

Abstract
Background:
Gastrointestinal stromal tumors (GISTs) and desmoid tumors (DTs) are two rare mesenchymal tumor. Anecdotal reports of individuals with both diseases led us to make the hypothesis that the association is a nonrandom event as the probability would be extremely low to observe such cases if they were independent events.
Patients and methods:
We evaluated the existence of patients with GIST and DT in a large multicenter cohort at 10 institutions in the United States, Australia and Europe. Data on gender, age at diagnosis, KIT, PDGFRA, CTNNB1 mutation status and follow-up time after diagnosis were collected.
Results:
We identified 28 patients diagnosed with both tumors. DT was diagnosed after GIST in 75% of patients and concomitantly in 21%. In only one case (4%), GIST was diagnosed after DT. KIT or PDGFRA mutations were detected in 12 of 14 GIST, 9 in KIT exon 11, 2 in KIT exon 9 and 1 in PDGFRA.
Conclusion:
A statistical analysis of these 28 cases suggests a nonrandom association between GIST and DT. Further studies may be able to elucidate the underlying biology responsible for this association.
Keywords: beta-catenin, deep fibromatosis, desmoid tumor, GIST, imatinib, KIT
introduction
Gastrointestinal stromal tumor (GIST) is the most common mesenchymal tumor of the gastrointestinal tract but remains rare compared with gastrointestinal carcinomas. The term ‘GIST’ was introduced to indicate a distinctive subgroup of gastrointestinal sarcomas [1]. Subsequently, the interstitial cell of Cajal, or a cell in that lineage, was implicated as the precursor of these tumors, further distinguishing them from other sarcomas [2].
Gain-of-function mutations of KIT play a key role in the oncogenesis of these tumors [3]. Approximately 80% of patients with GIST have primary KIT mutations. Most KIT mutations involve the juxtamembrane domain, exon 11. Other exons harbor KIT mutations with varying frequencies include exons 9, 13 and 17 [4]. Of the GIST not harboring KIT mutations, ∼30% have mutations in PDGFRA, which are mutually exclusive with KIT mutations [5]. A rare subset of GIST has been found to carry BRAF mutations in the absence of KIT or PGDFRA alterations [6]. Recently, mutations in the succinate dehydrogenase genes have been identified in tumors lacking a kinase gene mutation [7].
The standard treatment for patients with resectable GIST is surgery with the goal of achieving complete resection. For patients with metastatic or unresectable GIST, the introduction of molecularly targeted kinase inhibitors (e.g. imatinib, sunitinib) has significantly extended survival [8, 9]. The median time to disease progression is 18–24 months in patients with GIST who are receiving imatinib; however, imatinib has been shown to control disease for >6 years in a subset of individuals with GIST [10, 11].
As patients with GIST are living longer, their risk of developing other malignancies is now being evaluated and reported in the literature. Indeed, it is known that GIST can be associated with other neoplasms in conditions such as neurofibromatosis type 1, Carney’s triad and Carney–Stratakis syndrome [12–15]. However, outside of these syndromes, the associations between GIST and other tumors remain unclear. Most reports describe single cases or very small series of patients from a single institution [16–20]. GIST has been reported to coexist with gastric, breast, prostate, renal, esophagus, colorectal, lung and pancreatic carcinomas; carcinoids; lymphomas and melanomas [21, 22]. Additionally, two case reports of a patient with desmoid tumors (DTs, also known as deep or aggressive fibromatosis) and GIST in the same anatomic location were recently reported [23, 24]. DT is very rare fibroblastic proliferations with a tendency for slow local infiltrative growth. Their cell of origin is not clearly defined, but recent evidence suggests that DT originate from mesenchymal progenitor cells [25]. DT occurs sporadically or in association with Gardner's Syndrome and Familial Adenomatous Polyposis (FAP) [26]. These do not metastasize but can cause significant morbidity through their locally destructive effects. Prior trauma, such as previous surgery, is known to increase the risk of sporadic and FAP-associated DT [27, 28]. Complete surgical resection with a wide margin and/or radiation therapy constitute the mainstay of resectable DT therapy, while chemotherapy, anti-inflammatory agents and tyrosine kinase inhibitors are all treatment options for locally advanced DT. The Wnt/β-catenin signaling pathway seems to play an important role in the development of DT, and nuclear expression of β-catenin has increasingly been used in the differential diagnosis of spindle cell neoplasms, particularly in the abdomen [29]. Mutations in the CTNNB1 gene, which codes for β-catenin protein, have been found in the majority of DT patients, with most mutations occurring in exon 3 [30–32].
Anecdotal reports of individuals with GIST and DT led us to evaluate a larger cohort of patients with both tumors, in order to gain insight into whether their simultaneous occurrence is a coincidental event. In this study, we have found 28 patients from 10 institutions with a history of both GIST and DT.
materials and methods
patients and tumor tissues
This retrospective analysis included 28 patients with both GIST and DT. Clinical characteristics and formalin-fixed paraffin-embedded GIST and DT specimens accrued between 1978 and 2008 were retrieved from the patient records and pathology sample collections of The University of Texas M. D. Anderson Cancer Center, Helsinki University Central Hospital, Fox Case Cancer Center, Oregon Health & Science University Knight Cancer Institute, Cleveland Clinic, Italian National Tumor Institute, Prince of Wales Hospital, H. Lee Moffit Cancer Center, the Armed Forces Institute of Pathology and Memorial Sloan-Kettering Cancer Center using a protocol approved by the respective Institutional Review Boards. Specimens were further screened and evaluated by experienced soft-tissue pathologists at M. D. Anderson, the Armed Forces Institute of Pathology or Fox Chase who confirmed DT and GIST histology. Demographic and clinical information, including treatment, histology and outcome-related variables were tabulated for analyses.
genomic DNA isolation
When tissues were available genomic DNA was extracted from 10-μm-thick formalin-fixed paraffin-embedded tissue sections cut from blocks with at least 80% tumor using the QIAamp DNA mini kit (Qiagen, Valencia, CA) or the Easy-DNA kit (Invitrogen, Carlsbad, CA).
analysis of KIT, PDGFRA and CTNNB1 mutations
PCR amplification of DNA for KIT exons 9, 11, 13 and 17; PDGFRA exons 12, 14 and 18 and CTNNB1 (β-catenin) exon 3 was carried out. Each PCR amplification was carried out in a 50-μl volume containing 10 ng of genomic DNA, 15 μM concentrations of each primer, 0.2 mM deoxyribonucleotide triphosphate, 5 μl of 10× reaction buffer, 2.5 mM MgCl2 and 1 U of AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA). The KIT and PDGFRA primers were previously published [33, 34]. The PCR conditions were as follows: 30 s at 94°C; 30 s at 52°C and 1 min at 68°C for 36 cycles, followed by 10-min extension at 68°C. The primers for CTNNB1 exon 3 primers were as follows: forward: 5′-TTTGATGGAGTTGGACATGG-3′ and reverse: 5′-CTGAGAAAATCCCTGTTCCC-3′. The PCR conditions for CTNNB1 exon 3 were as follows: 30 s at 94°C; 30 s at 55°C and 1 min at 68°C for 36 cycles, followed by 10-min extension at 68°C. PCR products were analyzed in by gel electrophoresis and purified using a Qiaquick PCR purification kit (Qiagen). Direct sequencing was carried out from both directions using a BigDye Terminator v3.1 cycle sequencing kit on an ABI PRISM 3100 genetic analyzer (Applied Biosystems). The National Center for Biotechnology Information’s Basic Local Alignment Search Tool was used to analyze both strands to identify mutations.
statistical analysis
A standardized incidence ratio (SIR) was used. This ratio is calculated by the number of events observed divided by the number of events expected. In this study, SIR was calculated to compare the incidence of DT in patients with GIST to the incidence of DT in the general USA population from 2000 to 2008 [35]. We calculated the 95% confidence intervals (CIs) of SIRs using the formula proposed by Vandenbroucke [36]. Overall survival was calculated as the time between the date the first neoplasm was diagnosed and the date of death or the last known date at which the patient was alive. Survival curves were estimated using the Kaplan and Meier method and the log-rank test.
results
Patients with both GIST and DT were searched in a large cohort including 10 institutions. The total number of new patients per year in the USA institutions between 2000 and 2008 was 830 for GIST and 315 for DT. Twenty-eight patients harboring GIST associated with DT were identified as follows: M. D. Anderson Cancer Center (cases 1–4), Helsinki University Central Hospital (cases 5–6), Fox Case Cancer Center (cases 7–9), Oregon Health & Science University Knight Cancer Institute (case 10), Cleveland Clinic (case 11), Italian National Tumor Institute (case 12), Prince of Wales Hospital (case 13), H. Lee Moffit Cancer Center (cases 14–16), the Armed Forces Institute of Pathology (cases 17–23) and Memorial Sloan-Kettering Cancer Center (cases 24–28). Table 1 shows the clinical characteristics of this patient population, which included 19 men (68%) and 9 women (32%) whose ages ranged from 34 to 88 years. The majority of patients had sought medical advice because of vague symptoms such as epigastric pain, nausea or abdominal discomfort. Interestingly, no cases with a family history of FAP were observed.
Table 1.
Patient and tumor properties, summary of the 28 patients who developed GIST and DT
| Case no. | Age | Sex | Primary location of GIST | GISTsize (cm) | KIT or PDGFRAmutation (GIST) | Imatinib | Site of DT | CTNNB1 mutation (desmoid) | Interval of GIST with DT (months) | Length of follow-up (months) | Patient status |
| 1 | 62 | M | Gastric | 20 | KIT mutation exon 11: V560D | Yes | Perigastric/peripancreatic | Exon 3: WT | 39 | 58 | NED |
| 2 | 67 | M | Gastric | 10.5 | KIT exon 11 mutation: deletion | Yes | Gastric | Exon 3: T41A | 35 | 45 | AWD |
| 3 | 50 | M | Gastric | 0.5 | ND | No | Retrogastric | ND | 0 | 54 | NED |
| 4 | 45 | F | Omentum | 7 | KIT mutation exon 11: D579del | Yes | Colon | Exon 3: WT | 39 | 36 | AWD |
| 5 | 39 | F | Jejunum | 12.5 | WT | Yes | Small bowel | Exon 3: WT | 32 | 67 | Died of GIST |
| 6 | 62 | F | Gastric | 20 | KIT exon 11mutation : del Lys 550-Glu556+ insLeu | Yes | Right rectus abdominis | Exon 3: WT | 30 | 74 | NED |
| 7 | 59 | F | Gastric | 9.9 | KIT exon 11mutation: W557-K558del | Yes | Small bowel | Exon 3: T41A | 42 | 100 | AWD |
| 8 | 62 | F | Jejunum | 10 | KIT exon 11 mutation: del 556-573 | Yes | Small bowel | Exon 3: S45P | 16 | 75 | AWD |
| 9 | 74 | M | Gastric | 7.5 | ND | Yes | Omentum | Exon 3: T41A | 25 | 54 | NED |
| 10 | 75 | M | Gastric | 10 | PDGFRA exon 18 mutation: D842V | No | Peripancreatic | ND | 0 | 68 | NED |
| 11 | 88 | F | Small intestine | 5 | WT | No | Abdominal wall | Exon 3: WT | -7 | 118 | NED |
| 12 | 61 | M | Gastric | 30 | ND | Yes | Abdominal wall | ND | 28 | 72 | Died of GIST |
| 13 | 62 | M | Gastroduodenal | 15 | KIT exon 11 mutation: deletion | Yes | Infrapyloric mesenteric | ND | 36 | 95 | Died of GIST |
| 14 | 42 | F | Jejunal | 10.2 | ND | Yes | Pelvic | ND | 21 | 21 | NED |
| 15 | 71 | F | Gastric | 11.2 | ND | Yes | Caudate lobe liver | ND | 24 | 35 | AWD |
| 16 | 52 | M | Pelvic | 4 | KIT exon 9 mutation | Yes | Mesenteric | ND | 30 | 30 | AWD |
| 17 | 53 | M | Gastric | ND | ND | No | Spleen | ND | 33 | ND | ND |
| 18 | 75 | M | Castric | 16 | ND | No | Mesenteric | ND | 0 | ND | ND |
| 19 | 65 | F | Gastric | 1.8 | ND | No | Mesenteric | ND | 0 | 30 | Died cause unknown |
| 20 | 70 | M | Gastric | 6 | ND | No | Thigh | ND | 191 | 274 | NED |
| 21 | 66 | M | Gastric | 1.5 | ND | No | Mesenteric | ND | 0 | 212 | NED |
| 22 | 42 | F | Small intestine | 0.4 | ND | No | Mesenteric | ND | 0 | 307 | NED |
| 23 | 40 | M | Small intestine | 10 | ND | No | Mesenteric | ND | 12 | 12 | No follow-up |
| 24 | 34 | M | Proximal jejunum | 5.3 | KIT exon 9 mutation: 502-3 AY duplication | No | Terminal ileum | ND | 19 | 55 | NED |
| 25 | 58 | M | Gastric | 10.5 | ND | Yes | Mesentery | ND | 62 | 66 | AWD |
| 26 | 58 | M | Gastric | 5.4 | KIT exon 11 mutation: Del (6aaDEL 552-7 MYEVQW) | Yes | Mesentery | ND | 30 | 42 | AWD |
| 27 | 54 | M | Gastric | 6 | KIT exon 11 mutation: Del (557-8 WK del) | Yes | Small bowel/mesentery | ND | 33 | 37 | NED |
| 28 | 68 | M | Small intestine | 30 | ND | Yes | Proximal jejunum | ND | 6 | 6 | NED |
GIST, gastrointestinal stromal tumor; DT, desmoid tumor; WT, wild type; ND, no data; NED, no evidence of disease; AWD, alive with disease.
In 21 cases (75%), GIST was diagnosed before DT. The average time between these diagnoses was 30 months (range, 6 months–16 years). In 6 cases (21%), both tumors presented synchronously and in 1 case (case 11; 4%), GIST was diagnosed after DT. The primary GIST tumor site was gastric in 17 cases (60%), small intestinal in 9 cases (32%) and pelvic or mesenteric in 1 case each (4%). DT involved the extremity in only one patient, who developed DT of the thigh 16 years after treatment for gastric GIST. In three patients (11%), DT developed in the surgical incision site and mimicked a recurrence of GIST. Imatinib was used for 17 patients (61%) and in 10 cases (36%) GIST developed before imatinib therapy was available. A strong family history of GIST (father, brother and aunts) was found in case 4, caused by a rare germ line mutation of KIT that was previously described [37]. Interestingly, there are reports in the literature investigating the use of imatinib for treating DT. In particular, in our series, patient number 2, a 67-year-old man, received imatinib at 400 mg/day for gastric GIST and developed a DT on the posterior wall of the gastric antrum 35 months after the first diagnosis. His dose of imatinib was increased to 800 mg/day, resulting in a partial response in both tumors (Figure 1).
Figure 1.

Computed tomography of patient #2 shows that imatinib at 800 mg/day may induce a partial response in gastrointestinal stromal tumor (GIST) and desmoid tumor (DT). Arrows indicate tumor localization.
The frequency of mutations in KIT and PDGFRA in GIST was similar to that previously observed in large cohorts of patients with GIST [38]. KIT was mutated in 11 cases of 14 (79%), of which 9 cases (64%) were in exon 11 and 2 cases (14%) in exon 9. PDGFRA was mutated in one case (7%). Two cases (14%) lacked mutations in KIT or PDGFRA (wild type). β-catenin is commonly deregulated in DT indeed mutations in the CTNNB1 gene have been identified with a prevalence of 85% in a large cohort of patients with DT [31]. In our samples available for genotyping, CTNNB1 was mutated in 5 of 10 cases (50%) in exon 3 (4 type 41A and 1 type 45F; Table 1).
The annual incidence of GIST in the United States is estimated to be 10–15 cases/million [39], while the annual incidence of DT is 2–4 cases/million [40]. The expected number of DT events in patients with GIST was calculated by applying the DT rate for theoretical GIST population in the USA. To determine the statistical association between GIST and DT, the SIR was used. Analyses were restricted to cases from the USA from 2000 to 2008. In this case series, 13 patients were diagnosed with GIST and DT in the US between 2000 and 2008. This number was compared with 0.16, an estimated number of expected cases assuming the independence of events. The estimated risk of developing DT was significantly higher (SIR = 82; 95% CI 44–133) in GIST patients than in the general population. To assess the clinical outcomes of patients with GIST and DT, we displayed a Kaplan–Meier survival plots (Figure 2). The 5- and 10-year survival rates were 0.86 (95% CI 0.99–0.73) and 0.64 (95% CI 0.86–0.27), respectively.
Figure 2.
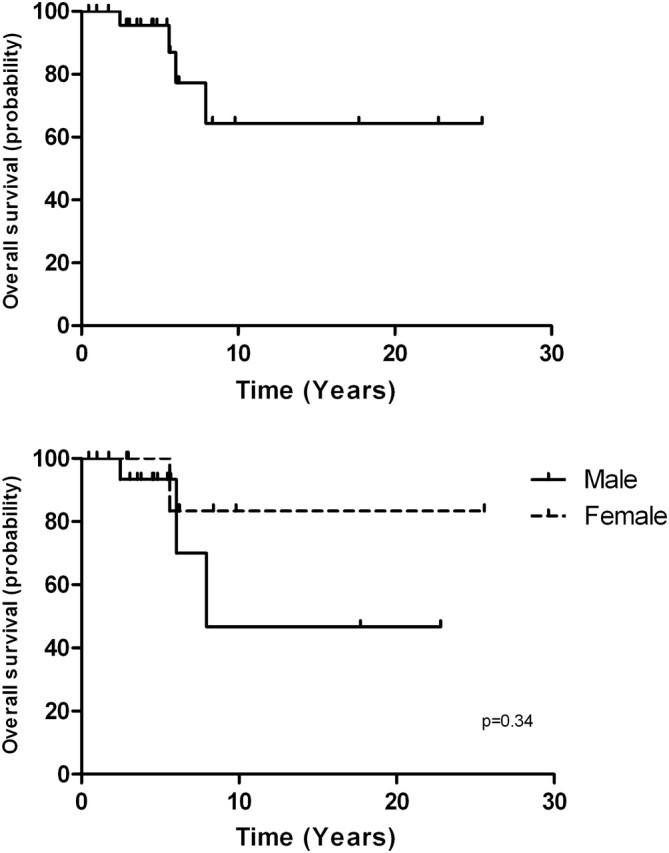
Overall survival analysis by the Kaplan–Meier method.
discussion
The development of tyrosine kinase inhibitors has lead to dramatic improvements in long-term survival of patients with GIST. The primary objective of this study was to determine the existence of the association between GIST and DT. Our analysis resulted in the identification of 28 cases. On a previous study, on a single institution-based tumor registry, we found that ∼20% of the GIST patients developed at least one additional tumor [20]. The occurrence of other malignancies appeared higher than that was expected in the general population. However, to our knowledge, the present study is the first report of a series of patients with coexisting GIST and DT. This relationship raises the questions about a potential link between these two neoplasms and the possibility of a cancer predisposition syndrome.
There were more men than women (19 versus 9) in our population of patients who developed both tumors. Accepting the possibility of a selection bias in this population, this gender distribution differs from that reported in previous DT studies, where female predilection is suggested; female to male ratios commonly ranged between 1.4 and 1.8 [41, 42]. In addition, a Kaplan–Meier survival analysis revealed a no significant difference in survival between men and women (Figure 2). Furthermore, the peak incidence of DT has been reported as 25–35 years; however, in our case series, DT occurred at an average of 59 (range 34–88 years) [40, 42]. Because these tumors are rare, the size of this series and the SIR suggest a nonrandom association between these histopathologically distinct tumors. At this stage, we can only speculate about this association.
Patients with GIST may be predisposed to developing DT or patients with DT might be prone to GIST. These possibilities should be considered when the patient has an occurrence of either of these tumors. Although there are no data to support a genetic predisposition syndrome, a germ line mutation might underlie predisposition for these two tumors. The possibility that activated KIT pathway is involved in the development of DT must also be considered. However, gain-of-function mutations of KIT were not found in DT tissues samples in our series. Most of the patients had abdominal surgery to remove their GIST and then developed a DT. Surgical trauma has been shown to induce desmoid growth, but this implication seems insufficient to explain all the cases of GIST and DT in the current series. Additionally, no history of FAP was observed in our patients that could explain the increase of risk for developing a post-operative desmoid. However, this series cannot conclude that GIST patients who undergo surgery are at higher risk for DT than other patients who undergo abdominal surgery. Furthermore, treatment with imatinib does not appear to play a role in the development of DT because 11 cases in our series were not treated with imatinib. Interestingly, there are reports in the literature investigating the use of imatinib for treating DT. Case reports have described responses to imatinib, and a series of 19 patients with DT showed a partial response in three patients [43–45]. Another possibility is that GIST patients under physical and radiological surveillance are more likely to have DT detected compared with ‘normal’ population. Finally, it is interesting to speculate that circulating stem cell factor (KIT ligand) or platelet-derived growth factor in patients with GIST could stimulate the growth of DT. Indeed, cross talk between the KIT and Wnt signaling pathways was recently described in mast cell leukemia. In this model, nuclear accumulation of β-catenin was increased by a gain-of-function mutation of KIT [46]. Recent clinical data regarding the effectiveness of sorafenib in both GIST and desmoid tumors highlight a possible therapeutic option when medical treatment of both conditions is necessary [47].
We show in the present study statistical evidences for a nonrandom relationship between the concomitant development of GIST and DT. Excluding a recurrence of GIST is fundamental for the clinical management of a patient that could potentially still benefit from targeted therapy, such as imatinib. Further investigation is needed to establish the link between these tumors and to evaluate possible risk factors for their association.
funding
This work was supported in part by grants from the GIST Cancer Research Fund.
disclosure
The authors have declared no conflicts of interest.
References
- 1.Mazur MT, Clark HB. Gastric stromal tumors. Reappraisal of histogenesis. Am J Surg Pathol. 1983;7:507–519. doi: 10.1097/00000478-198309000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152:1259–1269. [PMC free article] [PubMed] [Google Scholar]
- 3.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 4.Miettinen M, Lasota J. Gastrointestinal stromal tumors (GISTs): definition, occurrence, pathology, differential diagnosis and molecular genetics. Pol J Pathol. 2003;54:3–24. [PubMed] [Google Scholar]
- 5.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 6.Agaram NP, Wong GC, Guo T, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2008;47:853–859. doi: 10.1002/gcc.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 9.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 10.Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–1134. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 11.Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–625. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 12.Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30:90–96. doi: 10.1097/01.pas.0000176433.81079.bd. [DOI] [PubMed] [Google Scholar]
- 13.Carney JA, Sheps SG, Go VL, Gordon H. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N Engl J Med. 1977;296:1517–1518. doi: 10.1056/NEJM197706302962609. [DOI] [PubMed] [Google Scholar]
- 14.O'Riain C, Corless CL, Heinrich MC, et al. Gastrointestinal stromal tumors: insights from a new familial GIST kindred with unusual genetic and pathologic features. Am J Surg Pathol. 2005;29:1680–1683. doi: 10.1097/01.pas.0000173024.79852.08. [DOI] [PubMed] [Google Scholar]
- 15.Hirota S, Okazaki T, Kitamura Y, et al. Cause of familial and multiple gastrointestinal autonomic nerve tumors with hyperplasia of interstitial cells of Cajal is germline mutation of the c-kit gene. Am J Surg Pathol. 2000;24:326–327. doi: 10.1097/00000478-200002000-00045. [DOI] [PubMed] [Google Scholar]
- 16.Maiorana A, Fante R, Maria Cesinaro A, Adriana Fano R. Synchronous occurrence of epithelial and stromal tumors in the stomach: a report of 6 cases. Arch Pathol Lab Med. 2000;124:682–686. doi: 10.5858/2000-124-0682-SOOEAS. [DOI] [PubMed] [Google Scholar]
- 17.Wronski M, Ziarkiewicz-Wroblewska B, Gornicka B, et al. Synchronous occurrence of gastrointestinal stromal tumors and other primary gastrointestinal neoplasms. World J Gastroenterol. 2006;12:5360–5362. doi: 10.3748/wjg.v12.i33.5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruka W, Rutkowski P, Nowecki Z, et al. Other malignant neoplasms in patients with gastrointestinal stromal tumors (GIST) Med Sci Monit. 2004;10:LE13–LE14. [PubMed] [Google Scholar]
- 19.Agaimy A, Wuensch PH. Gastrointestinal stromal tumours in patients with other-type cancer: a mere coincidence or an etiological association? A study of 97 GIST cases. Z Gastroenterol. 2005;43:1025–1030. doi: 10.1055/s-2005-858378. [DOI] [PubMed] [Google Scholar]
- 20.Pandurengan RK, Dumont AG, Araujo DM, et al. Survival of patients with multiple primary malignancies: a study of 783 patients with gastrointestinal stromal tumor. Ann Oncol. 2010;21:2107–2111. doi: 10.1093/annonc/mdq078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goncalves R, Linhares E, Albagli R, et al. Occurrence of other tumors in patients with GIST. Surg Oncol. 2010;10:e140–e143. doi: 10.1016/j.suronc.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Liszka L, Zielinska-Pajak E, Pajak J, et al. Coexistence of gastrointestinal stromal tumors with other neoplasms. J Gastroenterol. 2007;42:641–649. doi: 10.1007/s00535-007-2082-4. [DOI] [PubMed] [Google Scholar]
- 23.Lee CK, Hadley A, Desilva K, et al. When is a GIST not a GIST? A case report of synchronous metastatic gastrointestinal stromal tumor and fibromatosis. World J Surg Oncol. 2009;7:8. doi: 10.1186/1477-7819-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan M, Bozas G, Cooke J, et al. Mesenteric desmoid tumor developing on the site of an excised gastrointestinal stromal tumor. Rare Tumors. 2010;2:e33. doi: 10.4081/rt.2010.e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu C, Nik-Amini S, Nadesan P, et al. Aggressive fibromatosis (desmoid tumor) is derived from mesenchymal progenitor cells. Cancer Res. 2010;70:7690–7698. doi: 10.1158/0008-5472.CAN-10-1656. [DOI] [PubMed] [Google Scholar]
- 26.Clark SK, Phillips RK. Desmoids in familial adenomatous polyposis. Br J Surg. 1996;83:1494–1504. doi: 10.1002/bjs.1800831105. [DOI] [PubMed] [Google Scholar]
- 27.Soravia C, Berk T, McLeod RS, Cohen Z. Desmoid disease in patients with familial adenomatous polyposis. Dis Colon Rectum. 2000;43:363–369. doi: 10.1007/BF02258303. [DOI] [PubMed] [Google Scholar]
- 28.Kersting S, Herbst H, Senninger N, Mittelkotter U. [Intra-abdominal fibromatosis after appendectomy as cause for ileus] Zentralbl Chir. 2004;129:317–320. doi: 10.1055/s-2004-820330. [DOI] [PubMed] [Google Scholar]
- 29.Kotiligam D, Lazar AJ, Pollock RE, Lev D. Desmoid tumor: a disease opportune for molecular insights. Histol Histopathol. 2008;23:117–126. doi: 10.14670/HH-23.117. [DOI] [PubMed] [Google Scholar]
- 30.Miyoshi Y, Iwao K, Nawa G, et al. Frequent mutations in the beta-catenin gene in desmoid tumors from patients without familial adenomatous polyposis. Oncol Res. 1998;10:591–594. [PubMed] [Google Scholar]
- 31.Lazar AJ, Tuvin D, Hajibashi S, et al. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol. 2008;173:1518–1527. doi: 10.2353/ajpath.2008.080475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tejpar S, Nollet F, Li C, et al. Predominance of beta-catenin mutations and beta-catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor) Oncogene. 1999;18:6615–6620. doi: 10.1038/sj.onc.1203041. [DOI] [PubMed] [Google Scholar]
- 33.Tarn C, Merkel E, Canutescu AA, et al. Analysis of KIT mutations in sporadic and familial gastrointestinal stromal tumors: therapeutic implications through protein modeling. Clin Cancer Res. 2005;11:3668–3677. doi: 10.1158/1078-0432.CCR-04-2515. [DOI] [PubMed] [Google Scholar]
- 34.Debiec-Rychter M, Dumez H, Judson I, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40:689–695. doi: 10.1016/j.ejca.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 35.Miettinen M, Kraszewska E, Sobin LH, Lasota J. A nonrandom association between gastrointestinal stromal tumors and myeloid leukemia. Cancer. 2008;112:645–649. doi: 10.1002/cncr.23216. [DOI] [PubMed] [Google Scholar]
- 36.Vandenbroucke J. A shortcut method for calculating the 95 percent confidence interval of the standardized mortality ratio. Am J Epidemiol. 1982;115:303–304. [Google Scholar]
- 37.Kleinbaum EP, Lazar AJ, Tamborini E, et al. Clinical, histopathologic, molecular and therapeutic findings in a large kindred with gastrointestinal stromal tumor. Int J Cancer. 2008;122:711–718. doi: 10.1002/ijc.23137. [DOI] [PubMed] [Google Scholar]
- 38.Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–1103. doi: 10.1016/j.ejca.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 39.Gutierrez JC, Perez EA, Franceschi D, et al. Outcomes for soft-tissue sarcoma in 8249 cases from a large state cancer registry. J Surg Res. 2007;141:105–114. doi: 10.1016/j.jss.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 40.Reitamo JJ, Hayry P, Nykyri E, Saxen E. The desmoid tumor. I. Incidence, sex-, age- and anatomical distribution in the Finnish population. Am J Clin Pathol. 1982;77:665–673. doi: 10.1093/ajcp/77.6.665. [DOI] [PubMed] [Google Scholar]
- 41.Posner MC, Shiu MH, Newsome JL, et al. The desmoid tumor. Not a benign disease. Arch Surg. 1989;124:191–196. doi: 10.1001/archsurg.1989.01410020061010. [DOI] [PubMed] [Google Scholar]
- 42.Lopez R, Kemalyan N, Moseley HS, et al. Problems in diagnosis and management of desmoid tumors. Am J Surg. 1990;159:450–453. doi: 10.1016/s0002-9610(05)81243-7. [DOI] [PubMed] [Google Scholar]
- 43.Folli F, Galimberti G, Pastore M, et al. Paraneoplastic insulin resistance syndrome in advanced aggressive fibromatosis (desmoid tumor) treated by imatinib mesylate. Diabetes Care. 2006;29:2178–2180. doi: 10.2337/dc06-0984. [DOI] [PubMed] [Google Scholar]
- 44.Mace J, Sybil Biermann J, Sondak V, et al. Response of extraabdominal desmoid tumors to therapy with imatinib mesylate. Cancer. 2002;95:2373–2379. doi: 10.1002/cncr.11029. [DOI] [PubMed] [Google Scholar]
- 45.Heinrich MC, McArthur GA, Demetri GD, et al. Clinical and molecular studies of the effect of imatinib on advanced aggressive fibromatosis (desmoid tumor) J Clin Oncol. 2006;24:1195–1203. doi: 10.1200/JCO.2005.04.0717. [DOI] [PubMed] [Google Scholar]
- 46.Kajiguchi T, Lee S, Lee MJ, et al. KIT regulates tyrosine phosphorylation and nuclear localization of beta-catenin in mast cell leukemia. Leuk Res. 2008;32:761–770. doi: 10.1016/j.leukres.2007.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gounder MM, Lefkowitz RA, Keohan ML, et al. Activity of Sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res. 2011;17:4082–4090. doi: 10.1158/1078-0432.CCR-10-3322. [DOI] [PMC free article] [PubMed] [Google Scholar]