

Abstract
The Iowa point mutation in apolipoprotein A-I (G26R; apoA-IIowa) leads to a systemic amyloidosis condition and the Milano mutation (R173C; apoA-IMil) is associated with hypoalphalipoproteinemia, a reduced plasma level of high density lipoprotein. To probe the structural effects that lead to these outcomes, we used amide hydrogen-deuterium exchange coupled with a fragment separation/mass spectrometry analysis (HX MS). The Iowa mutation inserts an arginine residue into the non-polar face of an α-helix that spans residues 7–44 and causes changes in structure and structural dynamics. This helix unfolds and other helices in the N-terminal helix bundle domain are destabilized. The segment encompassing residues 116–158, largely unstructured in wild type apoA-I, becomes helical. The helix spanning residues 81 to 115 is destabilized by 2 kcal/mol, increasing the small fraction of time it is transiently unfolded to 1% or more, which allows proteolysis at residue 83 in vivo over time, releasing an amyloid-forming peptide. The Milano mutation situated on the polar face of the helix spanning residues 147–178 destabilizes the helix bundle domain only moderately, but enough to allow cysteine-mediated dimerization which leads to the altered functionality of this variant. These results show how the HX MS approach can provide a powerful means for monitoring, in a non-perturbing way and at close to amino acid resolution, the structural, dynamic, and energetic consequences of biologically interesting point mutations.
Apolipoprotein A-I (apoA-I, 243-residues) is the principal protein component of high density lipoprotein particles (HDL). ApoA-I guides HDL formation, maintains HDL structure (1), and mediates its anti-atherogenic properties (2–4). In performing these functions, apoA-I interacts with the ATP binding cassette transporter A1 (ABCA1) to promote efflux of phospholipid and cholesterol from macrophages in the periphery, binds lecithin-cholesterol acyltransferase (LCAT) in plasma to convert cholesterol to cholesterol ester, and interacts with scavenger receptor class B type1 (SR-BI) on the surface of hepatocytes to mediate selective cholesterol uptake (5–7). Naturally occurring mutational variants in the N-terminal helix bundle domain (residues 1–180) of human apoA-I (8–10) affect functionality in ways that depend upon their position (11). Mutations between residues 1–90 are associated with amyloid formation whereas mutations within the central residues 140–170 are mostly associated with defective activation of LCAT. Few natural mutations have been found within the disordered C-terminal domain (residues 180–243) (11), probably because they have no physiological effect.
To investigate structure-function relationships, we earlier studied apoA-1 Milano (R173C; apoA-IMil) and apoA-I Nichinan (ΔE235) as examples of N-terminal domain and C-terminal domain mutations, respectively (12–15). The Milano mutation, the first reported natural variant of human apoA-I (16), leads to hypoalphalipoproteinemia (17) due to impaired LCAT activation (18). The Nichinan mutation is placed in the disordered region of the lipid-free apoA-I molecule but it inhibits lipid-dependent α-helix formation (12, 19–20).
ApoA-I mutants have so far been characterized mostly in terms of global changes in protein structure. One wants to understand the more local effects at high structural resolution and how they lead to the observed effects on function. Detailed structural and dynamic information cannot be readily obtained by crystallographic and NMR methods. We investigated the ability of hydrogen-deuterium exchange - mass spectrometry analysis (HX MS) to answer these questions using methods developed in our prior studies of wild-type apoA-I (10, 21).
Protein amide hydrogens, one in every amino acid (except proline) in every protein molecule exchange naturally with water hydrogens. HX rate and behavior are sensitive to protein structure, structure change, dynamics, energetics, and functional behavior and interactions, and this information is available at amino acid resolution to non-perturbing HX measurements. The HX capability has been widely exploited in HX NMR studies but routine NMR analysis is limited to relatively small, highly soluble proteins that are available in quantity. Investigations of larger and biologically more interesting protein systems can be achieved by a developing proteolytic fragmentation method followed by mass spectrometry analysis (22–26). In this method, protein samples taken from an H-D exchange experiment are proteolytically fragmented, the fragments are separated, and then subjected to MS analysis to determine the quantity and position of carried D-label at a fragment-resolved level. The comparison of high quality data for many overlapping fragments (27) can extend resolution to near the amino acid level (10, 21).
In previous work we used HX MS methods to define the positions, stabilities, and dynamic behavior of hydrogen-bonded structures in wild type lipid-free human apoA-IWT (10) and to characterize changes when the protein is wrapped around the periphery of larger and smaller discoidal HDL particles (21). The very slowly exchanging sites could be associated with α-helices since their number agreed very closely with CD results. In the lipid-free protein, the results located five helices at residues 7–44, 54–65, 70–78, 81–115 and 147–178, their connecting loops, and an unstructured region between residues 180–243. This agrees with known structure in other exchangeable apolipoproteins which display an N-terminal helical bundle domain. Here we ask how the Iowa mutation at amino acid position 26 makes the polypeptide chain labile to proteolysis at position 83 leading to release of an amyloidogenic peptide. This mechanism probably underlies amyloid formation by other apoA-I variants that contain mutations in the N-terminal region (11). We also examine how apoA-I structure is affected by the Milano mutation because knowledge of the mode of action of the mutation should be important for developing potential uses of this variant in the treatment of cardiovascular disease (28).
EXPERIMENTAL
Materials
Wild type (WT) human apolipoprotein A-I (apoA-IWT) (243 residues) was purified from human plasma, as described before (29–30). Human apoA-I cDNA cloned into the pET32a(+) vector from Novagen was used for expression of apoA-I as a His-tagged thioredoxin fusion protein (8, 31). cDNA inserts encoding either the human apoA-I Iowa G26R (apoA-IIowa) or Milano variant R173C (apoA-IMil) (14) were engineered from the pET32a(+) plasmid using the QuikChange site-directed mutagenesis kit (Stratagene, CA). The variants were expressed in E. coli strain BL21-DE3 and purified according to previously published procedures (8, 31). Cleavage of the thioredoxin fusion protein with thrombin leaves the target apoA-I with two extra amino acids, Gly-Ser, at the amino terminus. For consistency in the numbering of peptides from WT and mutant forms, the two residues preceding the normal apoA-I sequence are numbered −1 and −2. Preparations were at least 95% pure as assessed by SDS-PAGE. Protein concentrations were determined by a Lowry procedure (32) or absorbance at 280 nm (33).
Methods
Spectroscopy
Far-UV CD spectra to determine the α-helix contents from the molar ellipticity at 222 nm (14) were obtained as described before (8, 34) using a Jasco 810 spectropolarimeter. GdmCl melting experiments used molar ellipticity at 222nm to calculate the free energy of denaturation, as described before (14). To monitor the exposure of hydrophobic surface, 8-anilino-1-napthalenesulfonic acid (ANS) fluorescence spectra were collected from 400 to 600 nm at an excitation wavelength of 395 nm in the absence and presence of the apoA-I variants and analyzed as described previously (8).
H-D Exchange and Mass Spectrometry Analysis
For HX experiments, lipid-free protein samples in 6M GdmCl were dialyzed against 25 mM sodium phosphate buffer at pH7.3. Freshly dialyzed stock solutions were adjusted to 0.5 mg/ml and used within 12 h of dialysis. ApoA-IMil solutions contained 10 mM DTT to maintain the reduced monomeric state. H to D exchange was initiated by diluting solutions to 0.5 μM protein concentration in D2O buffer (pD 7.3, 5°C; 35-fold; final volume 500 μl). At appropriate time-points, the HX reaction was quenched to pD 2.5 by adding 8 μl 99% formic acid, and 30 μl was injected into an online fragmentation-separation system (maintained at 0°C) connected to an Orbitrap mass spectrometer (10, 27). Between 0–3 min, the protein sample was cleaved into small peptide fragments in an immobilized pepsin column, the peptide fragments were bound to a C18 trap column (2.5 × 0.5 mm; Higgins Analytical), and washed with aqueous buffer. Between 3–15 min, the fragments were eluted with a linear acetonitrile gradient (12–50%, 6 μl/min, 0.1% formic acid, pD 2.5, 0°C) and roughly separated on an analytical C18 HPLC column (75 × 0.3 mm; Agilent Technologies). For MS/MS identification of the proteolytic fragments, and as a zero-time control, a 100% protonated protein sample was injected into the fragmentation separation system. A 100% deuterated sample was prepared by incubating protein at 37°C for 5 h. The peptide fragments served as controls for correcting deuterium back-exchange (15 ± 5%; (35)) that occurs during sample preparation.
Each of the many fragments in the many MS spectra were identified and analyzed for carried D-label by the ExMS program (36). We used about 50 peptide fragments in this work. Each HX run used 25 pmol (0.7 μg) of protein. More details on methods and data analysis are in references (10, 21, 27, 36).
RESULTS
Results for H to D exchange of lipid-free apoA-IWT and for Iowa and Milano mutants are in Figs. 1 and 3. Each panel depicts the time-dependent exchange of deuterium into segments in the native protein, measured on the corresponding peptide fragment obtained in HX MS experiments, illustrated in Figs. 2 and 4. For comparison with measured HX curves, the dashed lines in Figs. 1 and 3 indicate the HX curve that is expected for each peptide fragment when the corresponding apoA-1 segment is solvent exposed and dynamically disordered. We express this condition in terms of a protection factor, Pf, equal to 1. Under the present conditions (pD 7.3, 5° C), an average unprotected amide HX rate is ~1 s−1. More complete results for the 51 peptides measured are in Table S1 in Supplementary Information.
Figure 1.

HX analysis of lipid-free apoA-IWT (◆) and apoA-IIowa (○) at pD 7.3, 5°C. HX kinetics for 16 peptides covering the apoA-I sequence are shown out of a total of 51 listed in Table S1. Each panel compares the measured H to D exchange time-course for the indicated peptide to its reference curve (dotted line) computed for the case of a dynamically disordered random coil with Pf = 1. Exposed but rigidly held segments tend to have Pf ~10 (37–38), as is seen for the C-terminal 180 to 243 residues in both proteins and for some inter-helical segments in the N-terminal domain. Mono-exponential HX behavior indicates a single cooperatively unfolding segment of secondary structure. A bi-exponential fit indicates a peptide that spans a helix terminus (21). Two time-courses (○, □) are shown for apoA-IIowa peptides between residues 114 to 158 because the HX MS envelopes measured these peptides exhibit bimodal HX behavior (see Fig. S1 for examples).
Figure 3.

HX analysis of lipid-free apoA-IWT (◆) and apoA-IMil (○, fast HX state; □, slow HX state) at pD 7.3, 5°C. See the legend to Fig. 1 for further explanation.
Figure 2.
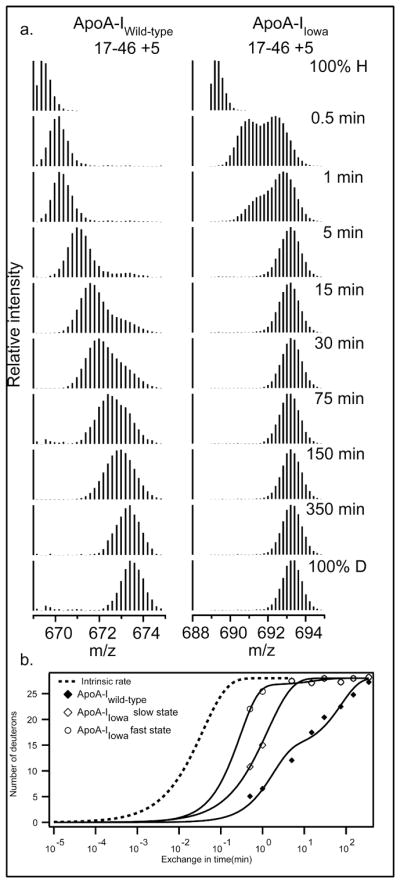
Mass spectra for peptide fragment 17–46 (charge state +5) and its increase in mass as a function of H to D HX time (pD 7.3, 5°C). (a) For the apoA-IIowa peptide, the 0.5 min and 1 min spectra reveal two populations of amide hydrogens with fast and slow HX rates and different degrees of incorporation of D. These mass spectra were fitted to a double Gaussian distribution (by non-linear regression using IGOR Pro (Wavemetrics Inc.)) and integrated to obtain peak intensities and the fraction of fast and slow states. (b) Comparison of the time-course for monomodal (but bi-exponential) H-to-D exchange for fragment 17–46 in apoA-IWT with the bimodal fast and slow HX states in apoA-IIowa. The data were fitted as in Fig. 1.
Figure 4.

Mass spectral envelopes versus H to D HX time for two peptides in apoA-IWT and apoA-IMil that exhibit bimodal HX kinetics (pD 7.3, 5°C). (a, b) Mass spectra at different times of D incorporation of peptide 17–46. (c, d) Mass spectra of peptide 114–126. The bimodal mass spectra were analyzed as described in the legend to Fig. 2.
Amide hydrogens that are protected by stable H-bonded structure can have Pf factors that are larger than 1 by orders of magnitude (10, 21). This occurs because HX can only proceed during the small fraction of time when the protecting H-bond is transiently separated in some dynamic “opening” reaction and the hydrogen is brought into contact with solvent (37–38). Opening can involve a local fluctuation of only one or a few residues, or it may involve the cooperative unfolding of a larger segment. Some of the time-dependent HX curves drawn through the measured data points in Figs. 1 and 3 describe a single (stretched) exponential because exchange in apoA-I is often determined by a sizeable cooperative helix unfolding that exposes a set of amides equally. Bi-exponential behavior occurs when the peptide measured crosses a helix boundary and thus reports on sets of residues with very different Pf.
Observable HX properties depend on the kinetic competition that ensues between chemical exchange and reclosing from the transiently open condition. If chemical exchange is faster than reclosing, exchange will occur on the first opening and the measured HX rate will equal the structural opening rate. This is called the EX1 case (monomolecular exchange). When a protein segment experiences pure EX1 exchange, peptide fragments that contain it will display a characteristic bimodal HX MS spectrum (Figs. 2 and 4). As H to D exchange progresses, the amplitude of the lighter isotopic envelope decreases and a higher mass envelope concurrently increases, while both envelopes do not move on the mass axis. If reclosing is faster than chemical exchange, opening and reclosing will occur many times before a successful HX event and the exchange rate measured will proportion to the fraction of time that structure is open, essentially the equilibrium constant for opening, Kop. This is called the EX2 case (bimolecular exchange) because exchange then depends on the bimolecular chemical HX rate catalyzed by OH− - ion. When protein residues experience EX2 exchange in an HX MS experiment, peptide fragments that contain them will slide continuously to higher mass as H to D exchange time increases (Figs. 2 and 4). In EX2 exchange, the structural stability against opening can be calculated as ΔGop = −RT ln Kop = +RT ln Pf. When HX versus time is plotted on a log (time) scale as in Figs. 1 and 3, the Pf factor can be read directly as the horizontal shift of the experimental HX curve compared to the reference curve.
When both EX1 and EX2 exchange occur within a given peptide fragment, both the see saw and the mass sliding behavior characteristic of EX1 and EX2 behavior will be seen, as in Figs. 2 and 4. A further complication is that bimodal, or even multi-modal, envelopes may reflect the presence of structural heterogeneity in which a given segment exists in different conformations with different HX protection.
ApoA-IIowa
Results obtained for lipid-free apoA-IWT (Figs. 1 and 3, Table S1) are fully consistent with previous results (10, 21). Fig. 1 shows that peptides corresponding to the C-terminal domain of apoA-I (residues 180–243) in both the wild type and Iowa proteins have essentially no protection. The Pf values of ≤10 are consistent with the absence of protecting H-bonded structure (10). HX In the well-structured N-terminal helical bundle is much slower and more complex.
The G26R mutation of apoA-IIowa, in the structured N-terminal domain (1–180), can be expected to have significant local effects and the HX MS results in Fig. 2 show that this is indeed the case. The residues 17–46 peptide in apoA-IIowa which contains the mutation is fully exchanged in 5 min whereas the same segment in the wild type protein requires ~6 h. With both apoA-IWT and apoA-IIowa, a bimodal distribution is apparent with behavior that reflects the presence of both EX1 and EX2 HX kinetics (21). In the isotopic envelopes of apoA-IWT the time-dependent slide to higher mass indicates EX2 behavior. The appearance of another envelope at heavier mass with increasing amplitude may indicate structural heterogeneity with a second more slowly exchanging conformation in this molecular region. Alternatively, the second envelope may result from EX1 behavior in which up to 20 residues participate in a large scale unfolding, with opening halftime ~2 h and reclosing that is slower than the chemical HX rate (<1 s). Similar behavior is seen for apoA-IIowa. If this represents EX1 behavior, the unfolding rate is much faster, ~ 30 s, and it dominates the exchange observed. In the alternative heterogeneous structure picture, the conformation that is more protected in wild type (halftime perhaps 30 min) exchanges here with halftime ~1 min. A more complex explanation in the heterogeneous structure view is that the slow and fast conformations interchange on the HX time scale. In either case, the region spanning residues 17–46 in apoA-IIowa is much less stable and more dynamic, and spends more time exposed to solvent exchange than in apoA-IWT.
The mass spectra in Fig. S1 show that isotopic envelopes for peptide fragments 114–126 and 127–158 in apoA-IIowa also exhibit bimodal distributions at HX times in the 30 s to 15 min range. As for the segment 17–46 discussed above, the data are consistent with the presence of both EX1 and EX2 HX behavior. In apoA-IWT, residues 114–126 have Pf = 10 (Table S1) reflecting the disordered structure that spans the segment 116–146. Interestingly, the G26R mutation induces slower exchange in 114–126. Similar behavior is exhibited by the segment corresponding to residues 127–158 (Fig. S1). The slowing is accompanied by the appearance of bimodal HX behavior in both peptides and is a consequence of helix formation through residues 116–146.
Overall, it is apparent that the G26R mutation induces widespread structural reorganization of residues in the central region of the apoA-I molecule, in addition to the local effects on the helix spanning residues 7–44 in which the mutation resides. The destabilization and partial unfolding is consistent with GdmCl denaturation results and an increase in hydrophobic surface exposure indicated by enhanced ANS binding (Table 1) (39).
TABLE 1.
Influence of Iowa and Milano mutations on apoA-I physical properties
| Parameter | Wild-type | Iowa | Milanoa |
|---|---|---|---|
| Change in α-helix content (%)b | - | −12 | −2 |
| Relative ANS binding | 1.0 | 1.5 | 1.6 |
| Free energy of denaturation (kcal/mol)c | 3.5 ± 0.1 | 2.4 ± 0.1 | 2.4 ± 0.2 |
The data for the monomer (reduced) form are from ref. (14).
The α-helix content of wild-type apoA-I was 49 ± 5% and varied somewhat with sample history.
CD was employed to monitor the loss of α-helix content when apoA-I was exposed to increasing concentrations of GdmCl.
ApoA-IMil
Time-courses of deuterium incorporation into apoA-IWT and apoA-IMil fragments that span the entire length of the apoA-I molecule are compared in Fig. 3 and detailed more completely in Table S2. In contrast to the situation with apoA-IIowa, the HX of fragments 159–169, 170–174 and 175–180 which are located close to the position of the R173C mutation is slow, indicating that the helix spanning this region (residues 147–178) in apoA-IWT is maintained in apoA-IMil. More broadly HX kinetics throughout both proteins, and therefore their helix stabilities, are similar (Table S2). In agreement, helix contents measured by CD are similar (Table 1). Thus the Milano protein appears relatively unperturbed by the R173C mutation.
However, there is an interesting difference from wild type behavior in that in HX of apoA-IMil several peptide fragments in the range spanning residues 17 to 158 (17–46, 51–71, 72–103, 114–126 and 125–158), which do not include the mutant position (R173C), exhibit bimodal HX kinetics (Fig. 3). Examples are in Fig. 4. About 10% of the amide hydrogens in residues 17–46 are fully deuterated at the 0.5 min HX time-point indicating their Pf is < 10 (Table S2). Similar considerations apply to the segments containing residues 51–71 and 72–103. Fragment 114–126 of apA-IMil also exhibits bimodal HX kinetics but in this case the minor population of amide hydrogens has a somewhat higher Pf and requires 30 min to achieve the fully deuterated state (Fig. 4). These protected amide hydrogens are not present in peptide 114–126 of apoA-IWT.
DISCUSSION
The present study demonstrates the ability of the HX MS methodology to monitor at close to amino acid resolution the structural consequences of point mutations in the apoA-I molecule. The amphipathic α-helix is the key structural and functional unit of apoA-I (40) and other exchangeable apolipoproteins, and knowledge of how mutations affect these structures can provide insight into the molecular basis for observed functional consequences.
ApoA-IIowa
The protection factors measured for all the apoA-IWT and apoA-IIowa fragments identified in the HX MS study of these proteins (Table S1) were used to construct the plot of helix stabilization free energy against apoA-I sequence position in Fig. 5. The profile for apoA-IWT agrees with prior results (10, 21). The helical segments are located in the N-terminal region spanning residues 1–180 and have free energy of stabilization of 3 to 5 kcal/mol. Major differences between the apoA-IWT and apoA-IIowa profiles occur in this region; the C-terminal domain (residues 180–243) remains disordered in both. The G26R mutation destabilizes the protein through the large region spanning residues 10–114 (Fig. 5). The residue 7–44 helix in apoA-IWT adopts a Pf = 15 to 35 (Table S1) (ΔG = 1.5 to 2.0 kcal/mol). Pf values in this range are consistent with the existence of either constrained random coil (38) or marginally stable hydrogen-bonded structure that spends about 5% of the time transiently unfolded.
Figure 5.

Summary of the HX-derived secondary structure stabilities for lipid-free apoA-IWT and apoA-IIowa. Detailed HX kinetic data (pD 7.3, 5°C) analyzed to obtain Pf values for 51 peptides are in Table S1. The corresponding free energies of stabilization are plotted as a function of apoA-I sequence position. The profile for apoA-IWT (solid grey line) is from data for peptide fragments obtained by proteolysis with pepsin; the results are consistent with our prior findings where both pepsin and a fungal protease were used to obtain shorter peptides, including some suggesting that residues 66–69 are disordered (10). For peptides encompassing apoA-IIowa residues 114–158 with bimodal HX kinetics (Fig. S1), the free energies of both states are included (solid black line, slow HX state; dashed black line, fast HX state). The similar free energies of the fragment 17–46 fast and slow HX states (Fig. 2b) are plotted as an average. The vertical arrows on the sequence position axis mark the mutation site at residue 26 and the proteolytic site at residue 83.
The helical wheel diagram in Fig. 6A shows that R26 is located on the non-polar face of the amphipathic α-helix, and probably at a helix-helix interface where it inhibits interaction within the N-terminal helix bundle structure. Also electrostatic repulsion with K23 and R27 on the same side of the helix (Fig. 6A) may contribute to the observed helix destabilization. These effects lead to the unfolding of the helix spanning residues 7–44. In agreement, an EPR study of the effects of the G26R mutation on the structure of residues 27–56, which are largely α-helical in apoA-IWT (41), found that residues 32–40 are in random coil in apoA-IIowa while residues 41–56 are converted from α-helix to β-sheet. The formation of β-structure may be favored by the apoA-I self-association that can occur at the higher concentration used in the EPR experiments.
Figure 6.

Helical wheel projections of the α-helical regions within the apoA-I sequence that contain the Iowa and Milano mutations. A. Residues 17–34 of apoA-IIowa with the mutated arginine residue at position 26 (cross-hatching) on the nonpolar face of the amphipathic α-helix. B. Residues 161–178 of apoA-IMil with the mutated cysteine residue at position 173 (cross-hatching) on the polar face of the amphipathic α-helix. The helical wheels are drawn with the Wheel program (43).
The disruption of the N-terminal helix bundle by the G26R mutation reflects extension of the local effect of the point mutation on helix structure to a more global effect. This global structural change includes a large destabilization (Fig. 5) of helix spanning residues 70–90, which is probably close in 3D space to the helix containing position 26 (9). The Pf values in Table S1 for fragment 72–91 indicate that the fraction of time this helix is unfolded is increased by a factor of ~ 40 and the helix is unfolded about 1% of the time. The helix unfolding induced by the G26R mutation may not occur when apoA-IIowa is associated with lipid. However, apoA-IIowa exhibits reduced lipid binding ability (41) so that its concentration in the lipid-free state is enhanced relative to apoA-IWT. This factor together with the longer existence of the segment spanning residues 72–91 as unfolded helix when in the lipid-free state enhances proteolysis in the plasma compartment to yield the N-terminal fragment encompassing residues 1–83 of apoA-IIowa. This fragment is able to form β-sheet-mediated amyloid fibrils (41–42).
In contrast to the destabilization in the region of residues 10–114, the G26R mutation induces an increase in helix content in the region spanning residues 115–158 (Fig. 5). Residues 70–180 in apoA-IIowa form α-helix presumably folded into a helix bundle. The average stability in this region is ~ 3.5 kcal/mol compared to an average of ~ 4.5 kcal/mol for the helices in apoA-IWT (Fig. 5). This decrease is consistent with the observed difference in free energy of denaturation (Table 1). The helix bundle disruption and reorganization in apoA-IIowa leads to exposure of more hydrophobic surface, reflected by an increase in ANS binding (Table 1). The HX data summarized in Fig. 2 and 5 indicate that the disordering of the segment spanning residues 7–70 and the ordering of residues 116–146 leads to a net loss of about 30 helical residues as a consequence of the G26R mutation. This agrees with the 12% decrease in α-helix content measured by CD (Table 1).
ApoA-IMil
The structural effects of the R173C mutation in apoA-IMil are quite different from those for the G26R Iowa mutation. Major destabilization of individual helices does not occur. Fig. 7 shows that the distribution of helix stabilization free energy along the molecular length is similar for apoA-IMil and apoA-IWT. The helical wheel diagram in Fig. 6B shows that C173 in apoA-IMil is located on the polar face of the amphipathic α-helix. The mutation does not destabilize the helix that contains it (residues 147–178). However, the data register a global perturbation of the helix bundle and a loss of overall stability (Table 1). This probably occurs as a consequence of the loss of stabilizing salt bridges due to the replacement of the basic arginine side chain with the acidic cysteine side chain at position 173 (14). The decreased activation of LCAT by lipid-bound apoA-IMil (18) is also likely due to altered charge distribution in this region. The major effects of the R173C mutation are probably a consequence of cysteine-linked dimer formation in apoA-IMil which is known to affect the interaction with lipid (14).
Figure 7.

Summary of the HX-derived secondary structure stabilities for lipid-free apoA-IWT and apoA-IMil. Detailed HX kinetic data (pD 7.3, 5°C) analyzed to obtain Pf values for each peptide are in (Table S2). The corresponding free energies of stabilization are plotted as a function of apoA-I sequence position (apoA-IWT, grey line; apoA-IMil slow and fast HX states are shown with solid and dashed black lines, respectively).
Summary and Conclusions
The HX MS method provides a powerful means of monitoring the structural consequences of point mutations in apoA-I. Structure, stability, and changes therein can be determined at close to amino acid resolution when sufficient overlapping peptide fragments are generated for MS analysis. The method can be applied readily to other apolipoproteins that are difficult to study by NMR and X-ray crystallography. Importantly, apolipoprotein variants in their native state can be screened rapidly in a non-perturbing manner and without the need to introduce chemical or mutational modifications. One simply measures the naturally occurring HX process.
Helix locations determined for apoA-IWT, apoA-IIowa and apoA-IMil are compared in Fig. 8. In the case of apoA-IIowa, HX MS shows that introduction of the G26R mutation into the non-polar face of an amphipathic α-helix induces it to unfold, thereby destabilizing the helix encompassing residue 83. This structural change promotes proteolysis at this location and leads to incorporation of the released peptide into amyloid fibrils. A similar mechanism probably underlies the formation of amyloid by other apoA-I variants with mutations in the N-terminal region (11). Helix unfolding is not a major consequence of the apoA-IMil R173C mutation. The mutation substitutes an acidic cysteine residue for a basic arginine residue on the polar face of an amphipathic α-helix; helix-helix interactions are perturbed leading to destabilization of the apoA-IMil molecule. The major functional differences between apoA-IMil and apoA-IWT seem to be due to the ability of the former to create cysteine-linked dimers. This observation is relevant to the development of apoA-IMil-containing HDL preparations that can be used to regress atherosclerotic plaques in individuals with cardiovascular disease (28).
Figure 8.

Comparison of the HX-derived helix locations for lipid-free (a) apoA-IWT (b) apoA-IIowa and (c) apoA-IMil. The cylinders represent α-helices and the lines indicate disordered secondary structure. Residues 17–46 (gray line in b) and 115–158 (cross-hatched in b) exhibit bimodal HX kinetics reflecting the existence of two states that undergo HX at different rates – see text for more description. The secondary structure of the predominant conformation of apoA-IMil is shown in (c). The positions of proline residues (P), whose presence leads to some perturbation of α-helix organization, are marked.
Supplementary Material
Acknowledgments
This research was supported by NIH grants HL22633 and GM031847, and Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science (No. 22590046)
ABBREVIATIONS
- ABCA1
ATP binding cassette transporter A1
- ANS
8-anilino-napthalenesulfonic acid
- apo
apolipoprotein
- GdmCl
guanidinium chloride
- HDL
high density lipoprotein
- HX
hydrogen-deuterium exchange
- LCAT
lecithin-cholesterol acyltransferase
- MS
mass spectrometry
- Pf
protection factor
- SR-BI
scavenger receptor class B type 1
Footnotes
SUPPORTING INFORMATION AVAILABLE
The fitting parameters for the HX time-courses of apoA-IWT and apoA-IIowa peptide fragments and the derived protection factors (Table S1), the equivalent data for apoA-IMil (Table S2), and comparisons of the mass spectra for apoA-IWT and apoA-IIowa peptide fragments 114–126 and 127–158 (Figure S1) are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Huang R, Silva RA, Jerome WG, Kontush A, Chapman MJ, Curtiss LK, Hodges TJ, Davidson WS. Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat Struct Mol Biol. 2011;18:416–422. doi: 10.1038/nsmb.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tall AR. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J Internal Med. 2008;263:256–273. doi: 10.1111/j.1365-2796.2007.01898.x. [DOI] [PubMed] [Google Scholar]
- 3.Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2009;50(Suppl):S189–194. doi: 10.1194/jlr.R800088-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rye KA, Bursill CA, Lambert G, Tabet F, Barter PJ. The metabolism and anti-atherogenic properties of HDL. J Lipid Res. 2009;50:S195–S200. doi: 10.1194/jlr.R800034-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84:276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- 6.Lund-Katz S, Phillips MC. High density lipoprotein structure-function and role in reverse cholesterol transport. Subcell Biochem. 2010;51:183–227. doi: 10.1007/978-90-481-8622-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothblat GH, Phillips MC. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr Opin Lipidol. 2010;21:229–238. doi: 10.1097/mol.0b013e328338472d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saito H, Dhanasekaran P, Nguyen D, Holvoet P, Lund-Katz S, Phillips MC. Domain structure and lipid interaction in human apolipoproteins A-I and E: A general model. J Biol Chem. 2003;278:23227–23232. doi: 10.1074/jbc.M303365200. [DOI] [PubMed] [Google Scholar]
- 9.Silva RAG, Hilliard GM, Fang J, Macha S, Davidson WS. A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence tracking. Biochemistry. 2005;44:2759–2769. doi: 10.1021/bi047717+. [DOI] [PubMed] [Google Scholar]
- 10.Chetty PS, Mayne L, Lund-Katz S, Stranz D, Englander SW, Phillips MC. Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2009;106:19005–19010. doi: 10.1073/pnas.0909708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorci-Thomas M, Thomas MJ. The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc Med. 2002;12:121–128. doi: 10.1016/s1050-1738(01)00163-3. [DOI] [PubMed] [Google Scholar]
- 12.Kono M, Tanaka T, Tanaka M, Vedhachalam C, Chetty PS, Nguyen D, Dhanasekaran P, Lund-Katz S, Phillips MC, Saito H. Disruption of the C-terminal helix by single amino acid deletion is directly responsible for impaired cholesterol efflux ability of apolipoprotein A-I Nichinan. J Lipid Res. 2010;51:809–818. doi: 10.1194/jlr.M002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weibel GL, Alexander ET, Joshi MR, Rader DJ, Lund-Katz S, Phillips MC, Rothblat GH. Wild-type apoA-I and the Milano variant have similar abilities to stimulate cellular lipid mobilization and efflux. Arterioscler Thromb Vasc Biol. 2007;27:2022–2029. doi: 10.1161/ATVBAHA.107.148403. [DOI] [PubMed] [Google Scholar]
- 14.Alexander ET, Tanaka M, Kono M, Saito H, Rader DJ, Phillips MC. Structural and functional consequences of the Milano mutation (R173C) in human apolipoprotein A-I. J Lipid Res. 2009;50:1409–1419. doi: 10.1194/jlr.M800578-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexander ET, Weibel GL, Joshi MR, Vedhachalam C, de la Llera-Moya M, Rothblat GH, Phillips MC, Rader DJ. Macrophage reverse cholesterol transport in mice expressing ApoA-I Milano. Arterioscler Thromb Vasc Biol. 2009;29:1496–1501. doi: 10.1161/ATVBAHA.109.191379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weisgraber KH, Rall SC, Jr, Bersot TP, Mahley RW, Franceschini G, Sirtori CR. Apolipoprotein A-IMilano. Detection of normal A-I in affected subjects and evidence for a cysteine for arginine substitution in the variant A-I. J Biol Chem. 1983;258:2508–2513. [PubMed] [Google Scholar]
- 17.Franceschini G, Sirtori CR, Capurso A, 2nd, Weisgraber KH, Mahley RW. A-IMilano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J Clin Invest. 1980;66:892–900. doi: 10.1172/JCI109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calabresi L, Franceschini G, Burkybile A, Jonas A. Activation of lecithin cholesterol acyltransferase by a disulfide-linked apolipoprotein A-I dimer. Biochem Biophys Res Commun. 1997;232:345–349. doi: 10.1006/bbrc.1997.6286. [DOI] [PubMed] [Google Scholar]
- 19.Han H, Sasaki J, Matsunaga A, Hakamata H, Huang W, Ageta M, Taguchi T, Koga T, Kugi M, Horiuchi S, Arakawa K. A novel mutant, apoA-I nichinan (Glu235 ->0), is associated with low HDL cholesterol levels and decreased cholesterol efflux from cells. Arterioscler Thromb Vasc Biol. 1999;19:1447–1455. doi: 10.1161/01.atv.19.6.1447. [DOI] [PubMed] [Google Scholar]
- 20.Huang W, Sasaki J, Matsunaga A, Han H, Li W, Koga T, Kugi M, Ando S, Arakawa K. A single amino acid deletion in the carboxy terminal of apolipoprotein A-I impairs lipid binding and cellular interaction. Arterioscler Thromb Vasc Biol. 2000;20:210–216. doi: 10.1161/01.atv.20.1.210. [DOI] [PubMed] [Google Scholar]
- 21.Chetty PS, Mayne L, Kan ZY, Lund-Katz S, Englander SW, Phillips MC. Apolipoprotein A-I Helical Structure and Stability in Discoidal High Density Lipoprotein (HDL) Particles by Hydrogen Exchange and Mass Spectrometry. Proc Natl Acad Sci USA. 2012;109:11687–11692. doi: 10.1073/pnas.1209305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Englander SW. Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom. 2006;17:1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaltashov IA, Bobst CE, Abzalimov RR. H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: is there a need for a top-down approach? Anal Chem. 2009;81:7892–7899. doi: 10.1021/ac901366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan X, Maier CS. Hydrogen/deuterium exchange mass spectrometry. Methods Mol Biol. 2009;492:255–271. doi: 10.1007/978-1-59745-493-3_15. [DOI] [PubMed] [Google Scholar]
- 25.Marcsisin SR, Engen JR. Hydrogen exchange mass spectrometry: what is it and what can it tell us? Analytical and Bioanalytical Chemistry. 2010;397:967–972. doi: 10.1007/s00216-010-3556-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Konermann L, Pan J, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2011;40:1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- 27.Mayne L, Kan ZY, Sevugan Chetty P, Ricciuti A, Walters BT, Englander SW. Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J Am Soc Mass Spectrom. 2011;22:1898–1905. doi: 10.1007/s13361-011-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calabresi L, Sirtori CR, Paoletti R, Franceschini G. Recombinant apolipoprotein A-IMilano for the treatment of cardiovascular diseases. Curr Atheroscler Rep. 2006;8:163–167. doi: 10.1007/s11883-006-0054-4. [DOI] [PubMed] [Google Scholar]
- 29.Lund-Katz S, Phillips MC. Packing of cholesterol molecules in human low-density lipoprotein. Biochemistry. 1986;25:1562–1568. doi: 10.1021/bi00355a016. [DOI] [PubMed] [Google Scholar]
- 30.Weisweiler P, Friedl C, Ungar M. Isolation and quantitation of apolipoprotein A-I and A-II from human high density lipoproteins by fast-protein liquid chromatography. Clin Chim Acta. 1987;169:249–254. doi: 10.1016/0009-8981(87)90325-1. [DOI] [PubMed] [Google Scholar]
- 31.Morrow JA, Arnold KS, Weisgraber KH. Functional characterization of apolipoprotein E isoforms overexpressed in escherichia coli. Protein Expr Purif. 1999;16:224–230. doi: 10.1006/prep.1999.1069. [DOI] [PubMed] [Google Scholar]
- 32.Markwell MAK, Haas SM, Bieber LL, Tolbert NE. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal Biochem. 1978;87:206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- 33.Vedhachalam C, Liu L, Nickel M, Dhanasekaran P, Anantharamaiah GM, Lund-Katz S, Rothblat G, Phillips MC. Influence of apo A-I structure on the ABCA1-mediated efflux of cellular lipids. J Biol Chem. 2004;279:49931–49939. doi: 10.1074/jbc.M406924200. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka M, Dhanasekaran P, Nguyen D, Ohta S, Lund-Katz S, Phillips MC, Saito H. Contributions of the N- and C-terminal helical segments to the lipid-free structure and lipid interaction of apolipoprotein A-I. Biochemistry. 2006;45:10351–10358. doi: 10.1021/bi060726t. [DOI] [PubMed] [Google Scholar]
- 35.Walters BT, Ricciuti A, Mayne L, Englander SW. Minimizing Back Exchange in the Hydrogen Exchange-Mass Spectrometry Experiment. J Am Soc Mass Spectrom. 2012 doi: 10.1007/s13361-012-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kan ZY, Mayne L, Sevugan Chetty P, Englander SW. ExMS: Data Analysis for HX-MS Experiments. J Am Soc Mass Spectrom. 2011;22:1906–1915. doi: 10.1007/s13361-011-0236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skinner JJ, Lim WK, Bedard S, Black BE, Englander SW. Protein dynamics viewed by hydrogen exchange. Protein Sci. 2012;21:996–1005. doi: 10.1002/pro.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skinner JJ, Lim WK, Bedard S, Black BE, Englander SW. Protein hydrogen exchange: testing current models. Protein Sci. 2012;21:987–995. doi: 10.1002/pro.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramella NA, Schinella GR, Ferreira ST, Prieto ED, Vela ME, Rios JL, Tricerri MA, Rimoldi OJ. Human apolipoprotein a-I natural variants: molecular mechanisms underlying amyloidogenic propensity. PLoS One. 2012;7:e43755. doi: 10.1371/journal.pone.0043755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 41.Lagerstedt JO, Cavigiolio G, Roberts LM, Hong HS, Jin LW, Fitzgerald PG, Oda MN, Voss JC. Mapping the structural transition in an amyloidogenic apolipoprotein A-I. Biochemistry. 2007;46:9693–9699. doi: 10.1021/bi7005493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gursky O, Mei X, Atkinson D. The crystal structure of the C-terminal truncated apolipoprotein A-I sheds new light on amyloid formation by the N-terminal fragment. Biochemistry. 2012;51:10–18. doi: 10.1021/bi2017014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones MK, Anantharamaiah GM, Segrest JP. Computer programs to identify and classify amphipathic alpha helical domains. J Lipid Res. 1992;33:287–296. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.