2 | M | OCA1A | TYR | p.D383N | p.R77Q | White | White | + |
| 5 | 27.0 | M | OCA1A | TYR | p.R77Q | c.1037-7T>A; c.1037-10delTT | White | White | + |
| 6 | 1.1 | F | OCA1B | TYR | c.929insC | c.1037-7T>A; c.1037-10delTT | Blond | White | + |
| 7 | 0.8 | F | OCA1B | TYR | c.1037-7T>A; c.1037-10delTT | Not identified | Blond | White | + |
| 8 | 29.0 | F | OCA1B | TYR | p.R52I | c.1037-7T>A; c.1037-10delTT | Blond | White | + |
| 9 | 4.5 | M | OCA4 | SLC45A2 | p.D93N | p.D157N | Blond | White | + |
| 10 | 5.9 | F | OCA | - | Not identified | Not identified | Blond | White | + |
| 11 | 1.0 | F | OCA | - | Not identified | Not identified | Blond | White | - |
| 12 | 29.0 | F | OCA | - | Not identified | Not identified | Blond | White | + |
OCA, oculocutaneous albinism; TYR, tyrosinase.
Spectrum of TYR and SLC45A2 mutations
Nine of 12 (75.0%) patients were identified using molecular analyses of TYR and SLC45A2. The remaining 3 (25.0%) patients were unable to be identified from the mutational screening for these 2 genes. There were 2 mutational alleles identified from 8 patients and 1 mutational allele from 1 patient.
OCA1 confirmed by mutations in the TYR gene was observed in 8 patients (66.7%) (Table II). OCA4 was diagnosed in 1 patient (8.3%) by SLC45A2 analysis. This patient had demonstrated improvement in pigment phenotype with age, and his hair color had changed to light yellow.
TYR analysis identified a total of 6 distinct mutations, consisting of 4 missense, 1 frameshift and 1 splice site, in 15 alleles of 8 patients (Table III). The mutations were located in exons 1, 2, and 3 and in intron 2. The 6 mutations had already been reported as pathological mutations. The most frequently detected mutation was c.929insC, which was found in 5 (31.3%) alleles of 4 patients. c.1037-7T>A/c.1037-10delTT, p.D383N, p.R77Q, p.R52I and p.R299H mutations were identified in 4 (25.0%) alleles of 4 patients, 2 (12.5%) alleles of 2 patients, 2 (12.5%) alleles of 2 patients, 1 (6.3%) allele of 1 patient, and 1 (6.3%) allele of 1 patient, respectively.
Table III.
Allelic frequencies of the TYR gene in 8 Korean OCA1 patients.
| Location | Nucleotide change | Amino acid change | No. (%) of alleles identified | ||
|---|---|---|---|---|---|
|
| |||||
| Korean patients (n=16) | Japanese patients (n=60) | Chinese patients (n=178) | |||
| Exon 1 | c.155G>T | p.R52I | 1 (6.3) | 0 (0) | 0 (0) |
| c.230G>A | p.R77Q | 2 (12.5) | 9 (15.0) | 7 (3.9) | |
| Exon 2 | c.896G>A | p.R299H | 1 (6.3) | 0 (0) | 26 (14.6) |
| c.929insC | p.R311LfsX7 | 5 (31.3) | 30 (50.0) | 25 (14.0) | |
| Intron 2 | c.1037-7T>A | Mis-splicing | 4 (25.0) | 5 (8.3) | 5 (2.8) |
| c.1037-10delTT | |||||
| Exon 3 | c.1147G>A | p.D383N | 2 (12.5) | 2 (3.3) | 1 (0.6) |
| Total | 15 (93.8) | 46 (76.7) | 64 (35.9) | ||
OCA1, Oculocutaneous albinism 1, TYR, tyrosinase.
There were 4 patients in whom mutations could not be identified in the coding sequences or exon/intron boundaries of TYR. These patients were screened for SLC45A2 mutations. A known disease-causing mutation, c.469G>A (p.D157N), and a novel sequence alteration, c.277G>A (p.D93N), was also detected (Fig. 1). p.D93N is considered to be a pathological mutation, since it was not detected in 50 unaffected controls and it is located in amino acid residues that are conserved in zebrafish, mice, rhesus monkeys and humans. The SIFT and PISC scores were 0.00 and 1.851, respectively, indicating that p.D93N may affect protein structure and possibly be a damaging mutation.
Figure 1.
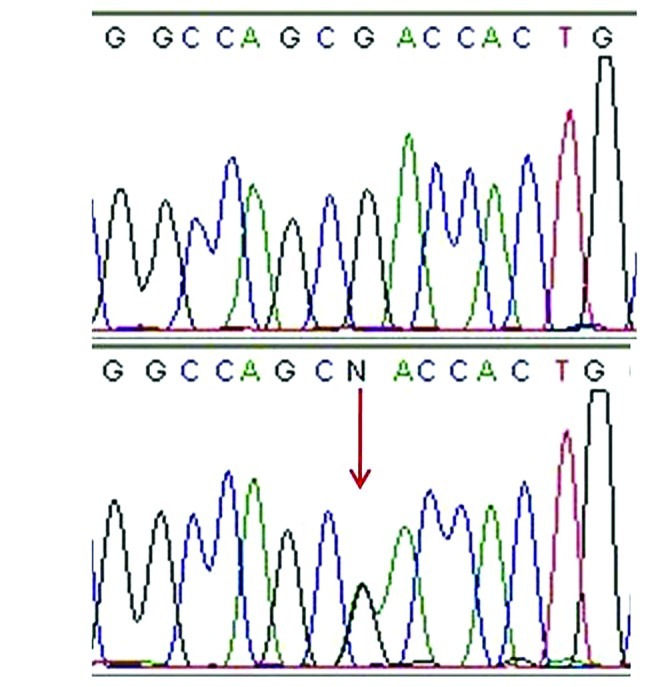
Partial sequence of the SLC45A2 reveals a novel mutation detected in the present study. (A) The wild form compared with (B) the heterozygous G>A mutation in exon 1 replaces an aspartate with an asparagine at codon 93.
Discussion
OCA1 is caused by mutations in the TYR gene on chromosome 11q14.3. The gene consists of 5 exons spanning approximately 65 kb of genomic DNA, and encodes a protein of 529 amino acids (12). Tyrosinase is a glycoprotein and a copper-containing oxidase that is involved in the formation of pigments, including melanins and other polyphenolic compounds. It catalyzes the rate-limiting conversions of tyrosine to dopa, dopa to dopaquinone, and possibly 5,6-dihydroxyindole to indole-5,6 quinone (13). During melanogenesis, tyrosinase may be released from the endoplasmic reticulum in the presence of a protonophore or proton pump inhibitors that increase the pH of intracellular organelles. Tyrosinase is then normally transported to the Golgi, and then to the melanosomes via the endosomal sorting system. For melanin synthesis, tyrosinase is sorted to the melanosomes via a dileucine-based signal (14). Mutations in the TYR gene appear to cause tyrosinase to be retained in the endoplasmic reticulum, with subsequent early degradation and destruction of tyrosinase activity (15).
At present, only 6 pathological TYR mutations have been detected in Korean patients (8,16), whereas more than 200 different mutations in TYR have been reported worldwide (17,18). We examined 24 alleles from the entire TYR gene and identified 6 different mutations in 15 (62.5%) alleles. These mutations had all been previously reported in Korean patients (Table III). According to a previous study, c.929insC was the most common of the 6 known Korean mutations, with an allele frequency of 0.5 (8). In this study, c.929insC was also the most common mutation, with an allele frequency of 0.31. This mutation is also known to be the most common TYR mutation in Japanese patients, with an allele frequency of 0.50 (19,20), and the second most common mutation in Chinese patients, with an allele frequency of 0.14 (7). c.929insC has only been identified in Far East Asian populations thus far, including Korean, Japanese and Chinese. This mutation is an insertion mutation, which causes a frameshift and premature termination of tyrosinase synthesis (p.R311LfsX7). It is regarded to be more commonly associated with OCA1A phenotypes than with OCA1B (19), and in this study 3 (1 homozygote and 2 compound heterozygotes) of 4 patients with this mutation also had OCA1A phenotypes. Another mutation, c.1037-7T>A/c.1037-10delTT, was identified in 4 alleles. Although c.1037-7T>A has been reported worldwide (21), c.1037-7T>A/c.1037-10delTT has only been identified in Korean, Japanese and Chinese patients, with allele frequencies of 0.25, 0.08 and 0.03, respectively. This mutation is located in intron 2 and causes mis-splicing of exon 3. In previous studies, it was found in patients with OCA1B phenotypes (8,19), and in this study 3 (compound heterozygotes) of 4 patients with this mutation had OCA1B phenotypes. p.R77Q and p.D383N mutations were found to be compound heterozygotes in 2 patients. These missense mutations have been reported in various ethnic groups, including Caucasian and Far East Asian populations, and both have been frequently found in patients with OCA1A phenotypes (19,21). All 3 patients (compound heterozygotes) harboring these mutations in our study were also classified with OCA1A. The other mutations, p.R52I and p.R299H, were each identified in 1 patient. p.R52I is an infrequent mutation and has been reported only in Caucasian patients with OCA1A phenotypes (17), even though two other mutations on the same amino acid location (p.R52K and p.R52T) were found in the Indian population (22). p.R299H has been identified worldwide (7,8,21) and is also the most frequent mutation in Chinese OCA1 patients, with an allele frequency of 0.15 (7). However, its allele frequency does not appear to be high in our Korean OCA1 patients and, there is currently no other study of an OCA1 Korean patient with this mutation.
Notably, the mutation p.R278X is the third most common in Japanese and Chinese patients, accounting for 11.6 and 11.8% of all reported TYR mutations, respectively. However, this mutant allele has yet to be identified in Korean OCA1 patients.
A comparison of the results of previous studies on Japanese and Chinese OCA1 patients and our results reveals that the identified mutations largely overlapped among these Far East Asian populations. The 6 mutations found in 93.8% of alleles in the Korean patients have been detected in 76.7% of Japanese patients and 35.9% of Chinese patients. The index of coincidence of this comparison suggests that the Korean population is genetically closer to the Japanese population than the Chinese one.
Mutations in the SLC45A2 gene (also known as MATP) on chromosome 5p13.3 cause OCA4. The SLC45A2 gene consists of 7 exons spanning approximately 40 kb of genomic DNA. The SLC45A2 protein of 530 amino acids, which contains 12 putative transmembrane domains, reveals sequence and structural similarity to the plant sucrose transporter, and is only expressed in melanocytes (23). SLC45A2 encodes a melanocyte differentiation antigen that is expressed in a high percentage of melanoma cell lines (24). Studies on the Japanese medaka fish reveal that this protein is significant in pigmentation and may function as a membrane transporter in melanosomes (23). Ultrastructural analysis has demonstrated that the vesicles secreted from OCA4 melanocytes are mostly early-stage melanosomes. In OCA4 melanocytes, tyrosinase processing and intracellular trafficking to the melanosome is disrupted, and the enzyme is abnormally secreted from the cells in immature melanosomes, which disrupts the normal maturation process of those organelles (25). SLC45A2 mutations were first found in a Turkish OCA patient in 2001 (26), since then more than 30 mutations have been reported (5,7,9,27,28).
OCA4 appears to be a rare form of OCA in Caucasians, and mutations in SLC45A2 have been reported as the cause of disease in 5–8% of German OCA patients (27). However, OCA4 is the second most common form in Japanese and Chinese populations, accounting for 24–27% and 13% of all OCA, respectively (5–7). Clinical phenotypes of OCA4 vary from complete depigmentation to partial pigmentation; and similarly in OCA1B certain patients improve during the first decade of life (5). Based on our results which reveal that the mutation spectrum of Korean OCA1 patients considerably overlaps with those of Japanese and Chinese patients, we examined SLC45A2 in 4 TYR mutation-negative patients. One known mutation (p.D157N) and 1 novel sequence alteration (p.D93N) were identified. Over 20 missense mutations have been reported in SLC45A2, and the majority of them are located within or in close proximity to the transmembrane domains, in areas that appear to be essential for the function of the SLC45A2 protein (23,26). The mutation p.D157N is known to be the most common mutation in Japanese OCA4 patients, with an allele frequency of 0.39 (6), and 1 Korean OCA4 patient was reported as a homozygote of this allele in a previous study (9). Therefore, this indicates a founder effect for p.D157N in Japanese and Korean populations (29). Of note, in Chinese OCA4 patients, the p.D160H allele accounted for approximately half of the mutational SLC45A2 alleles, and p.D157N was detected in just 1 allele with an allele frequency of 0.03 (7). This finding also suggests a founder mutation in the Chinese Han population, revealing a genetic disparity between the Japanese and Chinese populations. A novel sequence alteration, p.D93N, is considered to be a pathological mutation since this change is located close to the second transmembrane domain composed of amino acids 70 to 92, and it was not detected in 50 unaffected controls. It is also in an amino acid residue that is conserved among various species. We also predicted the functional impact of p.D93N using 2 in silico prediction algorithms, and this novel change was predicted to be a deleterious mutation.
There are certain limitations to the present study. Firstly, this study was performed at one institution, which may have caused selection bias. Secondly, the wide age distribution among the patients may have affected the phenotypes. Lastly, we did not perform any functional studies of the one putative novel mutation. Additional studies of Korean OCA patients, with molecular analyses of other OCA-associated genes, including OCA2 and TYRP, may aid in predicting more accurate genotype-phenotype correlations and provide information for genetic counseling. Additionally, anthropological studies are required to confirm ethnic differences in the mutational spectrum of OCA.
In conclusion, great progress in the detection and analysis of OCA-causative genes has been achieved in recent years; however, OCA remains a challenging disorder. Our study provides certain information concerning the genetic basis of OCA in Korean patients, and allows us to estimate the relative frequencies of OCA1 and OCA4 in Korea.
References
- 1.Tomita Y, Suzuki T. Genetics of pigmentary disorders. Am J Med Genet C Semin Med Genet. 2004;131:75–81. doi: 10.1002/ajmg.c.30036. [DOI] [PubMed] [Google Scholar]
- 2.Grønskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis. 2007;2:43. doi: 10.1186/1750-1172-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oetting WS, King RA. Molecular basis of albinism: mutations and polymorphisms of pigmentation genes associated with albinism. Hum Mutat. 1999;13:99–115. doi: 10.1002/(SICI)1098-1004(1999)13:2<99::AID-HUMU2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 4.Rooryck C, Roudaut C, Robine E, Musebeck J, Arveiler B. Oculocutaneous albinism with TYRP1 gene mutations in a Caucasian patient. Pigment Cell Res. 2006;19:239–242. doi: 10.1111/j.1600-0749.2006.00298.x. [DOI] [PubMed] [Google Scholar]
- 5.Inagaki K, Suzuki T, Shimizu H, et al. Oculocutaneous albinism type 4 is one of the most common types of albinism in Japan. Am J Hum Genet. 2004;74:466–471. doi: 10.1086/382195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki T, Tomita Y. Recent advances in genetic analyses of oculocutaneous albinism types 2 and 4. J Dermatol Sci. 2008;51:1–9. doi: 10.1016/j.jdermsci.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Wei A, Wang Y, Long Y, et al. A comprehensive analysis reveals mutational spectra and common alleles in Chinese patients with oculocutaneous albinism. J Invest Dermatol. 2010;130:716–724. doi: 10.1038/jid.2009.339. [DOI] [PubMed] [Google Scholar]
- 8.Park SK, Lee KH, Park KC, Lee JS, Spritz RA, Lee ST. Prevalent and novel mutations of the tyrosinase gene in Korean patients with tyrosinase-deficient oculocutaneous albinism. Mol Cells. 1997;7:187–191. [PubMed] [Google Scholar]
- 9.Suzuki T, Inagaki K, Fukai K, Obana A, Lee ST, Tomita Y. A Korean case of oculocutaneous albinism type IV caused by a D157N mutation in the MATP gene. Br J Dermatol. 2005;152:174–175. doi: 10.1111/j.1365-2133.2005.06403.x. [DOI] [PubMed] [Google Scholar]
- 10.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwon BS, Haq AK, Pomerantz SH, Halaban R. Isolation and sequence of a cDNA clone for human tyrosinase that maps at the mouse c-albino locus. Proc Natl Acad Sci USA. 1987;84:7473–7477. doi: 10.1073/pnas.84.21.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooksey CJ, Garratt PJ, Land EJ, et al. Evidence of the indirect formation of the catecholic intermediate substrate responsible for the autoactivation kinetics of tyrosinase. J Biol Chem. 1997;272:26226–26235. doi: 10.1074/jbc.272.42.26226. [DOI] [PubMed] [Google Scholar]
- 14.Watabe H, Valencia JC, Yasumoto K, et al. Regulation of tyrosinase processing and trafficking by organellar pH and by proteasome activity. J Biol Chem. 2004;279:7971–7981. doi: 10.1074/jbc.M309714200. [DOI] [PubMed] [Google Scholar]
- 15.Toyofuku K, Wada I, Spritz RA, Hearing VJ. The molecular basis of oculocutaneous albinism type 1 (OCA1): sorting failure and degradation of mutant tyrosinases results in a lack of pigmentation. Biochem J. 2001;355:259–269. doi: 10.1042/0264-6021:3550259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park KC, Park SK, Lee YS, et al. Mutations of the tyrosinase gene in three Korean patients with type I oculocutaneous albinism. Jpn J Hum Genet. 1996;41:299–305. doi: 10.1007/BF01913172. [DOI] [PubMed] [Google Scholar]
- 17.The Human Gene Mutation Database. [Accessed September 1, 2011]. http://www.hgmd.cf.ac.uk/ac/all.php.
- 18.Albinism database: Mutations of the tyrosinase gene associated with OCA1. [Accessed September 1, 2011]. http://albinismdb.med.umn.edu/oca1mut.html.
- 19.Goto M, Sato-Matsumura KC, Sawamura D, Yokota K, Nakamura H, Shimizu H. Tyrosinase gene analysis in Japanese patients with oculocutaneous albinism. J Dermatol Sci. 2004;35:215–220. doi: 10.1016/j.jdermsci.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Tomita Y, Miyamura Y, Kono M, Nakamura R, Matsunaga J. Molecular bases of congenital hypopigmentary disorders in humans and oculocutaneous albinism 1 in Japan. Pigment Cell Res. 2000;13(Suppl 8):130–134. doi: 10.1034/j.1600-0749.13.s8.23.x. [DOI] [PubMed] [Google Scholar]
- 21.King RA, Pietsch J, Fryer JP, et al. Tyrosinase gene mutations in oculocutaneous albinism 1 (OCA1): definition of the phenotype. Hum Genet. 2003;113:502–513. doi: 10.1007/s00439-003-0998-1. [DOI] [PubMed] [Google Scholar]
- 22.Ray K, Chaki M, Sengupta M. Novel human pathological mutations. Gene symbol: TYR Disease: tyrosinase deficiency. Hum Genet. 2007;122:556. [PubMed] [Google Scholar]
- 23.Fukamachi S, Shimada A, Shima A. Mutations in the gene encoding B, a novel transporter protein, reduce melanin content in medaka. Nat Genet. 2001;28:381–385. doi: 10.1038/ng584. [DOI] [PubMed] [Google Scholar]
- 24.Ray ME, Wistow G, Su YA, Meltzer PS, Trent JM. AIM1, a novel non-lens member of the betagamma-crystallin super-family, is associated with the control of tumorigenicity in human malignant melanoma. Proc Natl Acad Sci USA. 1997;94:3229–3234. doi: 10.1073/pnas.94.7.3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costin GE, Valencia JC, Vieira WD, Lamoreux ML, Hearing VJ. Tyrosinase processing and intracellular trafficking is disrupted in mouse primary melanocytes carrying the underwhite (uw) mutation. A model for oculocutaneous albinism (OCA) type 4. J Cell Sci. 2003;116:3203–3212. doi: 10.1242/jcs.00598. [DOI] [PubMed] [Google Scholar]
- 26.Newton JM, Cohen-Barak O, Hagiwara N, et al. Mutations in the human orthologue of the mouse underwhite gene (uw) underlie a new form of oculocutaneous albinism, OCA4. Am J Hum Genet. 2001;69:981–988. doi: 10.1086/324340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rundshagen U, Zuhlke C, Opitz S, Schwinger E, Kasmann-Kellner B. Mutations in the MATP gene in five German patients affected by oculocutaneous albinism type 4. Hum Mutat. 2004;23:106–110. doi: 10.1002/humu.10311. [DOI] [PubMed] [Google Scholar]
- 28.Gargiulo A, Testa F, Rossi S, et al. Molecular and clinical characterization of albinism in a large cohort of Italian patients. Invest Ophthalmol Vis Sci. 2011;14:1281–1289. doi: 10.1167/iovs.10-6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inagaki K, Suzuki T, Ito S, et al. OCA4: evidence for a founder effect for the p.D157N mutation of the MATP gene in Japanese and Korean. Pigment Cell Res. 2005;18:385–388. doi: 10.1111/j.1600-0749.2005.00261.x. [DOI] [PubMed] [Google Scholar]