

Abstract
Alzheimer's disease (AD) was first described by Alois Alzheimer in 1907. AD is the most prevalent dementia- related disease, affecting over 20 million individuals worldwide. Currently, however, only a handful of drugs are available and they are at best only able to offer some relief of symptoms. Acetylcholinesterase (AChE) inhibitors, antioxidants, metal chelators, monoamine oxidase inhibitors, anti-inflammatory drugs and NMDA inhibitors are usually used to attempt to cure this disease. AChE inhibitors are the most effective therapy for AD at present. Researchers have found that histamine H3 receptor antagonists decrease re-uptake of acetylcholine and the nervous transmitter substance acetylcholine increases. In this study, we designed compounds by using docking, de novo evolution and adsorption, distribution, metabolism, excretion and toxicity (ADMET) analysis to AChE inhibitors as well as histamine H3 receptor antagonists to forward drug research and investigate the potent compounds which can pass through the blood-brain barrier. The novel drugs may be useful for the treatment of AD, based on the results of this theoretical calculation study. We will subsequently examine them in future experiments.
Keywords: Alzheimer's disease, acetylcholinesterase inhibitors, histamine H3 receptor antagonists
Introduction
Alzheimer's disease (AD), a debilitating disease of the elderly, is the most common cause of senile dementia and is characterized by a progressive deterioration in cognitive function and loss of memory (1). According to statistics, in the next 20 years, the number of individuals suffering from AD will double according to the aging population. AD has become one of the most costly diseases which brings heavy social and financial burden to both society and families (2). Therefore, the development of effective therapies for AD is crucial at present and in the future. Recent research has revealed that the amyloid precursor protein (APP), a type of protein in central neural cell membranes, which is broken at the residues 672–687 in signal transduction, is the fragment β-amyloid protein (Aβ). Aβ is the main factor which causes AD (3–6). Unfortunately, at present, AD still cannot be thoroughly eliminated. The celebral cholinergic neurons of AD patients decrease approximately 30–95%, particularly in the celebral cortex and hippocampus.
Drugs for AD are shown in Fig. 1. These include donepezil (Aricept®), rivastigmine (Exelon®) (7) and galantamine (Reminyl®) (8). These drugs were developed based on the cholinergic hypothesis which consists of four concepts: i) cerebral cortex cholinergic neuroticism decreases in AD patients; ii) the celebral acetyltransferase activity decreases in AD patients; iii) cholinergic insufficient has some relation on cognition function; iv) these factors will be improved using acetylcholinesterase (AChE) inhibitors.
Figure 1.

Conformation of Evo27 in the binding site, in triple bonds will form a hydrophobic interactions, and hook the buttom of the tunnel. In the median of tunnel OH base of Evo27 f H-bond is formed with the residue Trp121, and an extended side chain also produces good performance at the top.
Two common cholinesterases are usually discussed in the treatment of AD: AChE, which is mainly found in cerebrum, striated muscle, skin and red blood cells; and butyrylcholinesterase (BChE) (9), which appears in the myocardium, smooth muscle, skin, gland and blood serum. Some drugs inhibit AChE and BChE together, but cause serious side effects, such as gastrointestinal discomfort and the risk of bradycardia.
According to the interaction of AChE-donepezil (PDB code 1EVE), donepezil interacts with various residues in the AChE active site, and interacts with the following groups, benzyl, pieridinic nitrogen and dimethoxy-indanone, which result in π stacking and hydrophobic interaction. The binding of donepezil with AChE is extremely dependent on interactions with Trp279 and Phe330, which does not exist in BChE. This phenomenon may successfully explain the reason why donepezil has higher specificity for AChE than BChE.
In this study, we designed new drugs,which exhibit stronger interactions with AChE than BChE by using docking and de novo evolution by using the basic structure of donepezil. We also discuss the novel drugs which have a higher dock score than donepezil, and analyze the phenomena from the point of physical chemistry.
HA is a cerebral neurotransmitter exerting its actions on target cells via three classes of molecularly and/or pharmacologically well-defined receptors designated H1, H2 and H3 (10–12). The H3 receptor is a presynaptic receptor regulating the synthesis and/or release of HA itself (13) as well as a variety of other aminergic or peptidergic neurotransmitters (14).
The binding of H3-agonists to H3 receptors in brain tissues has been shown to be regulated by guanine nucleotides, implying a linkage to heterotrimeric G-proteins (15–17). More recently, radiolabeled H3 receptor antagonists have become available. The first compound to be developed was iodophenpropit, which has been used to successfully label H3 receptors in rat brain membranes (18).
Considering the complexity of AD, the classic ‘one molecule, one target’ solution may not be effective enough (19–21). The novel multi-target-directed strategy has received attention, since single molecules simultaneously interact with multiple targets in complex neurotoxic cascades may achieve better efficacy by a complementary manner. Meanwhile, the hybrid would reduce individual toxicity by a special metabolic pathway compared with the combinational drugs (22). Regarding the histamine H3 receptor, its function as a heteroreceptor can be found on colocalized neurons, and H3R activation modulates the release of various important neurotransmitters, i.e. dopamine, acetylcholine, H3 receptor antagonists increasing acetylcholine levels. This approach might gain importance in the treatment of dementia. More recently, Bembenek et al reported the design and evaluation of the dual inhibitors of AChE and H3 receptor antagonists, which have had successful results (23). Furthermore, our aims focused on designing and synthesizing dual inhibitors that dock both AChE and histamine H3 receptors in this study
Materials and methods
Homology modeling using Accelrys software
Homology modeling was used to construct an atomic-resolution model of the target protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein. It relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence. The target structure is produced from the known sequence alignment and the template structure and its sequence. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.
Docking and score using Accelrys software
The score functions in the Discovery Studio 2.5 which we used were DockScore, PLP1, PLP2 and PMF. Candidate ligand poses were evaluated and prioritized according to the DockScore function. There are two types of DockScore. One is based on a forcefield approximation, the other on the piecewise linear potential function (PLP).
| Equation 1 |
| Equation 2 |
As shown in Equation 1, there are two energy terms in the forcefield version of DockScore: internal energy of the ligand and the interaction energy of the ligand with the receptor. The interaction energy is taken as the sum of the van der Waals energy and electrostatic energy. The computation of the interaction energy can be quite time consuming. To reduce the time needed for this calculation, a grid-based estimation of the ligand/receptor interaction energy was employed.
The van der Waals component of the forcefield interaction energy typically exhibits a steep rise at short interatomic distances, which can have undesirable consequences in the context of ligand-receptor docking. In particular, the combination of approximating the receptor structure as rigid and limited sampling of ligand conformational space tends to overly penalize poses with ‘mild’ short contacts between the ligand and receptor, due to the ‘hard’ nature of the van der Waals potential as defined in most standard forcefields.
To overcome this tendency, a softened form of the van der Waals potential is employed with the DockScore function. This softened potential rises to a large but finite value at zero interatomic separation. To maintain a proper balance between electrostatics and van der Waals, the electrostatic energy is also softened to prevent it from dominating the van der Waals energy at short separations.
The internal energy of the ligand is computed when using the forcefield version of DockScore. The purpose of including the internal energy is to avoid ligand conformations with bad internal nonbond clashes. By default, only the standard (not softened) van der Waals energy is used for the ligand internal energy. Including electrostatic energy as part of the ligand internal energy is optionally available.
The PLP version of DockScore uses the PLP1 function, as the functional form of PLP1 allows it to be readily represented with a grid-based approach. The PLP2 function has an angular dependence on hydrogen bonding interactions making its representation with a grid considerably more difficult.
In the PLP1 score function, there are four atom types as following, i) hydrogen bond donor only, ii) hydrogen bond acceptor only, iii) both hydrogen bond donor and acceptor, and iv) non-polar. When PLP1 is the docking score function, the internal energy is calculated for each ligand conformation that the ligand is in the binding site.
In the PLP2 score function, the atom typing remains the same as in the PLP1 score function. In addition, an atomic radius is assigned to each atom expected for hydrogen (24).
The PMF score function was developed based on statistical analysis of the 3D structures of the protein-ligand complex (25). They were found to correlate well with protein-ligand binding free energy while being fast and simple to calculate. The scores are calculated by summing pairwise interaction terms over all interatomic pairs of the receptor-ligand complex. The score function of DockScore is the default function in the Discovery Studio 2.5.
All the simulations were also applied by the forcefield of the chemistry at CHARMM (Chemistry at Harvard Macromolecular Mechanics). CHARMM was parameterized by experimental data. It has been widely used for simulations ranging from small molecules to solvated complexes of large biological macromolecules. CHARMM performs well over a broad range of calculations and simulations, including minima, time-dependent dynamic behavior, and barriers to rotation, vibrational frequencies, and free energy. CHARMM uses a flexible and comprehensive energy function:
where the out-of-plane (OOP) angle is an important torsion. The van der Waals term is derived from rare-gas potentials, and the electrostatic term can be scaled to mimic solvent effects. Hydrogen-bond energy is not included as a separate term as in AMBER. Instead, hydrogen-bond energy is implicit in the combination of van der Waals and electrostatic terms.
De novo evolution using Accelrys software
To design the new compounds from the potent drugs, we used the program ‘De novo evolution’ in the Discovery Studio 2.5 (Accelrys, San Diego, USA). We took the new compounds from the ‘De novo evolution’ and took them into the protein acceptor binding pocket and shown scores.
ADMET descriptors using Accelrys software
We computed the ADMET properties of small molecules by using Discovery Studio 2.5 (Accelrys) to estimate the following properties: aqueous solubility blood-brain barrier penetration (BBB), cytochrome P450 (CYP450) 2D6 inhibition, hepatotoxicity human intestinal absorption (HIA) and plasma protein binding. Furthermore, key issue was to calculate the BBB and other factors as the drugs should pass through the BBB to react with the receptor protein to cure AD.
Blood-brain barrier (BBB)
This model predicts blood-brain penetration (BBB) after oral administration. This model contains a quantitative linear regression model for the prediction of blood-brain penetration, as well as 95 and 99% confidence ellipses in the ADMET_PSA_2D, ADMET_AlogP98 plane. These ellipses are not the same as those associated with the ADMET-HIA, although they have an analogous interpretation. They were derived from over 800 compounds that are known to enter the CNS after oral administration.
Cytochrome P450
The cytochrome P450 2D6 model predicts CYP2D6 enzyme inhibition using 2D chemical structure as input. The model was developed from known CYP2D6 inhibition data on a diverse set of 100 compounds. An ensemble of recursive partitioning trees were trained against 2D descriptors and 1D similarity data. The model classifies compounds as either 0 or 1 for non-inhibitor or inhibitor and provides an average-class-value estimate of confidence.
Hepatotoxicity human intestinal absorption (HIA)
A computational model for predicting potential liver toxicity of a compound was recently reported by Cheng and Dixon (26). The model was developed from available literature data of 382 compounds known to exhibit liver toxicity (i.e., positive dose-dependent hepatocellular, cholestatic, neoplastic), or trigger dose-related elevated aminotransferase levels in more than 10% of the human population. From this, a model was generated from a classification structure activity relationship (SAR) technique that uses recursive partitioning. The model was tested against 54 compounds yielding >80% correct classification of ‘yes’ and ‘no’ questions about hepatotoxicity. Using only 2D information from the compounds provided, the model predicts, with greater than 80% accuracy, the potential occurrence of dose-dependent human hepatotoxicity of any compound. The model classifies compounds either as ‘toxic’ or ‘nontoxic’ and provides a confidence level indicator of the likelihood of the predictive accuracy of the model.
Each marker molecule has a characteristic 1D similarity threshold that is used to determine whether a given predicted compound is sufficiently similar to bind at that marker's associated level (90 or 95%). Thus, if a predicted compound exhibits a 1D similarity to any 90% marker molecule that meets or exceeds that marker's threshold, then the predicted compound is flagged as being likely to bind at 90% or higher. Binding at 95% or greater is predicted if the similarity threshold is exceeded for any marker in the 95% set. Binding level predictions are also supplemented by conditions on AlogP98 (27).
Result and discussion
The enzyme monomer of the T. californica AChE (PDB accession code: 1EVE) is an α/β serine hydrolase consisting of 537 residues with a 12-strand mixed b sheet surrounded by 14 α helices (28). The active site is approximately 20Å above the narrow tunnel. This tunnel penetrates halfway into the enzyme and widens out at the end. Fourteen highly conserved residues cover a substantial portion of the tunnel. The button of the tunnel approximately 4 A long is the catalytic machinery: Ser200, His440, and Glu327 formed a catalytic. In the region of the enzymatic cavity located between the choline binding site and the peripheral anionic site, the phenyl group of Phe330 of the enzymatic cavity interacts by cation-π with the piperidinic nitrogen of denepezil, which contains a positive charge, and Tyr334 forms a calix-domain with Phe330 and the piperidinic ring of donepezil. Hence, the interaction with Phe330 represents an additional site of quanternary binding with functional significance, inside the active site. In the peripheral anionic site, the indanone ring interacts with the indonic group of Trp279, via a classical parallel π-π interaction. Recent data suggest that the inhibitors interact with the peripheral anionic site and prevent the A from interacting with AChE. This could result in the elimination of AChE's role in the fibril formation process (29).
In order to make the structure of AChE more closer to the human type, the 3D homology model of the AChE of the human was established by using Discovery Studio 2.5 (Accelrys) from the crystal structure of the T. californica AChE and the known sequence (P04058 from Swiss-Prot). To proceed with the multiple sequence alignment, framework construction, energy minimization, structure reasonable analysis, finally we constructed human type AChE by homology modeling. The human type AChE has high sequence similarity with the crystal structure of the T. californica AChE. This explained successfully that we could simulate the structures precisely (30).
We built the structure of human named HUMAN.B99990001, which is a result of the homology model with the Discovery Studio 2.5, containing key residues. We found that Ser200, His440, Glu327, Trp84 and Trp279 are converted into Ser200, His444, Glu331, Trp83 and Trp283. Then we performed energy minimization, by using molecular mechanics energy minimization to correct unfavorable non-covalent touch and to reach ideal bond geometry and the lowest energy configuration. Ramchandran plot analysis shows 78.2% of the residues in the most favored region, 16.1% in the allowed and 5.7% in the disallowed region (30). We allowed donepezil to dock with HUMAN.B99990001, and revealed that donepezil can combine to the tunnel of AChE. Trp279 and Phe330 can still form π-π interaction with HUMAN.B99990001 (30).
New drug designed as an acetylcholinerase inhibitor
We used Discovery Studio 2.5 to design novel new drugs, which are not only more effective but also have no critical side effects. We found that there are five potent compounds for which the DockScores are higher than donepezil (3 are shown in Table I). We observed that Evo27 combines successfully to HUMAN.B99990001, which in triple bonds will form a hydrophobic interactions and hook the bottom of the tunnel; and in the median of the tunnel OH base of Evo27 to become a H-bond receptor which can form a H-bond with the residue Trp121, and that may give Evo27 higher interaction and extends the side chain that can also create constructive binding at the top (Fig. 1). Evo9 combined to HUMAN.B99990001 and different conditions were present at the top, which will be the H-bond donor in the NH base and residue Trp283 to form a hydrogen bond, such as putting an embolus at the opening. The benzyl group of Evo9 formulates a ring aromaticity just as donepezil and five rings of that formulate hydrophobic interaction resulting in better binding energy (Fig. 2). The structures of Evo27 and Evo9 express the same docking pose, but other side chains provide suitable interactions which cause a higher DockScore than donepezil (Fig. 3). These data demonstrated that Evo27 is the most suitable compound to dock in AChE.
Table I.
Score values of new AChE inhibitors and donepezil.
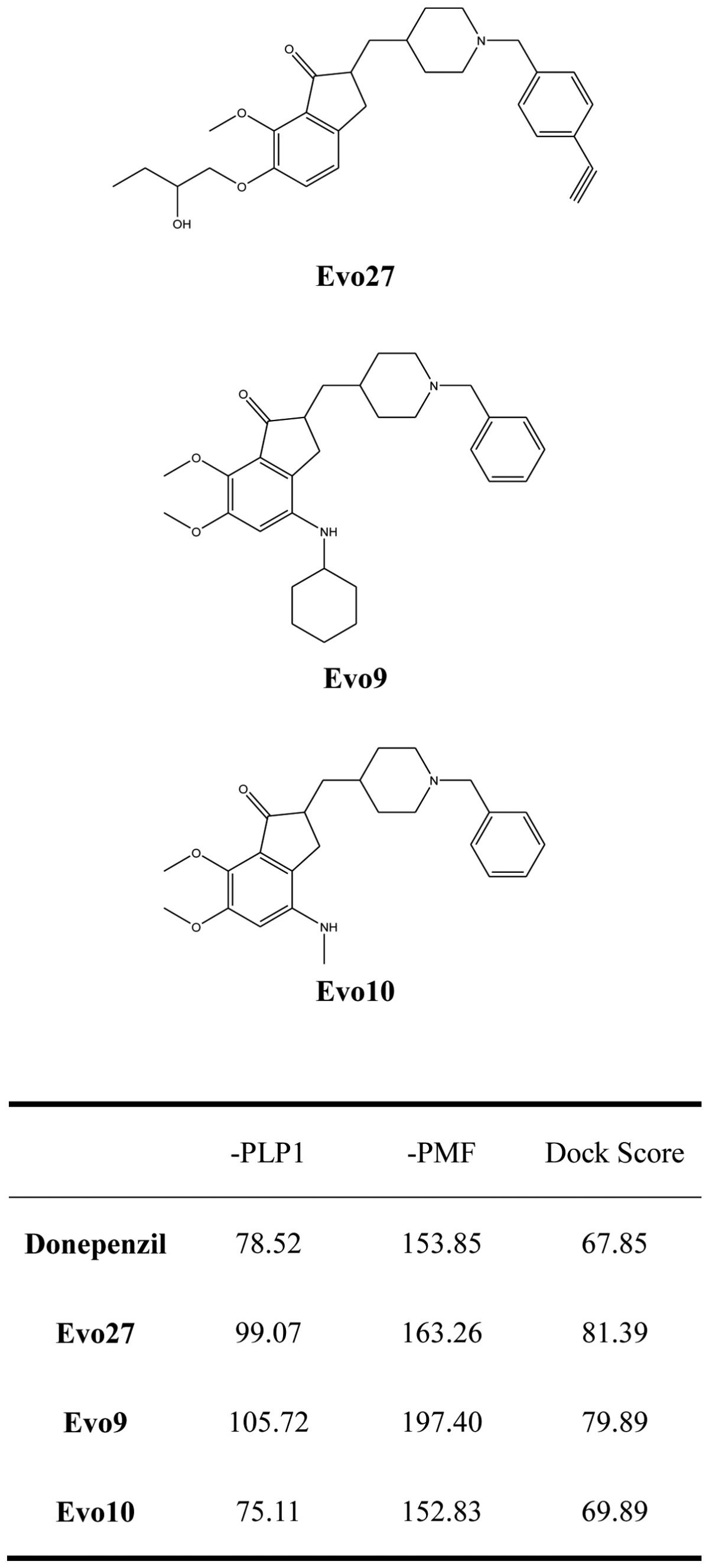
Figure 2.

Conformation of Evo9 in the binding site; Evo9 forms H-bonds with the residue Trp121 and Tyr338. Benzyl group formulates ring aromatic group and five rings of that formulate hydrophobic interaction.
Figure 3.

The principal structure of Evo27 and Evo9 express the same docking pose, but other side chains provide adequate interaction. Green, Evo27; blue, Evo9.
New drug designed as an histamine H3 receptor antagonist
Research indicates that histamine H3 receptors cause re-uptake of acetylcholine. In order to increase the quantities of acetylcholine in the brain, H3 receptor antagonists decrease the re-uptake of acetylcholine. There are various H3 receptor antagonists which may gain importance in the treatment of dementia since AChE inhibitors are commonly used in the therapy of AD. Therefore, affecting the pathology of AD by dual-acting compounds is a very key issue in designing more effective drugs. We used the pharmacophore of H3 receptor antagonists as a skeleton (31) to perform the program ‘De novo evolution’ in the Discovery Studio 2.5, and found four potent compounds (Table II). The benzyl group of Com1 resulting in the π-π stacking in the middle, and piperidinic nitrogen can form a cation-π interaction with residue Trp283, and create a key combination, that can prevent the β Aβ peptide from interacting with AChE (Fig. 4). Three inner triple bonds and double bond will interact with the buttom of the tunnel successfully. In the middle, the benzyl group of Com7 formed π-π stacking interactions with the top and bottom (Fig. 5). Their DockScore and donepenzil approach, but this type of compound contained dual-action, can reduce the drug uptake and attain a curing effect on AD. Finally, we used Discovery Studio 2.5 as a tool to perform ADMET. 2D ADMET Plot showed that Com1 and Com7 contained blood-brain barrier penetration potential, which implied that the compounds pass the BBB and have an effect on the brain (Fig. 6). Com1 and Com7 are the two best candidate compounds to cure AD, and will be investigated in the near future.
Table II.
Score values of new dual inhibitors and donepezil.
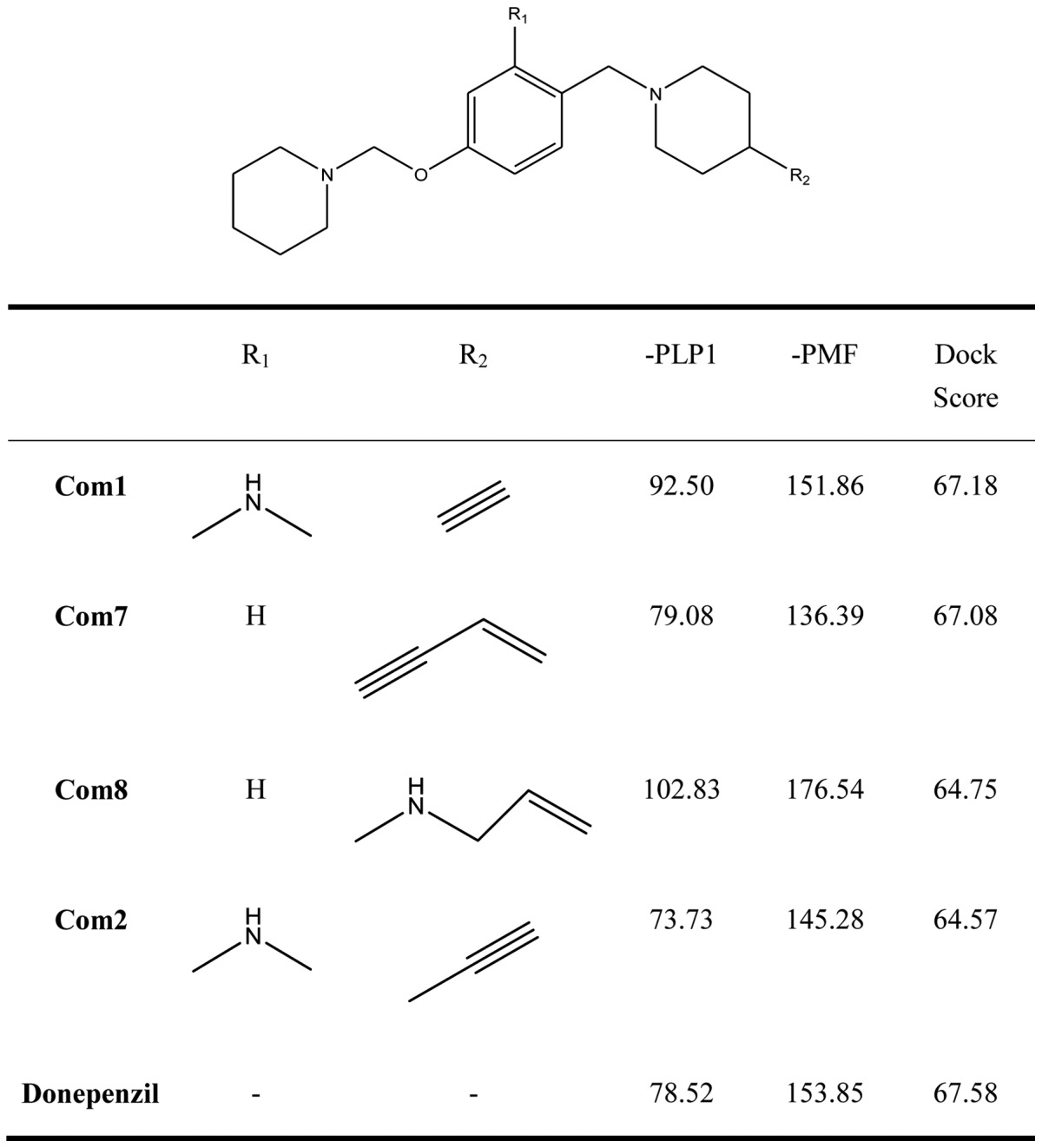
Figure 4.

Illustration of the ligand binding of side atoms. Benzyl group of Com1 to form π-π stacking in the middle, and piperidinic nitrogen can form cation-π interaction with residue Trp283.
Figure 5.

Illustration of the ligand binding side atoms. In the middle, benzyl group of Com7 forms π-π stacking interactions with the top and bottom.
Figure 6.

Evo27, Evo9, Com7 and Com1 perform ADMET tools. 2D ADMET Plot shows that Com7 and Com1 are in the 95 and 99% blood brain barrier (BBB) confidence ellipse region.
In conclusion, to explore novel effective drugs for the treatment of AD, two inhibitor compounds were designed and investigated. In this study, we aimed at homology modeling to simulate the characters of AChE. We used donepezil as a framework to design more effective chemical compounds for binding affinity. Our results suggest that Evo27 contains the highest DockScore, has stronger interactions with AChE, which we modeled using the crystal structure of the T. californica AChE and the human sequence than the other compounds including donepezil. Moreover, we designed the dual-acting compounds to lower the drug intake and increase neurotransmitters. Com1 not only had the characteristics of the antagonist of histamine H3 receptors and the ability of an AChE inhibitor but also contained BBB potential as well, and the DockScore was almost the same as donepezil.
Our results suggest that adding the hydrophobic functional group to chemical compounds such as Com1 and Evo27 resulted in interactions with the button of tunnel of AChE, which produce a higher DockScore. The docking results of three compounds may interact with the PAS and, based on current thinking, could potentially avoid the aggregation of β-amyloid and slow the progression of AD. However, these docking studies provide rational evidence that these inhibitors interact with target proteins, which offer additional direction to design more potent drugs for the treatment of AD.
Acknowledgements
This work was supported by the grants from China Medical University (CMU99-ASIA-22 and CMU100-TC-05). The authors would like to thank their parents, and all colleagues and friends who contributed to this study.
References
- 1.Dorronsoro C, Barbero S, Llorente L, Marcos S. On-eye measurement of optical performance of rigid gas permeable contact lenses based on ocular and corneal aberrometry. Optom Vis Sci. 2003;80:115–125. doi: 10.1097/00006324-200302000-00007. [DOI] [PubMed] [Google Scholar]
- 2.Guo T, Hobbs DW. Development of BACE1 inhibitors for Alzheimer's disease. Curr Med Chem. 2006;13:1811–1829. doi: 10.2174/092986706777452489. [DOI] [PubMed] [Google Scholar]
- 3.Wood KE, Becker BN, McCartney JG, D'Alessandro AM, Coursin DB. Care of the potential organ donor. N Engl J Med. 2004;351:2730–2739. doi: 10.1056/NEJMra013103. [DOI] [PubMed] [Google Scholar]
- 4.Jacobsen JS, Reinhart P, Pangalos MN. Current concepts in therapeutic strategies targeting cognitive decline and disease modification in Alzheimer's disease. NeuroRx. 2005;2:612–626. doi: 10.1602/neurorx.2.4.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mori F, Fukaya M, Abe H, Wakabayashi K, Watanabe M. Developmental changes in expression of the three ryanodine receptor mRNAs in the mouse brain. Neurosci Lett. 2000;285:57–60. doi: 10.1016/s0304-3940(00)01046-6. [DOI] [PubMed] [Google Scholar]
- 6.Barril X, Orozco M, Luque FJ. Towards improved acetylcholinesterase inhibitors: a structural and computational approach. Mini Rev Med Chem. 2001;1:255–266. doi: 10.2174/1389557013406828. [DOI] [PubMed] [Google Scholar]
- 7.Jann MW. Rivastigmine, a new-generation cholinesterase inhibitor for the treatment of Alzheimer's disease. Pharmacotherapy. 2000;20:1–12. doi: 10.1592/phco.20.1.1.34664. [DOI] [PubMed] [Google Scholar]
- 8.Zarotsky V, Sramek JJ, Cutler NR. Galantamine hydrobromide: an agent for Alzheimer's disease. Am J Health Syst Pharm. 2003;60:446–452. doi: 10.1093/ajhp/60.5.446. [DOI] [PubMed] [Google Scholar]
- 9.Hshieh TT, Fong TG, Marcantonio ER, Inouye SK. Cholinergic deficiency hypothesis in delirium: a synthesis of current evidence. J Gerontol A Biol Sci Med Sci. 2008;63:764–772. doi: 10.1093/gerona/63.7.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hill SJ, Ganellin CR, Timmerman H, et al. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol Rev. 1997;49:253–278. [PubMed] [Google Scholar]
- 11.Schwartz JC, Arrang JM, Garbarg M, Pollard H, Ruat M. Histaminergic transmission in the mammalian brain. Physiol Rev. 1991;71:1–51. doi: 10.1152/physrev.1991.71.1.1. [DOI] [PubMed] [Google Scholar]
- 12.Schwartz JC, Arrang JM, Garbarg M, Traiffort E, editors. Psychopharmacology: The Fourth Generation of Progress. Raven Press; New York: 1995. [Google Scholar]
- 13.Arrang JM, Garbarg M, Schwartz JC. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. 1983;302:832–837. doi: 10.1038/302832a0. [DOI] [PubMed] [Google Scholar]
- 14.Schlicker E, Malinowska B, Kathmann M, Gothert M. Modulation of neurotransmitter release via histamine H3 heteroreceptors. Fundam Clin Pharmacol. 1994;8:128–137. doi: 10.1111/j.1472-8206.1994.tb00789.x. [DOI] [PubMed] [Google Scholar]
- 15.Arrang JM, Garbarg M, Lancelot JC, et al. Highly potent and selective ligands for histamine H3-receptors. Nature. 1987;327:117–123. doi: 10.1038/327117a0. [DOI] [PubMed] [Google Scholar]
- 16.Arrang JM, Roy J, Morgat JL, Schunack W, Schwartz JC. Histamine H3 receptor binding sites in rat brain membranes: modulations by guanine nucleotides and divalent cations. Eur J Pharmacol. 1990;188:219–227. doi: 10.1016/0922-4106(90)90005-i. [DOI] [PubMed] [Google Scholar]
- 17.Clark EA, Hill SJ. Differential effect of sodium ions and guanine nucleotides on the binding of thioperamide and clobenpropit to histamine H3-receptors in rat cerebral cortical membranes. Br J Pharmacol. 1995;114:357–362. doi: 10.1111/j.1476-5381.1995.tb13234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jansen FP, Rademaker B, Bast A, Timmerman H. The first radiolabeled histamine H3 receptor antagonist, [125I]iodophenpropit: saturable and reversible binding to rat cortex membranes. Eur J Pharmacol. 1992;217:203–205. doi: 10.1016/0014-2999(92)90851-t. [DOI] [PubMed] [Google Scholar]
- 19.Youdim MB, Buccafusco JJ. Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol Sci. 2005;26:27–35. doi: 10.1016/j.tips.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 20.Marlatt MW, Webber KM, Moreira PI, et al. Therapeutic opportunities in Alzheimer disease: one for all or all for one? Curr Med Chem. 2005;12:1137–1147. doi: 10.2174/0929867053764644. [DOI] [PubMed] [Google Scholar]
- 21.Cavalli A, Bisi A, Bertucci C, et al. Enantioselective nonsteroidal aromatase inhibitors identified through a multidisciplinary medicinal chemistry approach. J Med Chem. 2005;48:7282–7289. doi: 10.1021/jm058042r. [DOI] [PubMed] [Google Scholar]
- 22.Zhu Y, Xiao K, Ma L, et al. Design, synthesis and biological evaluation of novel dual inhibitors of acetylcholinesterase and beta-secretase. Bioorg Med Chem. 2009;17:1600–1613. doi: 10.1016/j.bmc.2008.12.067. [DOI] [PubMed] [Google Scholar]
- 23.Bembenek SD, Keith JM, Letavic MA, et al. Lead identification of acetylcholinesterase inhibitors-histamine H3 receptor antagonists from molecular modeling. Bioorg Med Chem. 2008;16:2968–2973. doi: 10.1016/j.bmc.2007.12.048. [DOI] [PubMed] [Google Scholar]
- 24.Bouzida D, Arthurs S, Colson AB, et al. Thermodynamics and kinetics of ligand-protein binding studied with the weighted histogram analysis method and simulated annealing. Pac Symp Biocomput. 1999;1999:426–437. doi: 10.1142/9789814447300_0042. [DOI] [PubMed] [Google Scholar]
- 25.Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: a simplified potential approach. J Med Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 26.Cheng A, Dixon SL. In silico models for the prediction of dose-dependent human hepatotoxicity. J Comput Aided Mol Des. 2003;17:811–823. doi: 10.1023/b:jcam.0000021834.50768.c6. [DOI] [PubMed] [Google Scholar]
- 27.Dixon SL, Merz KM., Jr One-dimensional molecular representations and similarity calculations: methodology and validation. J Med Chem. 2001;44:3795–3809. doi: 10.1021/jm010137f. [DOI] [PubMed] [Google Scholar]
- 28.Sussman JL, Harel M, Frolow F, et al. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science. 1991;253:872–879. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- 29.Ferrari D, Yang LH, Miles EW, Dunn MF. Beta D305A mutant of tryptophan synthase shows strongly perturbed allosteric regulation and substrate specificity. Biochemistry. 2001;40:7421–7432. doi: 10.1021/bi002892l. [DOI] [PubMed] [Google Scholar]
- 30.Hong HJ, Chen PY, Shih TC, et al. Computational pharmaceutical analysis of anti-alzheimer's Chinese medicine: Coptidis Rhizoma alkaloids. Mol Med Rep. 2011;5:142–147. doi: 10.3892/mmr.2011.630. [DOI] [PubMed] [Google Scholar]
- 31.Axe FU, Bembenek SD, Szalma S. Three-dimensional models of histamine H3 receptor antagonist complexes and their pharmacophore. J Mol Graph Model. 2006;24:456–464. doi: 10.1016/j.jmgm.2005.10.005. [DOI] [PubMed] [Google Scholar]