

Abstract
Glyoxylate carboligase (GCL) is a thiamin diphosphate (ThDP)-dependent enzyme, which catalyzes the decarboxylation of glyoxylate and ligation to a second molecule of glyoxylate to form tartronate semialdehyde (TSA). This enzyme is unique among ThDP enzymes in that it lacks a conserved glutamate near the N1′ atom of ThDP (replaced by Val51), or any other potential acid-base side chains near ThDP. The V51D substitution shifts the pH optimum to 6.0-6.2 (pKa of 6.2) for TSA formation from pH 7.0-7.7 in wild type GCL. This pKa is similar to the pKa of 6.1 for the [1′,4′-iminopyrimidine (IP)]/[4′-aminopyrimidinium (APH+)] protonic equilibrium, suggesting that the same group(s) control both ThDP protonation and TSA formation. The key covalent ThDP-bound intermediates were identified on V51D GCL by a combination of steady-state and stopped-flow circular dichroism methods, yielding rate constants for their formation and decomposition. It was demonstrated that active center variants with substitution at I393 could synthesize (S)-acetolactate from pyruvate solely, and acetylglycolate derived from pyruvate as acetyl donor and glyoxylate as acceptor, implying that this substitutent favored pyruvate as donor in carboligase reactions. Consistent with these observations, the I393A GLC variants could stabilize the pre-decarboxylation intermediate analogs derived from acetylphosphinate, propionylphosphinate and methyl acetylphosphonate in their IP tautomeric forms notwithstanding the absence of the conserved glutamate. The role of the residue at the position occupied typically by the conserved Glu controls the pH dependence of kinetic parameters, while the entire reaction sequence could be catalyzed by ThDP itself, once the APH+ form is accessible.
Introduction
The enzyme glyoxylate carboligase (GCL) catalyzes the thiamin diphosphate-(ThDP) assisted ligation of two molecules of glyoxylate to form (R)-tartronate semialdehyde (TSA) and CO2.1-7 GCL is a member of the homologous family of ThDP-dependent enzymes catalyzing reactions which begin with the decarboxylation of a 2-oxoacid substrate.2 It is a homotetramer and requires FAD in addition to ThDP and Mg2+ for catalysis.1-3 Despite its structural similarity to enzymes whose physiological substrate is pyruvate (pyruvate decarboxylases, pyruvate oxidases and especially acetohydroxyacid synthases), the sole substrate of GCL is the 2-aldoacid glyoxylate and the enzyme is unreactive with 2-ketoacids (Scheme 1A). It is important to note that GCL lacks both the conserved glutamate residue which is found in all other known ThDP enzymes, where its carboxylate group is within hydrogen bonding distance of N1′ of the 4′-aminopyrimidine ring of ThDP5,6 and any other potential acid-base side chain near ThDP. This unique property among ThDP enzymes makes GCL particularly useful for mechanistic studies where interpretation of pH-dependent behavior is sought. In GCL, valine (residue V51 in the wild type Escherichia coli enzyme) appears in the position expected for this Glu residue. It had been assumed that the conserved glutamate is essential for activation of the coenzyme in ThDP-dependent enzymes by facilitating the tautomerization of the weakly basic 4′-aminopyrimidine (AP) to the more basic 1′,4′-iminopyrimidine (IP) tautomer (Scheme 1A). Surprisingly, steady-state kinetic analyses revealed that the V51D and V51E variants are less active than GCL despite having higher rates of activation of the coenzyme (deprotonation at C2 of the thiazolium moiety). The turnover rates are two orders of magnitude lower for the V51D and seven fold lower for the V51E variant.5 However, as the coenzyme analog N3′-pyridyl-ThDP (in which N′1 is replaced by CH) leads to an inactive GCL holoenzyme, the 1′,4′-iminopyrimidine tautomer of ThDP must have a role in catalysis despite the absence of the conserved Glu.5 Circular dichroism (CD) evidence suggested that the V51D variant can in fact stabilize the IP tautomer of ThDP. 5 In order to understand the mechanism of GCL in more detail, it is important to identify the intermediate species on the reaction pathway or analogs for such species (Scheme 1A). Covalent adducts of ThDP with the pyruvate analogs (Scheme 2) methyl acetylphosphonate (MAP) and acetylphosphinate (AcPhi) are stable analogs of the first pre-decarboxylation intermediate formed between pyruvate and ThDP, C2α-lactylThDP (LThDP). These adducts, as well as LThDP itself, have provided many insights into mechanisms of ThDP-dependent enzymes which act on pyruvate.9-12 Importantly, it has been demonstrated for several ThDP enzymes that incremental addition of MAP and AcPhi produced a positive CD band near 300-310 nm, a band assigned to the 1′,4′-iminophosphonolactylThDP (PLThDP) and 1′, 4′-iminophosphinolactylThDP, respectively.12-14 Unfortunately, no synthesis of phosphonyl or phosphinyl analogs of glyoxylate (e.g., formylphosphinate) is yet published. To gain further understanding of substrate binding and specificity in GCL, we used the I393A GCL and V51D/I393A GCL, predicted by modeling based on the crystal structure of GCL,5 to allow GCL to accept pyruvate or its analogs as donor substrates. Modeling indicates that the side chain of Ile393 creates steric hindrance for substrates larger than glyoxylate, similarly to the molecular determinants for substrate specificity identified on acetohydroxyacid synthase II from E. coli.36
Scheme 1.

A. Mechanism of GCL reaction, including tautomerization/ionization states of thiamin. The possible reactions of a decarboxylase-carboligase with glyoxylate. B. The possible reactions of a decarboxylase-carboligase with pyruvate as sole substrates, acetolactate is produced. With the I393A GCL, in the presence of pyruvate and glyoxylate the mixed product acetylglycolic acid is the expected product.
Scheme 2.

Formation of covalent adducts of ThDP with pyruvate and with the pyruvate analogs MAP and AcPhi.
In this study we employ kinetics studies using steady state and stopped-flow circular dichroism (CD) spectroscopy to further investigate the mechanistic ability of GCL to function despite the absence of the “conserved” glutamate, as well as the determinants of its specificity for glyoxylate as substrate. By using V51D GLC, we demonstrated that the pH optimum of the TSA formation, and the pKa for the [IP]/[APH+] equilibrium for this variant are both significantly shifted to the acid side by the same magnitude as compared with GLC. Using the unfavorable pH of 7.6 and low temperature in the CD experiments allowed us to identify the key ThDP-bound intermediates on V51D GCL. By using MAP, AcPhi and propionylphosphinate (PrPhi) we now present CD evidence that GCL variants with Ile393 substituted by Ala can stabilize the pre-decarboxylation intermediates derived from pyruvate and 2-oxobutanoate analogs even in the absence of V51D substitution.
Experimental Procedures
Materials
Sodium glyoxylate, pyruvate, FAD, ThDP, ampicillin, tetracycline, 2, 3-dimethoxy-5-methyl-1,4-benzoquinone (Coenzyme Q0), DTT and NADH were obtained from Sigma (St. Louis, MO). Ammonium sulfate and SDS were obtained from BDH Chemicals Ltd. (Poole, UK). Yeast extract, peptone and agar were from DIFCO (Detroit, MI). Restriction enzymes were from New England BioLabs Inc. (Beverly, MA), Novagen (Darmstadt, Germany) and Promega (Madison,WI). All reagents were of analytical grade.
Synthesis of AcPhi was carried out according to the procedure reported by Baillie et al.15 PrPhi was prepared as reported earlier.16 Methyl acetylphosphonate (MAP) was synthesized using trimethylphosphite and acetylchloride as reagents according to Kluger et al.17 Purity and correct synthesis of all compounds were confirmed by NMR spectroscopy and mass spectrometry.
Expression, purification and site-directed mutagenesis of GCL
The E. coli gene gcl encoding GCL was cloned as previously described.18 GCL and its variants were expressed from pQE60-GCL plasmid and purified by affinity chromatography using HiTrap Chelating HP Ni column (5 mL, Pharmacia) and the Pharmacia ÄKTA prime FPLC system.5 The single substitutions were introduced in pQE60-GCL using the overlap extension site-directed mutagenesis method with the FailSafe™ kit (EPICENTRE Biotechnologies). The primers used were designed to introduce a modification in the restriction site as an aid in screening for clones with mutated plasmids. The mutations were verified by sequencing of the coding region of each plasmid used for expression of variant proteins.
Enzyme assays
TSA activity measurements
The rate of steady state formation of TSA by GCL was measured in a coupled enzyme assay which follows the disappearance of NADH with tartronate semialdehyde reductase (TSAR).19 TSAR was cloned, expressed and purified as previously described.5 The TSA activity was also measured by direct detection of TSA formation using Chirascan CD Spectrometer at 290 nm (Applied Photophysics, Leatherhead, U.K.).18 The reaction medium in 2.4 mL contained: 50 mM KH2PO4 (pH 7.7), 0.06 M KCl, 0.1 mM ThDP, 5 mM MgCl2 and 50 μM FAD. The glyoxylate was added to a final concentration of 10 mM and reaction was started by addition of GCL (25 μg) or V51D GCL (30 – 50 μg) at 37 °C and monitored for 300 s. The linear part of the progress curves was used to calculate the initial velocity (slope/s) using the Pro-Data Viewer program supplied by the manufacturer.
Acetolactate determination
The modified Westerfeld creatine-naphthol colorimetric method was used for assaying acetolactate (AL) formation20,21 with absorbance measurements made with a Beckman DU640 spectrophotometer.
Reaction of I393A GCL with pyruvate monitored by reduction of 2,6-dichlorophenoindophenol
The 2,6-dichlorophenolindophenol (DCPIP) reaction was conducted at 5 °C by monitoring its reduction at 600 nm in a 1 mL reaction mixture containing 0.5 mM ThDP, 5 mM MgCl2, 80 μM DCPIP and 20 μg of protein in 50 mM KH2PO4 (pH 7.0). The reaction was initiated by addition of pyruvate (0.025 mM – 5 mM) after 5 min of equilibration of I393A in the reaction mixture because of the trace amounts of DTT present in the protein. The linear part of the progress curves was used to calculate the initial velocity (slope/min).
CD Spectroscopy
CD experiments were carried out on a Chirascan CD Spectrometer from Applied Photophysics (Leatherhead, U.K.) in 1 cm path length cell in the near-UV (250-450 nm) wavelength region. For each experiment, conditions are presented in the figure legend.
pH dependence of CD bands
To study the pH dependence of the CD bands pertinent to the IP or AP forms, the pH of the protein solution was adjusted using a sympHony pH electrode (VWR, Batavia IL) and spectra were recorded after each adjustment.22 The pKa was determined from the fit of the log CD302-304 versus pH according to eq (1) for a single ionizing group or according to eq (2) for two ionizing groups using SigmaPlot v.10.0,
| (1) |
| (2) |
where x is pH.
The pH dependence of the TSA activity catalyzed by V51D GCL and GCL as measured by direct CD detection of TSA was determined in 50 mM KH2PO4 adjusted to the desired pH (pH 5.9-7.5) and containing 0.06 mM KCl, 0.1 mM ThDP, 5 mM MgCl2 and 50 μM FAD. Glyoxylate was added to a final concentration of 10 mM and the reaction was initiated by addition of GCL (25 μg) or V51D GCL (30 – 50 μg). The CD290/s slope was calculated at different pH values and a plot of log CD290/s versus pH was prepared. Data were fitted using eq (1) or eq (2).
Titration of GCL with AcPhi, PrPhi and MAP
CD spectra of GCL and its variants in the absence and in the presence of pyruvate analogs and PrPhi were recorded in the near-UV region at 290-550 nm at 25 °C. Conditions of each experiment are quoted under figure legends. A plot of ellipticity at 302 nm vs. concentration of substrate analog was constructed by subtracting the ellipticity of the enzyme at the same wavelength, but recorded in the absence of substrate analog, as reported earlier.23 The Kd values were calculated using the Hill equation (3):
| (3) |
where nH is a Hill coefficient. When nH = 1.0, S0.5 = Kd.
CD titration of I393A GCL with pyruvate
The I393A GCL (2.5 mg/mL, concentration of active centers = 38.6 μM) in 0.10 M KH2PO4 (pH 7.6) containing 5 mM MgCl2 and 0.50 mM ThDP was titrated with pyruvate (0.02-4.0 mM) at 4 °C. After 15 h at 4 °C, the protein was removed from the reaction mixture with an Ultracel-30K Centrifugal Filter Unit (Millipore) and CD spectra were recorded at 250-400 nm for (S)-acetolactate detection.
Stopped-flow CD spectroscopy
Kinetic traces were recorded on a Pi *-180 stopped-flow CD spectrometer (Applied Photophysics, U.K.) using a 10 mm path length cell at the specified wavelengths. Temperature was maintained at 6 °C or at 15 °C as stated in the figure legends.
Pre-steady state formation of covalent 1′,4′-iminopyrimidylThDP-bound intermediates on V51D GCL
A solution of V51D GCL (3.6 mg/mL, concentration of active centers = 55.6 μM) in 0.1 M KH2PO4 (pH 7.6) containing 0.5 mM ThDP, 2.5 mM MgCl2, 1.0 mM DTT, 10 μM FAD and 1% glycerol in one syringe was mixed rapidly with an equal volume of glyoxylate (2.0 mM) in the same buffer placed in the second syringe at 6 °C. The data points were collected at 302 nm over a period of 50 s. Data from four-five repetitive shots were averaged and were fitted to a double exponential (4), single exponential (5) or triple exponential (6) model using SigmaPlot v.10. 0:
| (4) |
| (5) |
| (6) |
where k1, k2 and k3 are the apparent rate constants, c is CDmax302 in the exponential rise to maximum model or CDmin302 in the exponential decay model.
Results
Rationale for selection of active center residues V51 and I393 for substitution
According to the x-ray structure of GCL (Figure 1, PDB code 2PAN), residues I393, L478, and I479, are located close to the thiazolium ring of ThDP and apparently form a pocket proximate to C2 which can accommodate a glyoxylate moiety, but which cannot easily accommodate a pyruvate molecule, with an extra methyl group.5 To understand the structure-function relationship, these residues were substituted by alanine or by the amino acids conserved at the homologous positions in the AHAS (Table 1). The single I393A, I393V, L478A, and I479V substitutions led to a lower catalytic efficiency (kcat/Km) of GCL in TSA formation [approximately 10 – 36 -fold lower as compared with GCL, as detected, in a coupled enzyme assay with TSA reductase (TSAR)19] (Table 1). As a result of substitutions I393A or V51D/I393A, GCL is converted to an acetolactate synthase which can use pyruvate as a substrate with a catalytic efficiency (kcat/Km) of about twenty times higher than that of the wild type GCL (Table 1). Formation of acetolactate was detected by a colorimetric measurement,20,21 and by direct CD detection of (S)-acetolactate (see below).18 The I393V, L478A and I479V variants did not show marked increases in acetolactate synthesis compared to the GCL.
The Val51 residue in GCL is critical for TSA synthesis. Although the V51D variant catalyzes proton exchange at C2 of the thiazolium ring of ThDP about 6-fold more rapidly than GCL, the TSA-ThDP complex accumulates in this enzyme and its turnover rate is lower by two orders of magnitude.5 The x-ray structure of GCL suggested that aspartate can be easily accommodated in place of the V51 position in the active center of GCL with its carboxylate group positioned in a hydrogen bonding distance from N1′ of the 4′-aminopyrimidine ring. On the other hand, we suggested that the carboxylate in the V51E variant can only make such a hydrogen bond if the structure is distorted5. Hence, the V51D substitution appeared to be more fruitful for the present investigation.
Figure 1.
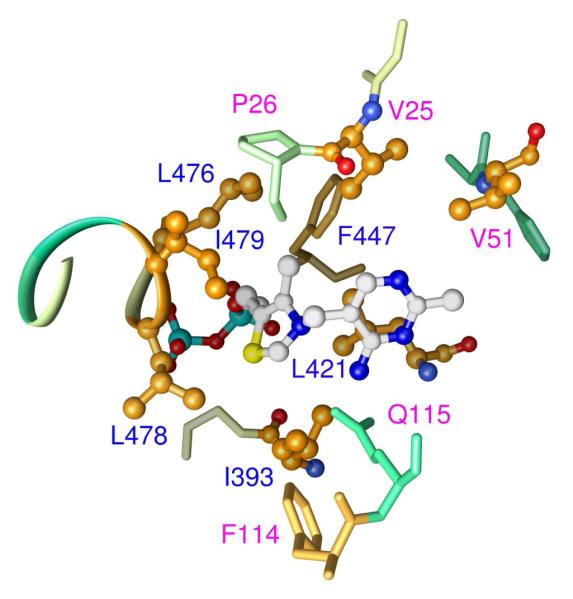
Structure of the active center of GCL demonstrating the hydrophobic environment of the thiazolium ring of ThDP. Figure was created using PyMOL based on the published structure of GCL (RCSB structure 2PAN) (5).
Table 1.
Kinetic properties of GCL and its active center variants.
| Tartronate semialdehyde formation a) |
Acetolactate formationb) | |||
|---|---|---|---|---|
| Enzyme | Specific activity μmol.min−1.mg−1 (%) |
kcat/Km (sec−1. mM−1) |
Specific activity nmol. min−1.mg−1 |
kcat/Km (sec−1.M−1) |
| Wild type | 17.5 ± 2.5 (100 %) |
21.0 | 8.2 ± 0.9 | 0.05 |
| I393A | 0.68 ± 0.02 (3.9 %) |
2.06 | 82.0 ± 3.4 | 0.86 |
| I393V | 0.93c) (5.3 %) |
0.65 | 3.0 ± 0.5 | 0.11 |
| L478A | 0.06c) (0.34 %) |
1.79 | 3.0 ± 0.14 | 0.37 |
| I479V | 0.84 ± 0.05 (4.8 %) |
0.58 | 4.0 ± 0.2 | 0.30 |
| V51D | 0.2 + 0.02d) (1.2%) |
1.3d) | - | - |
| V51D/ I393A |
n.d.e) | n.d.e) | 90.2 ± 3.0 | 0.94 |
The rate of TSA formation was measured in a coupled enzyme assay with TSAR.19
The modified colorimetric method by Westerfeld was used for measurement of acetolactate activity.20,21
For these two variants the activity was over a range of substrate levels and was measured once, so that the errors cannot be estimated.
Data from ref. 5 were used.
No activity could be detected.
The doubly substituted V51D/I393A GCL was created to address whether the combination of a carboxylate group, which can accelerate the activation of ThDP, with relief of the steric hindrance to the larger substrate pyruvate by replacement of Ile393 with Ala, might enable the variant to effectively stabilize the 1′,4′-iminopyrimidine tautomeric intermediates derived from pyruvate, and to effectively catalyze the synthesis of acetolactate. As shown in Table 1, the catalytic efficiency for acetolactate formation by V51D/I393A GCL is similar to that of the singly substituted I393A GCL despite the presence of the Asp residue at position 51. TSA formation by this doubly substituted variant cannot be detected in a coupled enzyme assay. The observation that the presence or absence of an aspartate residue at position 51 has little effect on the activity of a V51D/I393A variant in the formation of acetolactate (compared to the result with the single I393A substitution), implies that the effect of Asp51 on TSA formation is specifically related to the chemistry of the natural substrate glyoxylate.
Identification of ThDP-bound intermediates and their tautomeric forms on V51D GCL by CD
It was suggested that ThDP enzymes with a variety of active center environments can stabilize the IP form of ThDP, and that the presence of a glutamate residue within short hydrogen bonding distance of the N1′ atom of the 4′-aminopyrimidine ring is important for catalyzing the tautomeric equilibration and stabilizing the IP form12,13,24 It was further suggested that all C2α-tetrahedral ThDP adducts on the pathway for an enzymatic reaction will likewise exist as their 1′,4′-iminopyrimidine tautomers at pH values above the pKa of the APH+ ionization state. With these findings in mind, CD has now been applied to GCL and its variants.
The 1′,4′-iminopyrimidine tautomeric form of ThDP is stabilized on V51D GCL
It was demonstrated earlier that V51D GCL displayed a positive CD band at 302 nm in the resting state, suggested to correspond to the IP form of ThDP bound to this variant, not displayed on V51E variant.5 The following experimental evidence is presented here to confirm this suggestion: 1) The CD band at 302 nm was partially reduced on replacement of ThDP by thiamin 2-thiothiazolone diphosphate (ThTTDP), an analog of ThDP with the C2-H bond in the thiazolium ring substituted by a C2=S bond (not shown); 2) The amplitude of the CD band at 302 nm was reduced on addition of AcPhi, an analog of pyruvate, however, no signature for formation of 1′4′-iminophosphinolactylThDP was in evidence; 3) The positive CD band at 302 nm is replaced by a negative one at 290 nm on addition of glyoxylate, due to formation of the (R)-TSA product; 4) The intensity of the positive CD band at 302 nm depends on pH, in a manner similar to that reported earlier by the Jordan group for other ThDP-dependent enzymes.22
The effect of V51D substitution on equilibria of tautomeric and ionization states of ThDP
The AP form of ThDP on benzaldehyde lyase and benzoylformate decarboxylase, and both the AP and IP forms on pyruvate oxidase from Lactobacillus plantarum, as well as on human pyruvate dehydrogenase E1 component all exhibited pH-dependent behavior.22,25 Here, we carry out a pH titration to determine the pKa for the [IP]/[APH+] equilibrium (there is no evidence for the presence of a detectible concentration of the AP form on GCL so far) on V51D GCL. On reducing the pH from 7.95 to 5.72 the amplitude of the positive CD band at 302-304 nm observed on V51D GCL was reduced (Figure 2), indicating that the IP form of ThDP is dominant at pH 7.95, while the APH+ form is likely dominant at pH 5.72, with no CD band detected for this form so far. The pH titration data fits to a single proton titrating with a pKa of 6.1 for the [IP]/[APH+] equilibrium (Figure 2, bottom). On several ThDP enzymes studied so far, the pKa for the ([IP])/[APH+]) equilibrium is near the pH optimum of enzyme activity,22 which is pH 7.7 for GCL. The data thus suggest that the V51D substitution significantly shifted the pH optimum of enzyme activity to the acid side.
Figure 2.

The effect of pH on CD spectra of V51D GCL. (Top) Near-UV CD spectra of V51D GCL at different pH values. The V51D GCL from stock was diluted to a concentration of 2.6 mg/mL (concentration of active centers = 40 μM) in 0.1 M KH2PO4 containing 0.5 mM ThDP and 2.5 mM MgCl2. CD spectra were recorded after pH was adjusted to the desired value (pH 5.72 – pH 7.95). (Bottom) Dependence of the log CD304 versus pH. Data were fitted to a single ionizing group (eq 1).
To test this hypothesis, the rate of formation of (R)-TSA by GCL and V51D GCL was estimated directly by using CD290, as was reported earlier.18 Both GCL and V51D GCL displayed pH dependence for TSA formation. A plot of the log of the reaction slope detected from the progress curves (Figure 3A) (the molar ellipticity for (R)-TSA is not available in the literature) vs pH revealed a maximum activity at pH 7.0 – 7.7 for the GCL (Figure 3B), with the bell-shaped plot being described by two acid-base groups with apparent pK1 = 6.1 and pK2 above 8.0. For V51D GCL, the pH optimum of activity was shifted to pH 6.0 – 6.2 (Figure 4). The apparent pKa of 6.2 calculated from activity measurements correlates well with pKa of 6.1 determined for the [IP]/[APH+] equilibrium from the previous CD experiment, suggesting that the same group(s) control both events, probably the D51COO−—APH+ dyad (kinetically equivalent to, and indistinguishable from the D51COOH—IP dyad), perhaps assisting with release of product from the TSA-ThDP covalent complex (see later).
Figure 3.

The effect of pH on TSA activity of GCL. (A) Progress curves of TSA formation were recorded at different pH values using CD at 290 nm. Conditions of the experiment are presented under Experimental Procedures. The linear part of the progress curves was used to calculate the initial velocity (CD290/s) using the Pro-Data Viewer program. (B) Dependence of log (CD290/s) versus pH. The logCD290/s was plotted versus pH and data were fitted to eq (3) for two ionizing groups.
Figure 4.

The effect of pH on TSA activity of V51D GCL. (Top) Progress curves of TSA formation at different pH values using CD at 290 nm. Conditions of the experiment are presented under Experimental Procedure. The linear part of the progress curves was used to calculate the initial velocity (CD290/s) using the Pro-Data Viewer program. (Bottom) pH dependence of log (CD290/s). Data were fitted to eq. (2) for a single ionizing group.
Stabilization of the 1′,4′-iminopyrimidine form of ThDP-bound intermediates derived from glyoxylate on V51D GCL
Since V51D GCL is a slow variant (0.20 μmol.min−1.mg protein−1) compared to GCL (17.5 μmol.min−1.mg protein−1), the ThDP-bound intermediates could be detected by steady-state CD at low temperature (5 °C). On titration of V51D GCL by glyoxylate using concentrations in excess of the concentration of active centers (1 mM, 5 mM and 8 mM as compared with concentration of active centers of 9.5 μM), the positive CD band at 302 nm assigned to the IP form of ThDP on V51D GCL was reduced, and was finally replaced by a negative band on elevation of the temperature to 20 °C (Figure 5). This is largely due to the formation of (R)-TSA, which has a strong negative CD band at 290 nm.18 Formation of this product was confirmed by the CD spectrum of the reaction mixture after removal of protein: the negative CD band detected at 290 nm, is what was expected for (R)-TSA (not shown). A similar decrease in the amplitude of the positive CD band at 302 nm was observed when the experiment was carried out at pH 7.6 or pH 6.5 (Figure 5 presents data at pH 6.5) giving clear evidence of (R)-TSA (product) formation.
Figure 5.
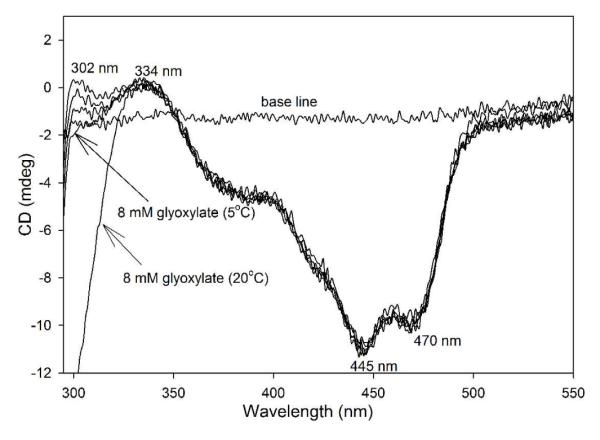
CD spectra of V51D GCL titrated by glyoxylate. The V51D GCL (1.23 mg/mL, concentration of active centers = 19.0 μM) in 0.1 M KH2PO4 (pH 6.5) containing 0.5 mM ThDP, 2.5 mM MgCl2,1.0 mM DTT, 10 μM FAD and 1% glycerol was titrated by glyoxylate (0.010-8 mM) at 5 °C. CD spectra were recorded in the near-UV region after each addition of glyoxylate. Observable changes in the intensity of the CD band at 302 nm were detected with 1 mM, 5 mM and 8 mM glyoxylate, but not with 0.01-0.5 mM glyoxylate. On increasing the temperature to 20 °C, formation of (R)-TSA was evident.
Time-resolved CD experiments on ThDP-bound intermediates on V51D GCL
Next, in a series of experiments, the reaction of V51D GCL with glyoxylate was monitored by stopped-flow CD at 302 nm at pH 7.6. In the first experiment conducted at 6 °C, the V51D GCL (concentration of active centers = 56 μM) in one syringe was mixed with 2 mM glyoxylate in the second syringe, resulting in a time dependent accumulation of a species with a positive ellipticity at 302 nm (Figure 6A). The reaction reached steady state within 5 s with rate constants of k1=6.2 s−1 and k1′ =0.58 s−1 (the data points were analyzed as a double exponential resulting in two rate constants k1 and k1′ for the fast and slow phases of the reaction), suggesting formation of a covalent glycolyl-ThDP pre-decarboxylation intermediate (Schemes 1A and 3 for rate constant assignment and Table 2 for values).
Figure 6.

ime course of the reaction of V51D GCL with glyoxylate monitored by stopped-flow CD. (A) Formation of 1′,4′-iminoglycolylThDP at pH 7.6 and 6 °C. V51D GCL (3.6 mg/mL, concentration of active centers = 55.6 μM) in 0.1 M KH2PO4 (pH 7.6) containing 0.5 mM ThDP, 2.5 mM MgCl2, 1.0 mM DTT and 10 μM FAD in one syringe was mixed with an equal volume of 2.0 mM glyoxylate in the same buffer in the second syringe. Data were fitted to a double exponential equation (eq 4). (B) The decarboxylation of 1′,4′-iminoglycolyl-ThDP and the formation of TSA-ThDP complex at pH 7.6 and 15 °C. V51D GCL (2.3 mg/mL, concentration of active centers = 35.5 μM) in one syringe was mixed with 2.0 mM glyoxylate in the second syringe at 15 °C. (C) Time-dependent decarboxylation of 1′,4′-iminoglycolylThDP with data from Figure 5 (B) expanded. Data were fitted to a single exponential (eq 5). (D) The formation of TSA-ThDP complex and TSA release by V51D GCL at pH 7.6. The V51D GCL (1.7 mg/mL, concentration of active centers = 26 μM) was pre-incubated with 0.5 mM glyoxylate for 15 min in one syringe, and was then mixed at 6 °C with 16 mM glyoxylate in the second syringe. The data were fitted to a triple exponential (eq 6).
Scheme 3.

Minimal mechanism and microscopic rate constants for V51D GCL
Table 2.
Kinetic analysis of the mechanism of V51D GCL by stopped-flow CD under different experimental conditions.
| Expta) | Conditions T° C, pH [glyoxylate] |
Glycolyl-ThDP formation k1 (s−1) |
Glycolyl-ThDP decarboxylation k2 (s−1) |
TSA-ThDP complex k3 (s−1) |
TSA release k4 (s−1) |
|---|---|---|---|---|---|
| 1 | 6 °C, pH 7.6 1 mM |
6.2 ± 0.6 b) 0.58 ± 0.03 |
n.d. c) | n.d. | n.d. |
| 2 | 15°C, pH 7.6 1 mM |
n.d. | 35 ± 4 | 2.9 ± 0.6 b) 0.56 ± 0.09 |
n.d. |
| 3 | 6°C, pH 7.6 8 mM |
n.d. | n.d. | 3.2 ± 1.1 b) 0.34 ± 0.13 |
0.16 ± 0.12 |
| 4 | 6 °C, pH 6.5 d) 1 mM |
n.d. | n.d. | 3.4 ± 0.4 b) 0.55 ± 0.04 |
0.04 ± 0.01 |
Experiments are numbered according to the text in Results. The experimental data are presented in Figures 6 A-D.
The experimental curves were treated as a double exponential leading to two rate constants fast and slow.
n.d., not detected.
Experiment was conducted at pH 6.5, favorable for V51D GCL as demonstrated in this publication.
The second experiment was conducted under similar conditions but at 15 °C. Initially, the CD signal at 302 nm decreased for at least the first 0.2 s (Figure 6B early times and Figure 6C for detail), followed by an increase in CD302 with exponential rise to maximum. We assign the final positive CD signal developed in this experiment to the TSA-ThDP covalent adduct, identified by a quench-NMR method.5 We assign the rate constant in Figure 6C (expansion of Figure 6B, the initial decrease) to decarboxylation of the glycolyl-ThDP pre-decarboxylation intermediate (k2= 35 s−1 in Figure 6C, see Scheme 1A, Scheme 3 and Table 2) and k3=2.9 s−1 and k3′ =0.56 s−1 for formation of TSA-ThDP complex on GCL (Figure 6B). The formation of glycolyl-ThDP pre-decarboxylation complex was too fast to be detected at 15 °C, but it most likely was detected at low concentration of glyoxylate at 6 °C in Figure 6A.
In the third experiment the V51D GCL was preincubated with 0.5 mM glyoxylate in one syringe to pre-form the covalent glycolyl-ThDP pre-decarboxylation intermediate and was then mixed with 16 mM glyoxylate in the second syringe at 6 °C. There was observed an initial fast rise (providing the rate constants for (R)-TSA-ThDP complex formation of k3=3.2 s−1 and k3′ =0.34 s−1) followed by a much slower decrease associated with the rate constant k4 = 0.16 s−1 for (R)-TSA (product) release from the enzyme of (Figure 6D).
This sequence of experiments could only be realized since pH 7.6 is unfavorable for the V51D GCL variant. At the same time, it allowed us to detect all key ThDP-bound intermediates on the reaction path, and calculate rate constants for formation of glycolyl-ThDP, its decarboxylation to the enamine, carboligation of the enamine with the second molecule of glyoxylate to provide the TSA-ThDP complex, and finally product release from GCL. Under these experimental conditions, product release is clearly the rate-limiting step.
Finally, when the experiment was conducted at pH 6.5 (more favorable pH for V51D GCL) and 6 °C, only the TSA-ThDP product accumulated, and its release was also detected (Figure 7); the positive ellipticity at 302 nm was developed within 5 s with rate constants k3 of 3.4 s−1 and k3′ of 0.55 s−1, followed by a slow decrease of the ellipticity with a rate constant k4 of 0.04 s−1, again confirming TSA release as the rate-limiting step in agreement with NMR data.5
Figure 7.
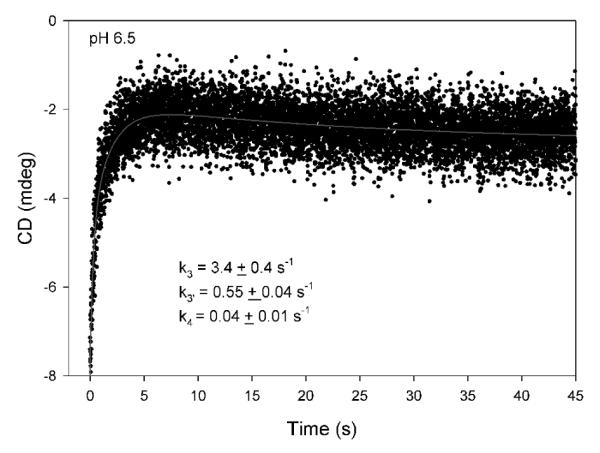
Reaction of V51D GCL with glyoxylate monitored by stopped-flow CD at pH 6.5. The V51D GCL (6.45 mg/mL, concentration of active centers = 99.6 μM) in 0.10 M KH2PO4 (pH 6.5) containing 0.5 mM ThDP, 2.5 mM MgCl2, 1.0 mM DTT and 10 μM FAD in one syringe was mixed with an equal volume of 2 mM glyoxylate in the second syringe. The reaction was monitored over 45 s at 6 °C. Data were fitted to a triple exponential (eq 7)
CD titration of V51D GCL with analogs of pyruvate
On addition of AcPhi (0.001-5 mM) or MAP (0.001-2.6 mM) to V51D GCL (concentration of active centers = 15.2 μM and 38 μM, respectively), the amplitude of the CD band at 302 nm assigned to the IP form of ThDP was reduced by approximately 57% in both cases but no 1′,4′-iminopyrimidine tautomeric forms of the pre-decarboxylation adduct resulting from addition of AcPhi and MAP were detected (not shown). The S0.5,AcPhi calculated for half of the reduction of the CD band at 302 nm was 0.18± 0.11 mM indicating that AcPhi is likely bound in the active centers of V51D GCL (Table 3). Similar experiment with MAP revealed an S0.5,MAP = 0.11 ± 0.09 mM for the CD band at 302 nm, indicating similar behavior with MAP (Table 3). Hence, the evidence for covalent complex formation with these substrate analogs is tentative.
Table 3.
Kinetic parameters for equilibrium binding of pyruvate and 2-oxobutanoate analogs to GCL variants.a)
| Substitutions in GCL |
Kd (S0.5) AcPhi (mM) |
Kd (S0.5), PrPhi (mM) |
Kd, MAP (mM) |
|---|---|---|---|
| I393A | 0.32 ± 0.02 (nH = 2.23) |
0.46 ± 0.03 (nH = 1.7) |
1.4 ± 0.09 |
| V51D | 0.18 ± 0.11b) | - | 0.11 ± 0.09b) |
| V51D/I393A | 0.37 ± 0.09 (nH = 1.29) |
0.28 ± 0.04 (nH = 1.1) |
0.11 ± 0.1 |
Kinetic parameters were determined in steady state CD experiments.
For V51D GCL no 1′,4′-iminopyrimidine tautomers from addition of AcPhi and MAP were detected. Instead, the intensity of the CD302 band was reduced by AcPhi and MAP with values of S0.5 presented in Table.
Interestingly, with the doubly substituted V51D/I393A GCL, we did observe changes in the CD spectrum on binding these pyruvate analogs (Table 3). We must emphasize, however, that while we have assigned CD spectroscopic signatures for the IP and AP forms of ThDP on the enzymes, we do not have such a characteristic signature for the APH+ form. It is therefore possible that addition of the pyruvate analogs to the V51D variant produces this ‘CD silent’ form [(now identified on three ThDP enzymes by solid-state NMR methods26,27]. Alternatively, an adduct may be formed whose thiazolium ring is sufficiently distorted to eliminate, or radically shift the CD band, a less likely scenario.
Stabilization of the 1′,4′-iminopyrimidine form of ThDP-bound intermediates derived from glyoxylate on GCL
On titration of GCL by glyoxylate in a steady-state CD experiment, formation of positive CD bands at 302 nm and 337 nm was observed. Maximum intensity of the CD band at 302 nm was reached at concentrations of glyoxylate equivalent or slightly higher than the concentration of active centers. Increasing the concentration of glyoxylate produced (R)-TSA (SI Figure S1, top).
In a single turnover experiment (with [GCL subunits]>[glyoxylate]) using stopped-flow CD at 302 nm, the reaction reached steady-state within approximately 5 s with rate constants of k1 = 1.13 s−1 and k1′ = 0.28 s−1 (SI Figure S1, bottom), at least five times slower than with V51D GCL (Figure 6A). The CD band at 302 nm can be assigned to the first covalent pre-decarboxylation intermediate, 1′,4′-iminopyrimidinylglycolylThDP. Support for this assignment comes from a comparison of the rate constants determined for the individual steps in GCL by NMR methods.5 It was demonstrated that formation of the first covalent pre-decarboxylation intermediate from glyoxylate was the rate-limiting step for GCL in TSA (product) formation.5 The CD band at 337 nm was not assigned so far. It developed simultaneously with the one at 302 nm on addition of low concentrations of glyoxylate, but it did not respond to increasing concentration of glyoxylate: the CD band at 302 nm was replaced by a negative CD band at 290 nm due to TSA release (SI, Figure S1, top).
Identification of ThDP-bound intermediates on GCL variants with I393A substitution
Stabilization of 1′,4′-iminopyrimidinylThDP pre-decarboxylation intermediates from pyruvate on I393A GCL and V51D/I393A GCL
It is evident from kinetic studies (Table 1) that on I393A substitution the GCL becomes an acetolactate synthase, accepting pyruvate as a substrate. In this study, by using pyruvate and 2-oxobutanoic acid analogs we were able to demonstrate that I393A GCL can stabilize the 1′,4′-iminopyrimidine ThDP pre-decarboxylation intermediates even in the absence of aspartate in position 51. The V51D/I393A GCL behaves similarly to I393A GCL.
A steady state CD titration of I393A GCL by pyruvate at 4 °C revealed two positive CD bands, one at 302 nm and the second at 335 nm (Figure 8A). The CD band at 335 nm was not assigned but it was similar to the CD band at 330 nm observed for V51D GCL (so far unassigned, but also seen at 337 nm on GCL). It was suggested5 that the weak CD band at 330 nm possibly originated from FAD.
Figure 8.

CD titration of I393A GCL by pyruvate at 6 °C. (A) CD spectra of I393A GCL (2.0 mg/mL, concentration of active centers = 30.8 μM) in 0.1 M KH2PO4 (pH 7.6) containing 0.5 mM ThDP, 2.5 mM MgCl2, 1.0 mM DTT and 10 μM FAD in the absence and in the presence of 0.020-0.70 mM pyruvate. Each spectrum was recorded after pre-incubation of I393A GCL with pyruvate for 20 min at 6 °C. (B) Dependence of the ellipticity at 302 nm on pyruvate concentration. Inset shows the plot of the CD302 data points versus [pyruvate] at concentrations less than 0.80 mM. Data were fitted to a Hill equation (3).
The CD band at 302 nm approached its maximum intensity at concentrations of pyruvate equivalent to the concentration of active centers (Figure 8A). A plot of ellipticity at 302 nm vs. [pyruvate] clearly shows a biphasic curve with saturation at concentrations of pyruvate below 1 mM (Kd, pyruvate = 8.2 μM) (Fig. 8B). At concentrations of pyruvate above 1 mM the plot was not saturated, rather the release of (S)-acetolactate was detected and was confirmed by a CD spectrum of the reaction mixture after protein was removed (SI Figure S2). Similar data were obtained for V51D/I393A GCL; a plot of ellipticity at 302 nm vs. [pyruvate] displayed saturation at concentrations of pyruvate < 500 μM with S0.5,pyruvate of 1.72 μM (not shown).
The production of (S)-acetolactate requires decarboxylation of pyruvate in the active centers of I393A GCL and V51D/I393A GCL resulting in formation of the enamine intermediate, then subsequent ligation to a second pyruvate (Scheme 1B, bottom). The intermediacy of the enamine was confirmed by the reaction of I393A GCL with pyruvate in the presence of DCPIP under conditions similar to those in the CD experiment. The plot of the initial velocity (slope/min) of DCPIP reduction vs. [pyruvate] was again biphasic, displaying saturation at low concentrations of pyruvate (Kd,pyruvate = 39 μM) (not shown).
Next, we demonstrated that glyoxylate could also be an acceptor of the enamine intermediate derived from pyruvate on I393A GCL. The positive CD band formed at 302-304 nm on I393A GCL on addition of stoichiometric amounts of pyruvate, was replaced by a negative CD band in the presence of glyoxylate indicating formation of acetylglycolic acid derived from pyruvate as acetyl donor and glyoxylate as acceptor (Scheme 1B, top) (Figure 9, Top). However, the acetylglycolate was not optically stable and could no longer be detected by CD after protein was removed from a reaction mixture. This is readily explained as the chiral carbon can undergo racemization being located in-between two carbonyl functions, a ketone and a carboxylic acid. The formation of acetylglycolate as the only carboligase product was proven by the absence of CD bands corresponding to (S)-acetolactate (positive band at 300 nm) or (R)-TSA (negative band at 290 nm). Both products are optically stable and should be detected if formed after removal of protein. Upon mixing I393A GCL incubated with pyruvate in one syringe with glyoxylate in the second syringe in a stopped-flow CD experiment, a positive ellipticity developed at 302 nm within 5 s with k3=2.69 s−1, and k3′ = 0.63 s−1, followed by a decrease in ellipticity with k4 = 0.02 s−1 suggesting formation of a acetylglycolate-ThDP complex on the enzyme (it being responsible for the CD band at 302 nm) and its release (Figure 9 middle, Scheme 1B top). On increasing the concentration of glyoxylate to 4 mM, only the release of acetylglycolate could be detected (k4 = 0.01 s−1) [Figure 9 bottom, Scheme 1B top]. These experiments suggest that the acetylglycolate-ThDP can be detected by stopped-flow CD at 302 nm and its release is the rate-limiting step. In a stopped-flow CD experiment with V51D/I393A GCL, the enzyme in one syringe was mixed with pyruvate in the second syringe but in the absence of glyoxylate (SI Figure S3). Again, the positive ellipticity at 302 nm was developed within 5 s with a k3= 2.22 s−1 and k3′ = 0.50 s−1 which can be assigned to formation of the acetolactate-ThDP complex. The origin of the CD band at 302 nm will be discussed under Discussion.
Figure 9.

Formation of an acetylglycolate product by I393A GCL as detected by CD. (Top) CD spectra of I393A GCL recorded in the presence of 1 mM pyruvate and on addition of 1 mM glyoxylate at 6 °C. The I393A GCL was diluted to a concentration of active centers of 30.8 μM in 0.1 M KH2PO4 (pH 7.6) containing 0.5 mM ThDP, 2.5 mM MgCl2, 1.0 mM DTT, 10 μM FAD and 1% glycerol. (Middle) Time-dependent formation of an acetylglycolate-ThDP complex and product release. The I393A GCL (4.69 mg/mL, concentration of active centers = 72.4 μM) in buffer as in (Top) was pre-incubated with 1 mM pyruvate in one syringe and was then mixed at 6 °C with 1.0 mM glyoxylate in the second syringe. Data were fitted to a triple exponential (eq 7). (Bottom) Time-dependent release of a acetylglycolate product. Conditions were the same as in (Middle) but 4 mM glyoxylate was present in the second syringe.
The I393A substitution enables GCL to stabilize the 1′,4′-iminopyrimidyl tautomer of stable pre-decarboxylation ThDP-bound intermediates from pyruvate analogs
On addition of AcPhi to I393A GCL (Figure 10) and V51D/I393A GCL (not shown), the positive CD band again developed at 302 nm and reached a plateau level, indicating formation of the 1′,4′-iminophosphinolactyl-ThDP intermediate, and providing half-saturation values of S0.5,AcPhi of 0.32 mM (I393A GCL) and 0.37 mM (V51D/I393A GCL) (Table 3). The I393A GCL displayed positive cooperativity on AcPhi binding (nH = 2.23). In contrast, for I393V GCL and for GCL, there was no significant change in the CD spectra on addition of AcPhi, most likely because there is little or no binding of AcPhi to the active centers (data not shown). Similar experiments carried out on I393A with MAP displayed the band at 302 nm, which we ascribe to the 1′,4′-iminophosphonolactylThDP (data not shown) and allowed calculation of Kd,MAP = 1.4 mM (Table 3). MAP thus also binds to I393A GCL, although it is a weaker substrate analog than AcPhi, as was found with other ThDP enzymes.24 For V51D/I393A GCL the Kd, MAP was 0.11 mM.
Figure 10.

CD spectra of I393A GCL titrated with AcPhi. (A)The I393A GCL (2.0 mg/mL, concentration of active centers = 30.8 μM) in 0.10 M KH2PO4 (pH 7.6) containing 5 mM MgCl2 and 0.50 mM ThDP, was titrated by AcPhi (0.050-4.0 mM) at 25 °C. (B) Dependence of the CD at 302 nm on concentration of AcPhi. Data were fitted to a Hill equation (eq 3).
CD evidence for formation of the 1′,4′-iminophosphinopropionylThDP from PrPhi
In addition to AcPhi, the I393A GCL and V51D/I393A GCL could also bind PrPhi, [CH3CH2C(=O)P(H)(=O)O−], an analog of 2-oxobutanoate, and form the 1′,4′-iminophosphinopropionylThDP, providing values of Kd, PrPhi of 0.46 mM (I393A GCL) and 0.28 mM (V51D/I393A GCL), similar to the value obtained with AcPhi (Table 3). The I393A GCL displayed positive cooperativity on PrPhi binding, similar to that observed with AcPhi. Apparently, the V51D substitution does indeed affect the cooperativity of pyruvate analog binding to the I393A variant (Table 3) for reasons unclear at this time.
A major conclusion from the above CD studies is that the pre-decarboxylation intermediate analogs derived from AcPhi, PrPhi and MAP can be stabilized on I393A and V51D/I393A variants in their 1′,4′-iminopyrimidine tautomeric forms. It is significant that the V51D/I393A variant behaves similarly to the I393A variant in this regard. The observation that the presence or absence of an aspartate at position 51 has little effect on acetolactate production by V51D/I393A GCL in comparison with a singly substituted I393A GCL, implies that its effect is specifically on the formation of TSA.
Discussion
Following the initial report on the structure of GCL,5 we here explored two important issues remaining to be elucidated on the enzyme: (1) control of substrate specificity and (2) role of the residue opposite the N1′ atom of ThDP, typically occupied by the highly conserved glutamate.
Control of substrate specificity at position 393
Experiments exploiting different techniques show that the putative higher reactivity of the aldehyde group of glyoxylate is not the important determinant of the substrate specificity of GCL, but rather, the structure of the active site pocket. Particularly, residue Ile393 restricts the ability of GCL to form covalent complexes of ThDP with donor substrates larger than glyoxylate.
Remarkably, once the size of the residue at position 393 is reduced to alanine, pyruvate is accepted as an alternate substrate, and evidence for ThDP-bound intermediates derived from pyruvate could be detected. The CD band at 302 nm observed on I393A at low pyruvate concentrations (Fig. 8A) indicates that a covalent intermediate (or intermediates) is formed on GCL at low pyruvate. We believe this intermediate is one with tetrahedral substitution at C2α, but is not the enamine. Since (R)-acetolactate is produced at higher pyruvate concentrations, the enamine must be present, albeit at a concentration too small to detect. A possible source of the band at 302 nm is the HEThDP, the conjugate acid of, and at protolytic equilibrium with the enamine, or the bound carboligase product acetolactylThDP (Scheme 1A, 1B). An increase in concentration of pyruvate to > 1 mM shifts this equilibrium to acetolactate release. Both the HEThDP and acetolactylThDP would be expected to exist in their 1′,4′-iminopyrimidyl tautomeric form on the bases of much precedent on several ThDP enzymes,12 and either species could be responsible for the CD band at 302 nm.
The V51D/I393A GCL behaves similarly to the I393A GCL according to a kinetic study of acetolactate formation, and from CD studies on stabilization of the pre-decarboxylation intermediate analogs derived from AcPhi, MAP and PrPhi. The pre-decarboxylation intermediate analogs derived from AcPhi, PrPhi and MAP could be stabilized on I393A GCL and V51D/I393A GCL in their 1′,4′-iminopyrimidine tautomeric forms.
The observation that the presence or absence of an aspartate at position 51 has little effect on the V51D/I393A GCL implies that it is specifically effective on TSA formation from the natural substrate glyoxylate.
With these observations on the I393 substituted GCLs, we suggest that variants engineered at this position would be expected to convert this enzyme to a useful tool in chiral synthesis. For example, both pyruvate and glyoxalate are excellent acceptors for the enamine derived from pyruvate, k3=2.2 s−1 for acetolactate formation and k3=2.7 s−1 for acetylglyoxalate formation (Scheme 1B).
As an aside, we also emphasize that CD has clearly been shown to provide an excellent direct kinetic assay for product formation, much simpler than any coupled assay, or the classical Westerfeld method for acetoin determination.
The role of the conserved glutamate
GCL has several properties, which set it apart from the other members of the pyruvate decarboxylase – pyruvate oxidase subfamily of ThDP-dependent enzymes.28-31 The conserved glutamate residue hydrogen bonded to N1′ of ThDP, found in all the other ThDP-dependent enzymes, is replaced by a valine residue, and the sole substrate of GCL is the 2-aldoacid glyoxylate rather than a 2-ketoacid (Scheme 1A). In addition, not only is the conserved glutamate missing, there are no potential acid-base side chains in proximity of the ThDP near the C2 thiazolium atom. The recently accepted paradigm regarding the mechanism of ThDP-dependent enzymes12,24 suggests that the conserved Glu residue is essential to the tautomerization of ThDP, and that the 1′,4′-iminopyrimidine-, but not the 4′-aminopyrimidine-tautomer, is sufficiently basic to deprotonate the thiazolium C2-H and thus lead to the active carbanion/ylide form of the coenzyme. The studies here reported provide convincing evidence that with GCL, 1) only the unusual 1′,4′-iminopyrimidine tautomer, rather than the canonical AP form is detected, and 2) the [IP]/[APH+] protolytic equilibrium functions in GCL, allowing the IP form to play its central role in the absence, as well as in the presence of a group which is hydrogen bonded to N1′.
The key ThDP-bound intermediates derived from glyoxylate were identified on the reaction path of V51D GCL. A comparison of the rate constants for 1′,4′-iminoglycolyl-ThDP formation, its decarboxylation to the enamine, TSA-ThDP complex formation, and finally TSA product release from GCL, indicates that under the experimental conditions, TSA product release is the rate-limiting step, in agreement with the NMR approach.5 In the NMR approach5, covalent ThDP-bound intermediates are released from the enzymes, while in this study, we observe enzyme-bound intermediates directly; both methods lead to the same conclusions.
The 1′,4′-iminoglycolyl-ThDP was detected on GCL when the enzyme was titrated by equivalent amounts of glyoxylate. The rate constant of 1′,4′-iminoglycolyl-ThDP formation was at least five times slower than that for V51D GCL.
A major surprise of our results in toto is that all steps with the exception of product release appear to have rates comparable or even better (when adjusted to the pH optimum of the variant) with Asp in place of Val at position 51 (Table 2).
There is also strong evidence that adding the acceptor substrate significantly increases the rate of decarboxylation, as seen in the glyoxylate concentration dependence of the kinetics of product formation. This may be yet another example of active sites communication where glyoxylate in the active site of one subunit increases the rate of decarboxylation in the active site of a second subunit in the ‘functional dimer’ typical of ThDP decarboxylases. Similar observations had already been reported on benzoylformate decarboxylase32 and yeast pyruvate decarboxylase,33 suggesting that the behavior reflects alternating active center mechanism in the functional dimer.
The presence of a water molecule in proximity to the N1′-atom might provide an explanation for the ability of GCL to catalyze the tautomeric equilibration required for the reaction. It should be noted that the structures of ThDP-dependent enzymes in the RCSB database show water molecules in proximity to the N1′ atom.5 Given the high concentration of water in all of these crystals, the presence of such water in GCL would not be surprising. The transient charge transferred to the 4′-aminopyrimidine moiety in the course of ThDP activation (Scheme 1A) and other steps in the mechanistic cycle of GCL can presumably be shared with a water molecule H-bonded to N1′ and with other groups in its hydrogen bonding network. Significant residual activity is seen in a variant of the homologous enzyme AHAS II when the conserved glutamate (Glu47) is replaced by alanine.34 When the conserved glutamates on the E1 component of the E. coli pyruvate dehydrogenase complex11 and of the yeast pyruvate decarboxylase35 are replaced by alanine, there is similar residual activity detected. It is reasonable to assume that a water molecule can fit into the gap created by such substitutions, and that this water might assist the tautomerization. It is relevant to mention that the pKa in models for the 4′-aminopyrimidinium ring of ThDP is about 4.85, so that only a modest pKa perturbation would be required in the active centers of ThDP enzymes to match this.22 It was pointed out that the ThDP binding site in GCL appears in fact to have a larger proportion of aliphatic, nonpolarizable residues in the region of the thiazolium and 4′-aminopyrimidine rings than is observed for other ThDP-dependent enzyme, which should favor the stability of the formally charge-neutral ylide on the thiazolium ring (in Scheme 1A).5 We suggest that the major need is for some manner of protonating N1′ so that the APH+ form can be generated, the source of the IP tautomer. The fact that the APH+ form was indeed observed by solid-state NMR experiments on each of the three enzymes examined (yeast pyruvate decarboxylase, and the E1 components of both the pyruvate and 2-oxoglutarate dehydrogenase complexes from Escherichia coli) supports this hypothesis.26
That said, the V51D substitution once more supports the suggestion that the pKa for the ([IP]+[AP])/[APH+] equilibrium coincides with the pH optimum of activity, ensuring the presence of all three forms for catalysis. This is the sixth ThDP enzyme to date supporting this notion. Having no other acid-base candidates near the thiazolium ring, allows us to conclude that (a) ThDP itself in its various forms can indeed catalyze all of the reactions of the GCL pathway and (b) the amino acid opposite the N1′ atom of ThDP (typically the conserved glutamate) is responsible for both the pKa of the APH+ form and the pH dependence of the activity profiles.
Perhaps the most interesting puzzle remaining concerning GCL is the selective advantage of the valine residue (Val51) in place of the “conserved” glutamate. It was previously shown that the V51D GCL and V51E GCL are less active than GCL in the synthesis of TSA from glyoxylate, despite having higher rates of activation (deprotonation at C2) of the coenzyme.5 With the results here presented, it would appear that it is in product release rate, rather than the rates of any intermediate steps where the valine has a dramatic effect. The structural basis of these findings remain to be elucidated.
Supplementary Material
Abbreviations
- GCL
wild type glyoxylate carboligase
- I393A GCL
V51D GCL and V51D/I393A GCL are GCL variants at the indicated positions
- TSAR
tartronate semialdehyde reductase
- AHAS
acetohydroxyacid synthase
- ThDP
thiamin diphosphate
- TSA
tartronate semialdehyde
- AL
acetolactate
- MAP
methyl acetylphosphonate
- AcPhi
acetylphosphinate
- PrPhi
propionylphosphinate
- LThDP
C2α-lactylthiamin diphosphate
- PLThDP
phosphonolactylthiamin diphosphate (the MAP – ThDP adduct)
- DCPIP
2,6-dichlorophenolindophenol
- PEG
polyethylene glycol
- AP
4′-aminopyrimidine tautomeric form of ThDP
- IP
1′,4′-iminopyrimidine tautomeric form of ThDP
- APH+
the N1′-protonated 4′-aminopyrimidinium form of ThDP
- CD
circular dichroism.
Footnotes
This work was supported by U.S. - Israel Binational Science Foundation Grant 2007-129 (to BS, DMC and FJ) and NIH grant GM050380 to FJ.
Figures S1-S3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Chang YY, Wang AY, Cronan JE., Jr Molecular cloning, DNA sequencing, and biochemical analyses of Escherichia coli glyoxylate carboligase. An enzyme of the acetohydroxy acid synthase - pyruvate oxidase family. J. Biol. Chem. 1993;268:3911–3919. [PubMed] [Google Scholar]
- 2.Gupta NK, Vennesland B. Glyoxylate Carboxylase of Escherichia coli: a Flavoprotein. J. Biol. Chem. 1964;239:3787–3789. [PubMed] [Google Scholar]
- 3.Gupta N, Vennesland B. Glyoxylate carboligase of Escherichia coli: some properties of the enzyme. Arch. Biochem. Biophys. 1966;113:255–264. doi: 10.1016/0003-9861(66)90185-8. [DOI] [PubMed] [Google Scholar]
- 4.Cromatie TH, Walsh CT. Escherichia coli glyoxalate carboligase. Properties and reconstitution with 5-deazaFAD and 1,5-dihydrodeazaFADH2. J. Biol. Chem. 1976;251:329–333. [PubMed] [Google Scholar]
- 5.Kaplun A, Binshtein E, Vyazmensky M, Steinmetz A, Barak Z, Chipman DM, Tittmann K, Shaanan B. Glyoxylate carboligase lacks the canonical active site glutamate of thiamin-dependent enzymes. Nat. Chem. Biol. 2008;4:113–118. doi: 10.1038/nchembio.62. [DOI] [PubMed] [Google Scholar]
- 6.Shaanan B, Chipman DM. Reaction mechanism of thiamin diphosphate enzymes: new insights into the role of a conserved glutamate residue. FEBS J. 2009;276:2447–2453. doi: 10.1111/j.1742-4658.2009.06965.x. [DOI] [PubMed] [Google Scholar]
- 7.Chipman DM, Barak Z, Shaanan B, Vyazmensky M, Binshtein E, Belenky I, Temam V, Steinmetz A, Golbik R, Tittmann K. Origin of the specificities of acetohydroxyacid synthases and glyoxylate carboligase. J. Molecular Catalysis B:Enzymatic. 2009;61:50–55. [Google Scholar]
- 8.O’Brien TA, Kluger R, Pike DC, Gennis RB. Phosphonate analogues of pyruvate. Probes of substrate binding to pyruvate oxidase and other thiamin pyrophosphate-dependent decarboxylases. Biochim. Biophys. Acta. 1980;613:10–17. doi: 10.1016/0005-2744(80)90186-2. [DOI] [PubMed] [Google Scholar]
- 9.Wille G, Meyer D, Steinmetz A, Hinze E, Golbik R, Tittmann K. The catalytic cycle of a thiamin diphosphate enzyme examined by cryocrystallography. Nat. Chem. Biol. 2006;2:324–328. doi: 10.1038/nchembio788. [DOI] [PubMed] [Google Scholar]
- 10.Tittmann K, Wille G. X-ray crystallographic snapshots of reaction intermediates in pyruvate oxidase and transketolase illustrate common themes in thiamin catalysis. J. Mol. Catal. B: Enzymatic. 2009;61:93–99. [Google Scholar]
- 11.Nemeria NS, Arjunan P, Chandrasekhar K, Mossad M, Tittmann K, Furey W, Jordan F. Communication between thiamin cofactors in the Escherichia coli pyruvate dehydrogenase complex E1 component active centers: evidence for a “direct pathway” between the 4′-aminopyrimidine N1′ atoms. J. Biol. Chem. 2010;285:11197–11209. doi: 10.1074/jbc.M109.069179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemeria NS, Chakraborty S, Balakrishnan A, Jordan F. Reaction mechanisms of thiamin diphosphate enzymes: defining states of ionization and tautomerization of the cofactor at individual steps. FEBS J. 2009;276:2432–2446. doi: 10.1111/j.1742-4658.2009.06964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nemeria N, Chakraborty S, Baykal A, Korotchkina LG, Patel MS, Jordan F. The 1′,4′-iminopyrimidine Tautomer of thiamin diphosphate is poised for catalysis in asymmetric active centers on enzymes. Proc. Natl. Acad. Sci. USA. 2007;104:78–82. doi: 10.1073/pnas.0609973104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baykal AT, Kakalis L, Jordan F. Electronic and nuclear magnetic resonance spectroscopic features of the 1′,4′-iminopyrimidine tautomeric form of thiamin diphosphate, a novel intermediate on enzymes requiring this coenzyme. Biochemistry. 2006;45:7522–7528. doi: 10.1021/bi060395k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baillie AC, Wright BJ, Wright K. U.S. Patent No 4,339,443. 1982
- 16.Shim DJ, Nemeria NS, Balakrishnan A, Patel H, Song J, Wang J, Jordan F, Farinas ET. Assignment of function to histidines 260 and 298 by engineering the E1 component of the Escherichia coli 2-oxoglutarate dehydrogenase complex; substitutions that lead to acceptance of substrates lacking the 5-carboxyl group. Biochemistry. 2011;50:7705–7709. doi: 10.1021/bi200936n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kluger R, Pike DC. Active site generated analogues of reactive intermediates in enzymic reactions. Potent inhibition of pyruvate dehydrogenase by a phosphonate analogue of pyruvate. J. Amer. Chem. Soc. 1977;99:4504–4506. doi: 10.1021/ja00455a052. [DOI] [PubMed] [Google Scholar]
- 18.Vinogradov M, Kaplun A, Vyazmensky M, Engel S, Golbik R, Tittmann K, Uhlemann K, Meshalkina L, Barak Z, Hübner G, Chipman DM. Monitoring the acetohydroxy acid synthase reaction and related carboligations by circular dichroism spectroscopy. Anal. Biochem. 2005;342:126–133. doi: 10.1016/j.ab.2005.03.049. [DOI] [PubMed] [Google Scholar]
- 19.Chung ST, Tan RT, Suzuki I. Glyoxylate carboligase of Pseudomonas oxalaticus. A possible structural role for flavine-adenine dinucleotide. Biochemistry. 1971;10:1205–1209. doi: 10.1021/bi00783a017. [DOI] [PubMed] [Google Scholar]
- 20.Epelbaum S, Chipman DM, Barak Z. Determination of products of acetohydroxy acid synthase by the colorimetric method, revised. Anal. Biochem. 1990;191:96–99. doi: 10.1016/0003-2697(90)90393-n. [DOI] [PubMed] [Google Scholar]
- 21.Engel S, Vyazmensky M, Geresh S, Barak Z, Chipman DM. Acetohydroxyacid synthase: a new enzyme for chiral synthesis of R-phenylacetylcarbinol. Biotechnology and Bioengineering. 2003;83:833–840. doi: 10.1002/bit.10728. [DOI] [PubMed] [Google Scholar]
- 22.Nemeria N, Korotchkina L, McLeish MJ, Kenyon GL, Patel MS, Jordan F. Elucidation of the chemistry of enzyme-bound thiamin diphosphate prior to substrate binding: defining internal equilibria among tautomeric and ionization states. Biochemistry. 2007;46:10739–10744. doi: 10.1021/bi700838q. [DOI] [PubMed] [Google Scholar]
- 23.Nemeria NS, Korotchkina LG, Chakraborty S, Patel MS, Jordan F. Acetylphosphinate is the most potent mechanism-based substrate-like inhibitor of both the human and Escherichia coli pyruvate dehydrogenase components of the pyruvate dehydrogenase complex. Bioorg. Chem. 2006;34:362–379. doi: 10.1016/j.bioorg.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kluger R, Tittmann K. Thiamin Diphosphate Catalysis: Enzymic and Nonenzymic Covalent Intermediates. Chem. Rev. 2008;108:1797–1833. doi: 10.1021/cr068444m. [DOI] [PubMed] [Google Scholar]
- 25.Nemeria NS, Chakraborty S, Balakrishnan A, Jordan F. Reaction mechanisms of thiamin diphosphate enzymes: defining states of ionization and tautomerization of the cofactor at individual steps (minireview) FEBS J. 2009;276:2432–2446. doi: 10.1111/j.1742-4658.2009.06964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balakrishnan A, Paramasivam S, Chakraborty S, Polenova T, Jordan F. Solid-State Nuclear Magnetic Resonance Studies Delineate the Role of the Protein in Activation of Both Aromatic Rings of Thiamin. J. Am. Chem. Soc. 2012;134:665–672. doi: 10.1021/ja209856x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paramasivam S, Balakrishnan A, Dmitrenko O, Gobert A, Begley TP, Jordan F, Polenova T. Solid-state NMR and density function theory studies of ionization states of thiamin. J. Phys. Chem. 2011;115:730–736. doi: 10.1021/jp109765b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller YA, Schulz GE. Structure of the thiamin- and flavin-dependent enzyme pyruvate oxidase. Science. 1993;259:965–967. doi: 10.1126/science.8438155. [DOI] [PubMed] [Google Scholar]
- 29.Lindqvist Y, Schneider G, Ermler U, Sundstrom M. Three-dimensional structure of transketolase, a thiamine diphosphate dependent enzyme, at 2.5 Å resolution. EMBO J. 1992;11:2373–2379. doi: 10.1002/j.1460-2075.1992.tb05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dyda F, Furey W, Swamibathan S, Sax M, Farrenkopf B, Jordan F. Catalytic centers in the thiamin diphosphate dependent enzyme pyruvate decarboxylase at 2.4 Å resolution. Biochemistry. 1993;32:6165–6170. doi: 10.1021/bi00075a008. [DOI] [PubMed] [Google Scholar]
- 31.Arjunan P, Umland T, Dyda F, Swaminathan S, Furey W, Sax M, Farrenkopf B, Gao Y, Zhang D, Jordan F. Crystal structure of the thiamin diphosphate-dependent enzyme pyruvate decarboxylase from yeast Saccharomyces cerevisiae at 2.3 Å resolution. J. Mol. Biol. 1996;256:590–600. doi: 10.1006/jmbi.1996.0111. [DOI] [PubMed] [Google Scholar]
- 32.Sergienko EA, Wang J, Polovnikova L, Hasson MS, McLeish MJ, Kenyon GL, Jordan F. Spectroscopic Detection of Transient Thiamin Diphosphate-Bound Intermediates on Benzoylformate Decarboxylase. Biochemistry. 2000;39:13862–13869. doi: 10.1021/bi001214w. [DOI] [PubMed] [Google Scholar]
- 33.Sergienko EA, Jordan F. Yeast pyruvate decarboxylase tetramers can dissociate into dimers along two interfaces. Hybrids of low-activity D28A (or D28N) and E477Q variants, with substitution of adjacent active center acidic groups from different subunits, display restored activity. Biochemistry. 2002;41:6164–6169. doi: 10.1021/bi0121712. [DOI] [PubMed] [Google Scholar]
- 34.Bar-Ilan A, Balan V, Tittmann K, Golbik R, Vyazmensky M, Hubner G, Barak Z, Chipman DM. Binding and activation of thiamin diphosphate in acetohydroxyacid synthase. Biochemistry. 2001;40:11946–11954. doi: 10.1021/bi0104524. [DOI] [PubMed] [Google Scholar]
- 35.Balakrishnan A, Gao Y, Moorjani P, Nemeria N, Tittmann K, Jordan F. Bifunctionality of the thiamin diphosphate cofactor: assignment of tautomeric/ionization states of the 4′-aminopyrimidine ring when various intermediates occupy the active sites during the catalysis of yeast pyruvate decarboxylase. J Am Chem Soc. 2012;134:3873–3885. doi: 10.1021/ja211139c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steinmetz A, Vyazmensky M, Meyer D, Barak Z, Golbik R, Chipman DM, Tittmann K. Valine 375 and Phenylalanine109 Confer Affinity and Specificity for Pyruvate as Donor Substrate in Acetohydroxy Acid Synthase Isozyme II from Escherichia coli. Biochemistry. 2010;49:5188–5199. doi: 10.1021/bi100555q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.