

Abstract
Background: Personalizing non-small-cell lung cancer (NSCLC) therapy toward oncogene addicted pathway inhibition is effective. Hence, the ability to determine a more comprehensive genotype for each case is becoming essential to optimal cancer care.
Methods: We developed a multiplexed PCR-based assay (SNaPshot) to simultaneously identify >50 mutations in several key NSCLC genes. SNaPshot and FISH for ALK translocations were integrated into routine practice as Clinical Laboratory Improvement Amendments-certified tests. Here, we present analyses of the first 589 patients referred for genotyping.
Results: Pathologic prescreening identified 552 (95%) tumors with sufficient tissue for SNaPshot; 51% had ≥1 mutation identified, most commonly in KRAS (24%), EGFR (13%), PIK3CA (4%) and translocations involving ALK (5%). Unanticipated mutations were observed at lower frequencies in IDH and β-catenin. We observed several associations between genotypes and clinical characteristics, including increased PIK3CA mutations in squamous cell cancers. Genotyping distinguished multiple primary cancers from metastatic disease and steered 78 (22%) of the 353 patients with advanced disease toward a genotype-directed targeted therapy.
Conclusions: Broad genotyping can be efficiently incorporated into an NSCLC clinic and has great utility in influencing treatment decisions and directing patients toward relevant clinical trials. As more targeted therapies are developed, such multiplexed molecular testing will become a standard part of practice.
Keywords: carcinoma, non-small cell, genotype, molecular targeted therapy
introduction
Certain genetically defined cancers are ‘oncogene addicted’ to activated kinases and are thereby highly sensitive to drugs that selectively inhibit the corresponding kinase. Employing genotype-based therapy has been highly successful in chronic myelogenous leukemia, gastrointestinal stromal tumors, non-small-cell lung cancer (NSCLC) and melanoma, and in many instances, the targeted agent is far more effective than traditional chemotherapy [1–9]. This shifting paradigm has dramatically impacted lung cancer treatments. Until recently, therapeutic options for advanced NSCLC were limited to chemotherapies that were ‘personalized’ only by considering the side-effect profiles of a number of similar modestly effective regimens. Response rates were typically 20%–30% and progression-free survival (PFS) was 3–5 months [10–13]. But now, we know that determining NSCLC genotype can inform the most effective personalized therapies. Patients with mutations in the epidermal growth factor receptor (EGFR) gene benefit from EGFR tyrosine kinase inhibitors (TKIs) with a response rate of ∼75%, PFS of 9–13 months and improved quality of life compared with chemotherapy [8, 14–16]. Similarly, patients with EML4-ALK translocations have a 60% response rate, 9-month PFS and a low degree of toxicity when treated with crizotinib, an ALK TKI [6].
Although these landmark studies have focused on a single or small number of genetic mutations, there is an increasing motivation to develop technologies that can simultaneously determine the mutational status of many genes. Responding to the need for real-time, effective, multiple-gene tumor genotype analysis, our group developed a clinical genotyping test (SNaPshot) based on a commercially available platform. SNaPshot is a multiplexed PCR-based assay designed to test >50 hot-spot mutation sites in 14 key cancer genes. The development of the SNaPshot platform focused on capturing somatic events with known or putative implications for molecularly targeted therapy and has previously been described in detail [17]. We began using SNaPshot routinely in our clinic in March 2009. This report constitutes our experience screening 589 patients during the initial 15 months of test availability.
methods
patients
NSCLC patients seen at Massachusetts General Hospital and Mass General/North Shore Cancer Center (a satellite location) between March 2009 and May 2010 underwent clinical genotype testing at the discretion of their treating physician. The cut-off for this analysis was set to coincide with our site opening the Lung Cancer Mutation Consortium genotype testing (NCT01014286), a USA collaborative genotyping effort. When SNaPshot was initiated, only patients with adenocarcinoma were eligible. In August 2009, as the laboratory became more efficient at handling high throughput, any patient with NSCLC could be tested. All patients signed a clinical consent form and test results were entered into the medical record. Records from patients who had been successful genotyped were reviewed for demographic, clinical and pathological data under an Institutional Review Board-approved protocol. Smoking status was categorized as ‘never’ if <100 cigarettes were consumed per lifetime, ‘former’ if >100 cigarettes and smoking cessation was >1 year before lung cancer diagnosis, otherwise ‘current’. Pack-years of smoking were calculated as packs per day multiplied by years of smoking. Patients undergoing a repeat biopsy at the time of acquired resistance to EGFR TKIs were excluded from this analysis as they have been reported elsewhere [18].
genotype screening
All specimens submitted for clinical genotyping were prescreened by a pathologist to confirm sufficient tumor in the sample. Genotyping was carried out on DNA derived from formalin-fixed paraffin-embedded (FFPE) tumor specimens using SNaPshot, a targeted mutational analysis assay designed by our group [17]. The SNaPshot platform from Applied Biosystems consists of multiplexed PCR and single-base extension reactions that generate fluorescent labeled probes designed to interrogate hot-spot mutation sites. The SNaPshot products are then resolved and analyzed using capillary electrophoresis. During the first year of this study, tumor genotyping was carried out with our original SNaPshot panel (SNaPshot Version 1, Table 1), as previously described [17]. In April 2010, the assay was expanded to accommodate a broader range of tumor types being genotyped, leading to the addition of some additional tests including IDH1 and HER2 genotyping (SNaPshot Version 2, Table 1), see Supplemental Methods (available at Annals of Oncology online).
Table 1.
Summary of findings from SNaPshot assay versions 1 and 2.
| Gene | Loci tested, amino acid–nucleotide | Mutations identified, n (%) |
| AKT1V2 | E17–49G | — |
| APC | R1114–3340C | — |
| Q1338–4012C | ||
| R1450–4348C | ||
| T1556fs-4666_4667insA | ||
| BRAF | 9 (100) | |
| V600–1798G | ||
| V600–1799T | V600E, 9 (100) | |
| CTNNB1 (β-catenin) | 11 (100)a | |
| D32–94G | ||
| D32–95A | D32A, 1 (9) | |
| S33–98C | S33Y, 2 (18) | |
| G34–101G | G34V, 1 (9) | |
| S37–109T | ||
| S37–110C | S37C, 2 and S37F, 3; total, 5 (45) | |
| T41–121A | T41A, 1 (9) | |
| T41–122C | T41I, 1 (9) | |
| S45–133T | ||
| S45–134C | ||
| EGFR | 73 (100) | |
| G719–2155G | G719C, 2 (3) | |
| T790–2369C | ||
| L858–2573T | L858R, 24 (33) | |
| Exon 19 deletionsb | 45 (62) | |
| Exon 20insert/delbV2 | 2 (3) | |
| ERBB2 (HER2)V2 | 2 (100) | |
| Exon 20 insertionsb | 2 (100) | |
| FLT3V1 | D835–2503G | — |
| IDH1V2 | 1 (100) | |
| R132–394C | R132C, 1 (100) | |
| R132–395G | ||
| JAK2V1 | V617–1849G | — |
| KIT | D816–2447A | — |
| KRAS | 134 (100) | |
| G12–34G | G12S, 5; G12R, 4; G12C, 58; total, 67 (50) | |
| G12–35G | G12V, 26; G12D, 19; G12A; 10; total, 57 (41) | |
| G13–37G | G13C, 6 (4) | |
| G13–38G | G13D, 6 (4) | |
| NOTCH1 | L1575–4724T | — |
| L1601–4802T | ||
| NRAS | 6 (100) | |
| G12–34G | G12S, 1 (17) | |
| G12–35G | ||
| G13–37G | ||
| G13–38G | ||
| Q61–181C | ||
| Q61–182A | Q61L, 3; Q61R, 2; total, 5 (83) | |
| Q61–183A | ||
| PIK3CA | 22 (100) | |
| R88–263G | ||
| E542–1624G | E542K, 6 (27) | |
| E545–1633G | E545K, 8 (36) | |
| Q546–1636C | Q546K, 1 (5) | |
| Q546–1637A | ||
| H1047–3139C | ||
| H1047–3140A | H1047R, 6; H1047L, 1; total, 7 (32) | |
| G1049–3145G | ||
| PTEN | R130–388C | — |
| R173–517C | ||
| R233–697C | ||
| K267fs–800delA | ||
| TP53 | 26 (100) | |
| R175–524G | R175H, 1; R175L, 3; total, 4 (15) | |
| G245–733G | G245C, 3 (12) | |
| R248–742C | R248W, 5 (19) | |
| R248–743G | R248Q, 2; R248L, 3; R248P, 1; total, 6 (23) | |
| R273–817C | R273C, 3; R273S, 2; total, 5, (19) | |
| R273–818G | R273L, 2 (8) | |
| R306–916C | R306X, 1 (4) |
Tested loci are listed and differences between versions 1 and 2 are indicated. V1—this assay was included in SNaPshot version 1 only V2—this assay was included in SNaPshot version 2 only. The number and frequency of observed genotype alterations are listed (percent refers to the frequency of a particular mutation among all mutations identified for that gene).
One patient was found to have two separate β-catenin mutations, one at locus S33–98C (S33Y) and one at S37–110C (S37F).
Sizing assays were used to identify these mutations.
FISH was carried out on FFPE tumor sections using a break apart probe to the ALK gene (Vysis; Abbott Molecular, Downers Grove, IL). Samples were classified positive for ALK rearrangement if >15% of scored tumor cells had split ALK 5′ and 3′ probe signals or isolated 3′ signals [6]. Though technically separate tests, ALK FISH analyses and EGFR and HER2 sizing assays are referred to in conjunction with the mutational analyses of SNaPshot collectively as ‘SNaPshot’ for convenience throughout the manuscript.
All genotyping tests were carried out in our hospital's Clinical Laboratory Improvement Amendments-certified Translational Research Laboratory. Turnaround time (TAT) was calculated as the interval from genotype requisition to result finalization. Thus, TAT includes the time required to obtain the pathology specimen, which had to be requested from outside institutions in some cases. Also, when the initial genotyping data did not meet clinical quality control standards due to limiting tissue amount or integrity, genotyping was repeated on DNA re-extracted from either the same tumor specimen or from an alternative paraffin block, which significantly prolonged the TAT.
statistical considerations
Summary statistics are provided regarding the demographic characteristics of the 546 patients tested. Demographic and disease characteristics were compared among patients with mutant and wild-type status for each gene using Wilcoxon, χ2 and Fisher's exact tests as appropriate. In these analyses, the demographics of the patient corresponding with each tested tumor specimen were included; hence, the six patients with two distinct specimens genotyped were accounted for twice. Survival was analyzed via the Kaplan–Meier method and compared between groups with a log-rank test.
results
patients
From March 2009 to May 2010, 1016 patients with NSCLC were seen at Massachusetts General Hospital and 589 were referred for clinical genotyping (supplemental Figure S1, available at Annals of Oncology online). Pathology prereview of the submitted FFPE specimen(s) identified adequate material in 552 (94%) cases, with 37 (6%) having insufficient tissue for genotyping; all cases passing pathology prereview were successfully tested. The 552 genotyped samples were from 546 patients, with 6 patients having two samples tested. The majority of samples (n = 431; 78%) were tested with SNaPshot version 1, while those sent after 19 April 2010 (n = 100; 18%) were tested with SNaPshot version 2, which included AKT1, HER2 and IDH1 mutations (Table 1). Nearly all samples (n = 528; 96%) also underwent ALK FISH testing. A minority of cases (n = 21, 4%) had ALK analysis only.
Patients had a median age of 64 years (range 22–89), and included 58% females, and 92% white patients, reflecting our clinic's racial homogeneity (Table 2). Twenty-four percent were never smokers. Histology was predominantly adenocarcinoma (81%), due to the initial restriction of testing to adenocarcinoma only and the overrepresentation of this tumor type in our clinic.
Table 2.
Demographics of the tested patients
| Overall group, n = 546 | EGFR status | KRAS status | Less frequent mutations (only positive columns shown) | |||||||
| Positive (n = 73) | Wild type (n = 453) | Positive (n = 134) | Wild type (n = 395) | ALK pos. (n = 27) | B-cat pos. (n = 11) | PIK3CA pos. (n = 22) | BRAF pos. (n = 9) | NRAS pos. (n = 6) | ||
| Median age (range) | 64 (22–89) | 61(39–89) | 64 (22–86) | 65 (26–83) | 63 (22–89) | 57 (37–86) | 61 (45–85) | 62 (44–79) | 64 (50–72) | 67 (49–85) |
| Gender | ||||||||||
| Male | 228 (42) | 20 (27) | 197 (44) | 49 (37) | 169 (43) | 13 (48) | 3 (27) | 7 (32) | 3 (33) | 3 (50) |
| Female | 318 (58) | 53 (73) | 255 (56) | 85 (63) | 225 (57) | 14 (52) | 8 (73) | 15 (69) | 6 (66) | 3 (50) |
| Racea | ||||||||||
| White | 503 (92) | 60 (82) | 424 (94) | 131 (98) | 357 (91) | 26 (96) | 10 (90) | 21 (95) | 8 (89) | 6 (100) |
| Black | 7 (1) | 1 (1) | 5 (1) | 0 | 6 (2) | 0 | 0 | 0 | 1 (11) | 0 |
| Asian | 22 (4) | 10 (14) | 11 (2) | 0 | 20 (5) | 1 (4) | 0 | 0 | 0 | 0 |
| Smokinga | ||||||||||
| Never | 128 (24) | 35 (48) | 84 (19) | 5 (4) | 113 (29) | 18 (67) | 8 (72) | 2 (9) | 4 (50) | 0 |
| Former | 278 (51) | 30 (41) | 241 (54) | 81 (61) | 192 (49) | 7 (26) | 3 (27) | 12 (54) | 2 (25) | 1 (17) |
| Current | 137 (25) | 8 (11) | 125 (28) | 47 (35) | 88 (22) | 2 (7) | 0 | 8 (36) | 2 (25) | 5 (83) |
| Median pack-yearsa (range) | 24 (0–180) | 1 (0–76) | 30 (0–180) | 30 (0–158) | 20 (0–180) | 0 (0–50) | 0 (0–80) | 40 (0–158) | 5 (0–51) | 78 (15–163) |
| Histology | ||||||||||
| Adenocarcinoma | 440 (81) | 66 (90) | 357 (79) | 120 (90) | 306 (77) | 23 (85) | 11 (100) | 11 (50) | 8 (89)c | 4 (67) |
| Squamous | 50 (9) | 1 (1) | 49 (10) | 3 (2) | 47 (12) | 0 | 0 | 6 (27) | 0 | 1 (17) |
| Adenosquamous | 9 (2) | 3 (4) | 6 (1) | 1 (1) | 8 (2) | 0 | 0 | 1 (5) | 1 (11) | 0 |
| NSC-NOS | 47 (9) | 3 (4) | 41 (9) | 10 (7) | 34 (9) | 4 (15) | 0 | 4 (18) | 0 | 1 (17) |
| Stagea | ||||||||||
| IA | 107 (20) | 16 (22) | 89 (20) | 38 (28) | 70 (18) | 2 (7)b | 2 (18) | 5 (23) | 2 (22) | 2 (33) |
| IB | 58 (11) | 10 (14) | 47 (10) | 14 (10) | 42 (11) | 1 (4) | 1 (9) | 3 (14) | 1 (11) | 0 |
| IIA | 11 (2) | 0 | 11 (2) | 3 (2) | 9 (2) | 2 (7) | 0 | 0 | 0 | 0 |
| IIB | 21 (4) | 1 (1) | 20 (4) | 10 (7) | 11 (3) | 0 | 1 (9) | 1 (5) | 0 | 0 |
| IIIA | 58 (11) | 3 (4) | 52 (12) | 14 (10) | 41 (10) | 4 (15) | 0 | 2 (9) | 1 (11) | 2 (33) |
| IIIB | 47 (9) | 7 (10) | 38 (8) | 5 (4) | 40 (10) | 3 (11) | 2 (18) | 3 (14) | 0 | 0 |
| IV | 241 (44) | 36 (49) | 193 (43) | 50 (37) | 179 (46) | 15 (56) | 5 (45) | 8 (36) | 5 (56) | 2 (33) |
| Metastatic pattern | ||||||||||
| Not metastaticc | 193 (35) | 25 (34) | 164 (36) | 58 (43) | 133 (34) | 7 (26)b | 3 (27) | 7 (32) | 4 (44) | 3 (50) |
| Lungs only | 97 (18) | 12 (16) | 80 (18) | 14 (10) | 78 (20) | 7 (26) | 4 (36) | 3 (14) | 1 (11) | 1 (17) |
| CNS only | 34 (6) | 2 (3) | 31 (7) | 6 (4) | 27 (7) | 2 (7) | 1 (9) | 1 (5) | 0 | 2 (33) |
| Bone only | 25 (5) | 5 (7) | 19 (4) | 8 (6) | 16 (4) | 3 (11) | 1 (9) | 0 | 0 | 0 |
Characteristics of the entire group (n = 546) and of the patients with tumors testing positive and wild type for each mutation are shown. Note that the overall group (data column one) includes a small number of patients who have two separate tumors accounted for in the other columns of the table. All gene mutations were tested in 552 tumors, while ALK FISH was tested in 549 tumors. Numbers in parentheses indicate percentages. Bolded data indicates that a characteristic varied significantly (P-value ≤ 0.05) among those tested for that genotype comparing the mutated with the wild-type cohorts.
A small number of patients have unknown values for this variable.
A characteristic varied with borderline significance (P-value >0.05 to 0.09) among those tested for that genotype comparing the mutated with the wild-type cohorts.
Not metastatic implies that there was no metastatic disease at baseline nor did it develop during follow-up. All others developed metastatic disease but are listed as a specific pattern only if spread was confined to either lungs only, brain only or bones only.
genotypes
Among the 552 genotyped cases, TAT (defined in ‘Methods’) was 2.8 weeks (range 1.0–8.9 weeks). Samples with longer TAT were more likely to have required DNA re-extraction to confirm initial test results not meeting quality control standards (7% required re-extraction among cases with TAT ≤ 2.8 weeks compared to 35% among those with TAT > 2.8 weeks; P < 0.001).
Mutations in at least one tested gene loci and/or translocations involving ALK were identified in 282 (51%) samples, while 270 (49%) had a negative screen (Table 1, Figure 1A). Twenty-five (5%) samples were positive for two mutations while two tumors had three simultaneous mutations (Figure 1B). Overall, we observed 73 (13%) EGFR mutations, 134 (24%) KRAS mutations, 27 (5%) ALK translocations, 26 (5%) TP53 mutations, 22 (4%) PIK3CA mutations, 11 (2%) β-catenin mutations, 9 (2%) BRAF mutations, 6 (1%) NRAS mutations, 2 HER2 mutations and 1 IDH1 mutation. Of note, IDH1 and HER2 were assessed only in SNaPshot version 2 (n = 100) and hence, their frequencies are not necessarily representative.
Figure 1.
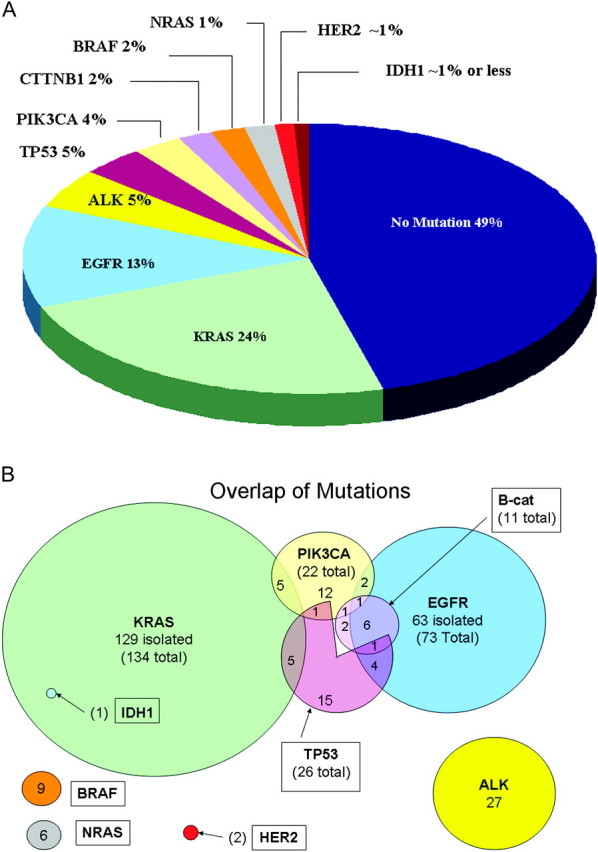
Distribution frequency (A) and overlap (B) of the genotypes observed. Genotypes observed are depicted in a pie chart showing frequency of each mutation with regard to all patients tested (A) as well as in a Venn diagram showing the overlap of patients with more than one mutation (B). Note that only 100 patients were screened for IDH1 and HER2 mutations, so the frequency depicted here may not be truly representative. Also note that TP53 screening only encompassed a minority of the ‘hot-spot’ mutations described in NSCLC for TP53.
Examining demographic and other clinical correlations with genotype (Table 2), we observed both expected and novel associations. As anticipated, patients with EGFR mutations were significantly more likely to be female, Asians, and have adenocarcinoma than EGFR wild-type patients. In our cohort, KRAS mutation positivity was associated with white race (P = 0.02), adenocarcinoma histology (P = 0.004) and earlier stage disease (P = 0.002). ALK translocations correlated with young age (P < 0.001) and possibly more advanced stage (P = 0.06), while PIK3CA mutations occurred in squamous cell cancers (P = 0.003). Smoking history seemed to be one of the most discriminating clinical features (Table 2, Figure 2), as low smoking was strongly associated with EGFR mutations (P < 0.001), ALK translocations (P < 0.001) and β-catenin mutations (P = 0.03), while heavier smoking history was significantly associated with mutations in KRAS (P < 0.001) and NRAS (P = 0.05).
Figure 2.
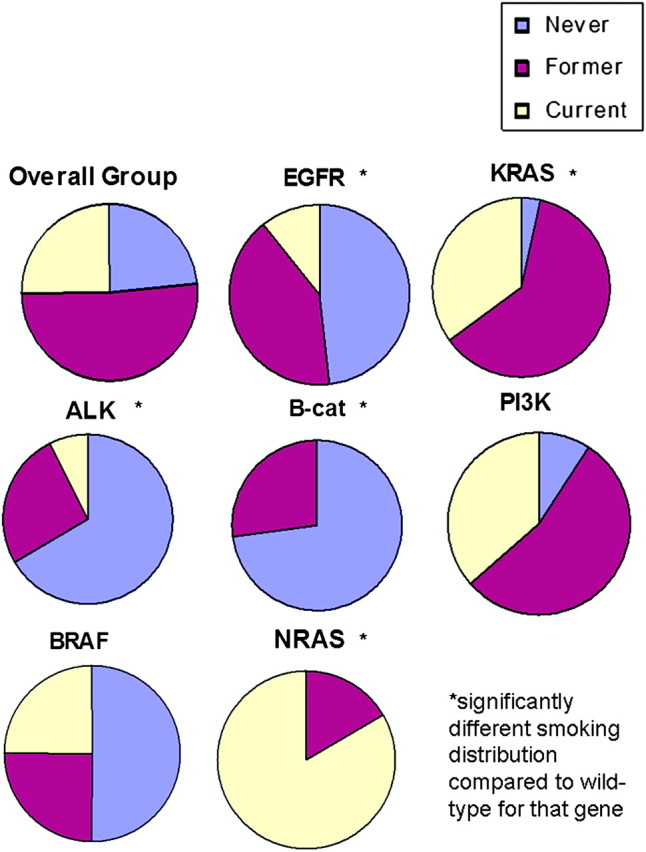
Smoking status distribution by genotype. The proportion of patients that were never, former and current smokers are depicted in separate pie charts representing the overall study cohort and the subset positive for each of the major mutation types.
We identified two patients with HER2 and one with an IDH1 mutation. The HER2-mutant tumors were both stage IV adenocarcinomas in never smokers; one a 68-year-old white male and the other a 50-year-old white female. The IDH1-mutant tumor also harbored KRAS and was a stage IIIA adenocarcinoma in a 77-year-old white male former smoker with a 100 pack-year history.
survival
We examined survival estimates among the 346 patients diagnosed with advanced NSCLC (defined in this analysis as stage III or IV) and divided the analysis by genotype if there were >40 patients in each genotype (mutant and wild type), which in this cohort included KRAS and EGFR. The median follow-up time was 16.1 months, with 212 deaths observed, and the median overall survival (OS) among all 346 patients was 21.7 months (supplemental Table S1, available at Annals of Oncology online). There was a detriment in survival (P = 0.04) for those with KRAS mutations compared with KRAS wild type, with a median OS of 16.4 and 22.5 months for the two groups, respectively (Figure 3A) and an improvement in survival (P = 0.04) for those with EGFR mutations compared with EGFR wild type, with a median OS of 34.3 and 20.0 months, respectively, (Figure 3B). Due to lack of full information about treatment administration and responses, no multivariable adjusted analyses were carried out.
Figure 3.
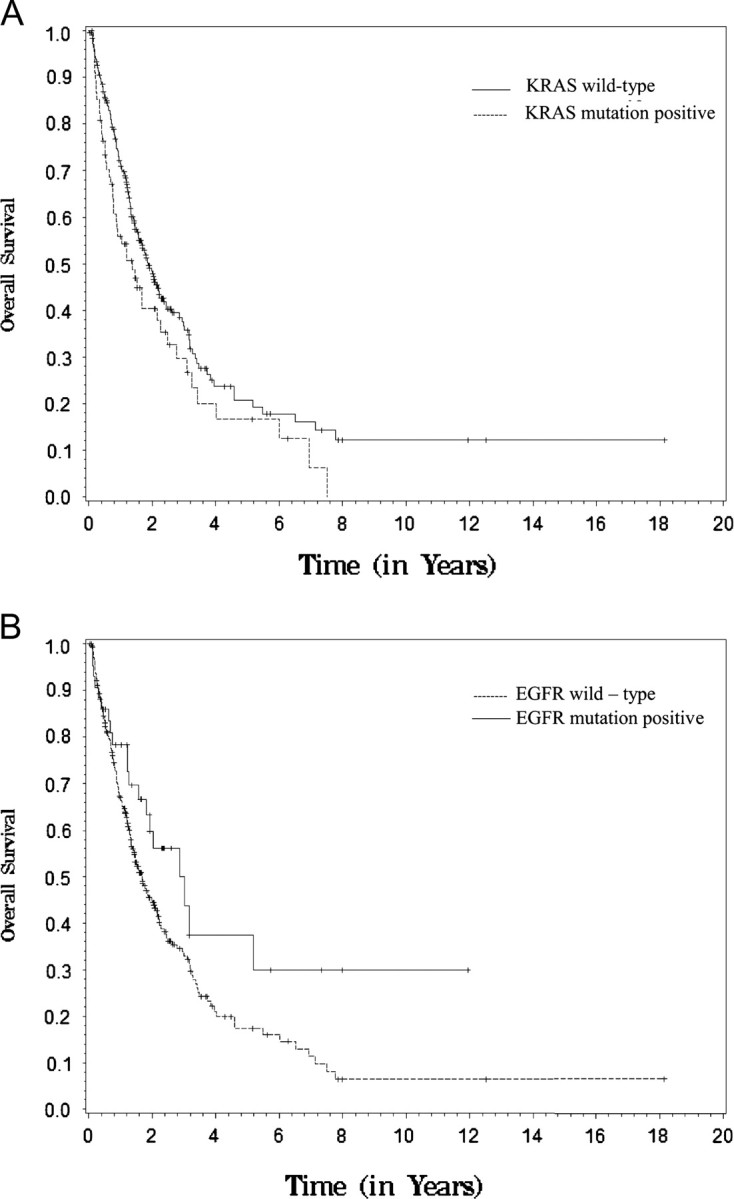
Survival among patients with stage III and IV non-small-cell lung cancer by KRAS mutation status (A) and EGFR mutation status (B). Mutant patients are depicted with a solid line and wild-type patients with a dashed line.
clinical implications and genotype-directed clinical trials
Six patients had two distinct tumor specimens genotyped; often, the information was useful in establishing the correct stage. Two patients underwent concurrent surgical resections of T1 tumors in different lobes and had each tumor genotyped. Distinct mutations in the resection specimens (KRAS G12A and G12C in one case, KRAS G12C and BRAF V600E in the other) suggested synchronous stage IA primaries (as opposed to metastatic disease) in both patients. Similarly, a third patient with stage I resected NSCLC developed a contralateral lung lesion 2 years later and underwent a biopsy. The initial tumor had KRAS G12R, while the subsequent NSCLC was wild type for all tested loci, supporting a second primary, and the patient was treated aggressively. Three additional patients had similar scenarios, but genotyping did not definitively affect clinical care.
Overall, 353 (65%) patients were diagnosed with stage IV NSCLC or recurred during the study follow-up period (through July 2011). Of these, 170 (48%) were found to have a mutation or translocation in either EGFR (n = 48), KRAS (n = 76), ALK (n = 25), BRAF (n = 5), PIK3CA (n = 14) or HER2 (n = 2), which we classified as ‘potentially targetable’ genotypes since we had appropriate clinical trials open during the study period. Sixty-four (38%) of the patients with a potentially targetable genotype enrolled in at least one study utilizing a targeted therapy (Figure 4). The trials included examined drugs that blocked EGFR, ALK, HER2, BRAF or PI3K, or closely related downstream pathways integral to driver mutation signaling (i.e. MEK inhibitors for KRAS mutations). The majority (n = 48, 75%) of study accruals resulted directly from genotype results (in most cases, the trials were genotype specific), including 14 EGFR, 19 ALK, 8 KRAS, 3 BRAF, 3 PIK3CA and 1 HER2 mutation-positive patients. Furthermore, 30 additional EGFR-mutant patients were treated with erlotinib ‘off protocol’ because of genotyping results, suggesting that a total of 78 (22%) patients (48 on trial, 30 off protocol) with advanced NSCLC had therapies initiated as a direct result of genotype findings. Note that an additional five patients with early-stage EGFR-mutant NSCLC were enrolled on a genotype-specific trial of adjuvant erlotinib. It was not possible to assess how many additional patients were directed ‘away’ from therapies due to genotype findings (e.g. KRAS-mutant patients directed away from erlotinib), though we suspect that this occurred.
Figure 4.

Flow of patients with advanced or recurrent non-small-cell lung cancer onto genotype-directed therapies.
discussion
Genotyping for ‘driver mutations’ is becoming increasingly central to oncology care. Over the course of 15 months, we tested 552 NSCLC tumors for genotype abnormalities using a multiplexed PCR-based SNaPshot assay plus FISH for ALK translocations as part of routine clinical practice. To our knowledge, our center was the first in the United States to offer this type of broad screening for NSCLC patients as part of standard care [19]. We found genotype testing to be feasible within the clinical workflow, with a median TAT of 2.8 weeks, which includes the time necessary to acquire FFPE samples from outside hospitals. A full 51% of cancers tested were positive for a driver mutation, most commonly mutations in KRAS (24%) and EGFR (13%) and translocations involving ALK (5%). While widely agreed that it is important to identify patients with EGFR and ALK given the availability of effective therapeutics, it is also noteworthy that in a short time frame at a single institution, we identified over 30 patients with less common mutations like BRAF, PIK3CA and HER2, which also have relevant candidate targeted therapies [20]. Among the patients with advanced or recurrent NSCLC seen within these 15 months, 22% began a genotype-specific therapy in response to SNaPshot results. We anticipate that this proportion should increase further in the future, as the scope of genotype-specific clinical trial efforts is rapidly broadening. Furthermore, SNaPshot provided strong evidence of multiple primary cancers in half of patients who had more than one tumor sample screened. This type of testing could significantly affect treatment decisions, especially when considering whether to pursue surgery or other therapy with curative intent versus treatment of metastatic disease. Other groups have similarly described the power of genotyping multiple lesions from the same patient [21]. Overall, we have demonstrated that broad clinical genotyping with SNaPshot can be tightly integrated into clinical practice and we believe it can make a real difference for patients.
A recent study from China examined a research-based genotyping panel in a smaller cohort of early-stage adenocarcinomas from exclusively never-smoking Asian patients [22]. They found that an impressive 90% of patients had a mutation in EGFR, KRAS, ALK or HER2. While adenocarcinoma in Asian nonsmokers appears to be almost completely defined by oncogenic driver mutations, it is quite remarkable that 51% of patients in our clinic, made up primarily of white patients with a positive smoking history, also had mutations defined on SNaPshot. A North American lung cancer genome collaboration reported their sequencing effort of nearly 200 adenocarcinomas and found several recurrent oncogenes and tumor suppressor mutations [23]. Deep sequencing will likely represent the future of clinical genotyping; however, this option is currently neither feasible nor affordable for clinical use.
In addition to the genotypes well associated with NSCLC, we made the novel observation of an IDH1 mutation in one patient. IDH1 mutations have been mainly associated with glioblastoma, lower grade gliomas and acute myeloid leukemia and will likely have possible therapeutic implications in the near future [24–26]. According to compiled data from published reports, IDH1 mutations appear to be rare in NSCLC [27]. We added IDH1 genotyping to our panel when we moved from SNaPshot version 1 to 2 primarily for its predicted utility in glioma patients, but since we utilize a single genotyping assay for all tumor types at our hospital, we were able to serendipitously observe an IDH1 mutation in one lung cancer specimen. We also observed β-catenin mutations in 2% of patients, commonly in conjunction with EGFR mutations. β-Catenin has been associated with lung tumorigenesis and pulmonary blastomas, but to our knowledge has not been related to EGFR-mutant NSCLC [28, 29]. We found that PIK3CA mutations also tended to be found in combination with other driver mutations, confirming other reports [30].
As with other mutation-specific assays, SNaPshot testing is most suitable for genotyping oncogenes, which are usually affected at a very limited number of loci. Tumor suppressor genotyping is more challenging. While the SNaPshot panel was designed to capture the most commonly mutated sites in TP53, these represent only a fraction of the many variants reported to occur in this tumor suppressor. Thus, the 5% incidence of TP53 mutants detected in our cohort is far lower than the reported frequency expected in NSCLC [31, 32].
A point of discussion recently has been the utility of clinical characteristics in referring patients for genotyping [33]. Our patient cohort is in line with prior literature showing that many genotypes have associated clinical features. Smoking history was one of the more discriminating demographics with low smoking correlated with EGFR and ALK, while heavier smoking was associated with KRAS, consistent with prior literature [22, 34–36]. We made the novel observations that low smoking is correlated with β-catenin mutations and heavy smoking with NRAS. Unlike Riely et al., who identified KRAS mutations in 15% of never smokers with adenocarcinoma, we saw KRAS in only 5 of 128 (4%) never-smoking patients [37]. We also observed known histology associations, such as adenocarcinoma among EGFR and KRAS mutants and squamous cell among PIK3CA mutants [38–41]. ALK translocations were associated with younger age and more advanced stage, while KRAS mutations were seen preferentially in early-stage cancers [35]. However, given the growing panel of relevant genotypes in NSCLC, clinical characteristics are no longer an efficient method for selecting which patients to test. The ability to order a single comprehensive genotyping panel, rather than specific tests á la carte, is crucial since clinical features do not correlate perfectly with genotypes and trends for clinical associations often diverge for different gene mutations. Furthermore, as clinicians become more adept at incorporating genotype information into treatment-making algorithms, they may wish to know not only what genotypes are positive but also what mutations are absent. For example, we know that EGFR TKIs are most active in EGFR mutation-positive patients, but there is growing evidence that KRAS mutations predict for non-benefit from EGFR TKIs; hence, many clinicians are becoming hesitant to administer erlotinib-known KRAS mutants [39, 42].
The results of our study should be interpreted within the context of the retrospective observational study design, and its limitations acknowledged, including selection biases introduced by the population of patients seeking care at our institution and those in which SNaPshot was ordered. We saw interesting survival differences among stage III and IV patients by EGFR and KRAS genotype, though this analysis is crude and not corrected for other prognostic factors or treatment information. In addition, while SNaPshot provided an improvement in molecular testing over conventional molecular strategies (which have typically focused on EGFR and KRAS sequencing only), it still required a 2- to 3-week turnaround, which in some cases was prolonged by the need to retest or identify an alternative sample because the initial specimen was of poor quality. Moving forward toward a more comprehensive genetic picture of these tumors may involve expansion of the SNaPshot panels to include additional hot-spot sites and the adoption of further complementary platforms to capture not only a myriad of point mutations but also translocation events and copy number changes.
In summary, in our experience, SNaPshot tumor genotyping is a viable clinically feasible approach to support diagnostic and treatment decisions and to facilitate clinical trial enrollment. It is uncovering new therapeutic opportunities for a growing number of patients and advancing NSCLC management at our institution.
disclosure
Lecia Sequist has consulted for Clovis Oncology, Merrimack Pharmaceuticals, Daiichi-Sankyo and Celgene. Alice Shaw has consulted for Pfizer, Ariad and Chugai. Tom Lynch is a joint holder for a patent for EGFR mutation testing. John Iafrate has consulted for Pfizer and Abbott Molecular. Jeff Engelman has consulted for Agios. Leif Ellisen, Darrell Borger, John Iafrate and Dora Dias-Santagata are consultants for Bioreference Laboratories, which has licensed the SNaPshot technology. John Iafrate and Dora Dias-Santagata submitted a patent for the SNaPshot tumor genotyping assay (pending).
None for all other authors have anything relevant to declare, which includes RSH, PF, RR, JST, ITL, SD, BAW, EB, ST, LM, AMuz, SBG, JG, CLC, JCW, HG, DMD, AMun, CW, HW, DJM, NCC, JB, ML, and MM-K.
Acknowledgments
This work was made possible by philanthropic supporters of lung cancer research at Mass General Hospital.
The authors would like to acknowledge Nick Jessop, Diane Davies, Nancy French, Kathy Vernovsky, Michele Myers and Sachiko Grimes for their invaluable help with coordinating patient specimens and Arjola Cosper, Kenneth Fan, Hector Lopez, Vanessa Scialabba, Mai Nitta and Anhthu Nguyen for their technical assistance with genotyping.
References
- 1.Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344(14):1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 2.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 3.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 4.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 5.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 9.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346(2):92–98. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 11.Fossella F, Pereira JR, von Pawel J, et al. Randomized, multinational, phase III study of docetaxel plus platinum combinations versus vinorelbine plus cisplatin for advanced non-small-cell lung cancer: the TAX 326 study group. J Clin Oncol. 2003;21(16):3016–3024. doi: 10.1200/JCO.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 12.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 13.Scagliotti GV, Parikh P, von Pawel J, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26(21):3543–3551. doi: 10.1200/JCO.2007.15.0375. [DOI] [PubMed] [Google Scholar]
- 14.Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 15.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 16.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 17.Dias-Santagata D, Akhavanfard S, David SS, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2(5):146–158. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pao W, Kris MG, Iafrate AJ, et al. Integration of molecular profiling into the lung cancer clinic. Clin Cancer Res. 2009;15(17):5317–5322. doi: 10.1158/1078-0432.CCR-09-0913. [DOI] [PubMed] [Google Scholar]
- 20.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12(2):175–180. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 21.Girard N, Deshpande C, Azzoli CG, et al. Use of epidermal growth factor receptor/Kirsten rat sarcoma 2 viral oncogene homolog mutation testing to define clonal relationships among multiple lung adenocarcinomas: comparison with clinical guidelines. Chest. 2010;137(1):46–52. doi: 10.1378/chest.09-0325. [DOI] [PubMed] [Google Scholar]
- 22.Sun Y, Ren Y, Fang Z, et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol. 2010;28(30):4616–4620. doi: 10.1200/JCO.2010.29.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wellcome Trust Sanger Institute. COSMIC v55. 2011. http://www.sanger.ac.uk/genetics/CGP/cosmic/ (14 September 2011, date last accessed) [Google Scholar]
- 28.Shigemitsu K, Sekido Y, Usami N, et al. Genetic alteration of the beta-catenin gene (CTNNB1) in human lung cancer and malignant mesothelioma and identification of a new 3p21.3 homozygous deletion. Oncogene. 2001;20(31):4249–4257. doi: 10.1038/sj.onc.1204557. [DOI] [PubMed] [Google Scholar]
- 29.Nakatani Y, Miyagi Y, Takemura T, et al. Aberrant nuclear/cytoplasmic localization and gene mutation of beta-catenin in classic pulmonary blastoma: beta-catenin immunostaining is useful for distinguishing between classic pulmonary blastoma and a blastomatoid variant of carcinosarcoma. Am J Surg Pathol. 2004;28(7):921–927. doi: 10.1097/00000478-200407000-00012. [DOI] [PubMed] [Google Scholar]
- 30.Pietanza MC, Arcila ME, Chaft MF. Clinical, pathologic, and molecular characteristics of patients with non-small cell lung cancer harboring mutations in PIK3CA. Paper presented at: American Society of Clinical Oncology, 2011; Chicago, IL (Abstr 7587) [Google Scholar]
- 31.Toyooka S, Tsuda T, Gazdar AF. The TP53 gene, tobacco exposure, and lung cancer. Hum Mutat. 2003;21(3):229–239. doi: 10.1002/humu.10177. [DOI] [PubMed] [Google Scholar]
- 32.Bamford S, Dawson E, Forbes S, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91(2):355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D'Angelo SP, Pietanza MC, Johnson ML, et al. Incidence of EGFR exon 19 deletions and L858R in tumor specimens from men and cigarette smokers with lung adenocarcinomas. J Clin Oncol. 2011;29(15):2066–2070. doi: 10.1200/JCO.2010.32.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pham D, Kris MG, Riely GJ, et al. Use of cigarette-smoking history to estimate the likelihood of mutations in epidermal growth factor receptor gene exons 19 and 21 in lung adenocarcinomas. J Clin Oncol. 2006;24(11):1700–1704. doi: 10.1200/JCO.2005.04.3224. [DOI] [PubMed] [Google Scholar]
- 35.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian J, Govindan R. Molecular genetics of lung cancer in people who have never smoked. Lancet Oncol. 2008;9(7):676–682. doi: 10.1016/S1470-2045(08)70174-8. [DOI] [PubMed] [Google Scholar]
- 37.Riely GJ, Kris MG, Rosenbaum D, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14(18):5731–5734. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25(5):587–595. doi: 10.1200/JCO.2006.07.3585. [DOI] [PubMed] [Google Scholar]
- 39.Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc. 2009;6(2):201–205. doi: 10.1513/pats.200809-107LC. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto H, Shigematsu H, Nomura M, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008;68(17):6913–6921. doi: 10.1158/0008-5472.CAN-07-5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okudela K, Suzuki M, Kageyama S, et al. PIK3CA mutation and amplification in human lung cancer. Pathol Int. 2007;57(10):664–671. doi: 10.1111/j.1440-1827.2007.02155.x. [DOI] [PubMed] [Google Scholar]
- 42.Roberts PJ, Stinchcombe TE, Der CJ, Socinski MA. Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for epidermal growth factor receptor-targeted therapy? J Clin Oncol. 2010;28(31):4769–4777. doi: 10.1200/JCO.2009.27.4365. [DOI] [PubMed] [Google Scholar]