

Abstract
Oxidative stress contributes to many disease etiologies including ageing, neurodegeneration, and cancer, partly through DNA damage induction (genotoxicity). Understanding the i nteractions of free radicals with DNA is fundamental to discern mutation risks. In genetic toxicology, regulatory authorities consider that most genotoxins exhibit a linear relationship between dose and mutagenic response. Yet, homeostatic mechanisms, including DNA repair, that allow cells to tolerate low levels of genotoxic exposure exist. Acceptance of thresholds for genotoxicity has widespread consequences in terms of understanding cancer risk and regulating human exposure to chemicals/drugs. Three pro-oxidant chemicals, hydrogen peroxide (H2O2), potassium bromate (KBrO3), and menadione, were examined for low dose-response curves in human lymphoblastoid cells. DNA repair and antioxidant capacity were assessed as possible threshold mechanisms. H2O2 and KBrO3, but not menadione, exhibited thresholded responses, containing a range of nongenotoxic low doses. Levels of the DNA glycosylase 8-oxoguanine glycosylase were unchanged in response to pro- oxidant stress. DNA repair–focused gene expression arrays reported changes in ATM and BRCA1, involved in double-strand break repair, in response to low-dose pro-oxidant exposure; however, these alterations were not substantiated at the protein level. Determination of oxidatively induced DNA damage in H2O2-treated AHH-1 cells reported accumulation of thymine glycol above the genotoxic threshold. Further, the H2O2 dose-response curve was shifted by modulating the antioxidant glutathione. Hence, observed pro- oxidant thresholds were due to protective capacities of base excision repair enzymes and antioxidants against DNA damage, highlighting the importance of homeostatic mechanisms in “genotoxic tolerance.”
Key Words: Pro-oxidants, DNA damage, reactive oxygen species, DNA repair, OGG1, antioxidants, glutathione, genotoxicology, thresholds.
Assessing the genotoxic threat of chemicals is essential in gaining a better understanding of their carcinogenic potential to reduce any deleterious effects that may be produced through occupational and recreational exposures (Doak et al., 2007; Sedelnikova et al., 2010). Traditionally, regulatory authorities utilize a linear model to assess the safety of direct-acting genotoxins, whereby a mutagenic response at high doses is extrapolated to lower doses. The so-called single hit, single target hypothesis infers that there is no minimum safe exposure limit for such agents (Jenkins et al., 2005). This view, however, does not account for the plethora of homeostatic mechanisms, which allow mammalian cells to tolerate low levels of genotoxins. The application of a threshold mechanism in toxicology is not new; it is widely accepted that indirect-acting genotoxins, which have non-DNA targets, may exhibit a threshold mode of action (MOA) (Elhajouji et al., 2011). To date, however, the effect has been established experimentally for a limited number of DNA reactive compounds (Doak et al., 2007; Jenkins et al., 2005; Platel et al., 2009). We recently demonstrated that genotoxic thresholds induced by the alkylating agents ethyl methane sulfonate (EMS) and methyl methane sulfonate (MMS) are due to DNA repair by the base excision repair (BER) enzyme N-methylpurine DNA glycosylase (MPG; GenBank ID:4350) and DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT; GenBank ID:4255), selectively removing DNA adducts at low doses but becoming saturated (or repressed) at higher doses (Doak et al., 2008; Zair et al., 2011). The acceptance of dose-response thresholds for genotoxins may have substantial regulatory implications, which aid our understanding of genotoxic and neoplastic risk more fully.
An important class of DNA reactive agents present in the human environment is pro-oxidants. Mammalian cells are constantly exposed to potentially damaging reactive oxygen species (ROS) arising from multiple sources (Evans et al., 2004; Loft et al., 2008). Cellular defenses exist to combat attack from ROS, including the antioxidant glutathione (GSH) and the scavenging enzyme superoxide dismutase (Forman et al., 2009). Despite this, oxidative stress and damage to cellular macromolecules can arise when the number of ROS produced exceeds the antioxidant capacity of the cell. Numerous types of DNA damage can potentially arise upon exposure to ROS, with thymine bases being the most susceptible to modification and thymine glycol (TG) representing an important thymine lesion formed after treatment with oxidizing agents such as hydrogen peroxide (H2O2) (Basu et al., 1989).
A network of complex DNA repair pathways has evolved to avoid the perpetuation of such damage. Oxidized base lesions are repaired predominantly by the BER pathway. BER is mediated by damage-specific DNA glycosylases, with 8-oxoguanine DNA glycosylase (OGG1; GenBank ID:4968) repairing one of the most common forms of oxidatively generated DNA base damage, that is, 8-oxo-7,8-dihydroguanine (8-oxoG) (Boiteux and Radicella, 2000; Wallace, 2002). The role of repair in maintaining cellular homeostasis is highlighted by reports of BER gene activation in response to genotoxic exposure (Powell et al., 2005). Further, mutation and abnormal expression of DNA repair genes, such as ATM (ataxia telangiectasia mutated; GenBank ID:472) and BRCA1 (breast cancer 1, early onset; GenBank ID:672), have been directly linked with genomic instability and cancer development (Hartman and Ford, 2003; Smirnov and Cheung, 2008).
The present study focuses on DNA-pro-oxidant interaction and generates genotoxic dose responses for three pro-oxidant chemicals, H2O2, menadione, and potassium bromate (KBrO3). Two different mutagenic endpoints were assessed in vitro as recommended by Committee on Mutagenicity: the cytokinesis block micronucleus (CBMN) assay and HPRT (GenBank ID:3251) forward mutation assay, examining the induction of chromosomal damage and frequency of point mutations, respectively (Great Britain. Committee on Mutagenicity of Chemicals in Food, 2000). The principal aims were to analyze low-range dose-response curves, to establish the existence of possible genotoxic thresholds, and to explore MOA of any observed thresholds (tolerance). Compounds were chosen due to their known capacity to generate DNA damage through differential generation of various ROS.
H2O2 is a physiological constituent of living cells, continually produced by a variety of cellular pathways, and has a wide range of external applications, for instance, in bleaching and in treatment of water and sewage (Jeong et al., 2010; Naik et al., 2006). H2O2 is a nonradical ROS but can react via radical- mediated routes; for example, in the presence of ferrous ions, H2O2 undergoes Fenton’s reaction to form the highly reactive hydroxyl radical (HO•) (Pryor, 1986). Menadione is a multivitamin component and a therapeutic agent for hypothrombinemia and cancer. In the presence of flavoenzymes, menadione may undergo reduction to a semiquinone, an extremely unstable compound that reacts rapidly with oxygen forming superoxide anion radical (O2 •–) and other ROS (Chung et al., 1999; Nutter et al., 1992). KBrO3 has been used as a food additive primarily in bread-making processes. European Union (EU) countries now prohibit this application due to its proven carcinogenicity. The mechanism by which KBrO3 generates damaged DNA is not fully elucidated but is believed to involve reduction of bromate by thiols such as cellular GSH to reactive intermediates including bromine radicals (Br•) or oxides (BrO•, BrO2 •, etc.) (Ballmaier and Epe, 1995, 2006).
MATERIALS AND METHODS
Chemicals
Buthionine sulfoximine (BSO), H2O2, KBrO3, menadione, and N- acetylcysteine (NAC) were all purchased from Sigma (Dorset, U.K.). All chemical dilutions were freshly prepared from stock solutions with water.
Cell Culture
The human male near-diploid lymphoblastoid cell line AHH-1 (ATCC, Middlesex, U.K.) was cultured in RPMI 1640 (Life Technologies, Paisley, U.K.) and supplemented with 1% L-glutamine (Life Technologies) and 10% donor horse serum (BD Gentest, Oxford, U.K.) in 80-cm2 flasks at 37°C, 5% CO2. The cells were maintained at a concentration of 1–2 X 105/ml. AHH-1 cells were utilized in the study as they have been widely used in genetic toxicology and represent a versatile and reproducible system for examining genotoxic agents and the induction of gene locus mutation, including the analysis of damage response pathways and cellular defenses upon exposure to free radicals. AHH-1 contains native CYP1a1 activity, and despite harboring a heterozygous p53 mutation at the codon 281/282 interface within exon 8, it retains the ability to undergo DNA damage–induced apoptosis and has been reported to express phospho-p53 and p21 (Doak et al., 2008; Guest and Parry, 1999). Further, previous studies on thresholds have been completed utilizing AHH-1 and have described stable background levels of chromosomal damage and point mutations (Doak et al., 2007; Zair et al., 2011).
Forward Mutation Assays
We employed the in vitro HPRT assay to study induced point mutations. The assay was performed as previously described (Doak et al., 2007), with the following modifications: AHH-1 cell suspensions (10 ml), at 5 X 105/ml, were exposed to the test chemical in 80-cm3 flasks at the appropriate concentration for 24 h at 37°C, 5% CO2. For each dose, fifteen 96-well plates for assessing mutation frequency and another five for plating efficiency were set up. Each dose was performed in triplicate.
Micronucleus Assay
Micronuclei (MN) frequency was utilized to assess the level of chromosome aberration induction. AHH-1 (10 ml) suspensions of cells at 1 X 105/ml were seeded for 24 h at 37°C, 5% CO2. Replicate flasks (n = 3, independently produced on different days) were dosed with appropriately diluted test chemical (in duplicate) for 4 h, after which cells were centrifuged, washed once in PBS, and resuspended in 10-ml fresh media containing 6-µg/ml cytochalasin B for one cell cycle (22 h). Treated cells were harvested, resuspended in 10 ml of hypotonic solution (0.56% KCl), and centrifuged immediately. Cell suspension was resuspended in fixative 1 (methanol:acetic acid:0.9% NaCl [5:1:6 parts]) and centrifuged after a 10-min incubation period. Cells were transferred to fixative 2 (methanol:acetic acid [5:1 parts]) for a 10-min incubation, centrifuged, washed four times, and maintained in the final fixative 2 wash at 4°C for 16 h. Fixed cells were centrifuged, and 100 µl was dropped onto polished, fixed, and hydrated slides, stained with DAPI (4′,6-diamidino-2-phenylindole; 0.15 µg/ml final concentration), and viewed under a Carl Zeiss Axio Imager fluorescence microscope. Slides were scored utilizing the Metafer 4 software version 3.5 (MetaSystems, Altlussheim, Germany). An optimal scoring criterion was achieved from the development of specific classifiers adjusted to accommodate the particular lymphoblast cell line (AHH-1) and to identify binucleate cells containing MN. The criteria for identifying MN were as previously described (Fenech, 2007). A minimum of 2000 binucleated cells were scored per replicate, and each dose was performed in triplicate (an average of 6000 binucleates per dose).
RNA Isolation and Gene Expression Analysis
Following exposure of AHH-1 cells to the test pro-oxidants for 0, 2, 4, and 24 h time points, total cellular RNA was isolated from AHH-1 cells followed by DNase treatment using the RNeasy mini kit (Qiagen, West Sussex, U.K.) and TURBO DNA-free kit (Ambion, Huntingdon, U.K.), respectively, according to the manufacturer’s instructions. Quantitative PCR of mRNA was performed as a one-step reaction, where the entire reaction from cDNA synthesis to real-time PCR amplification was performed in a single well, on a MyIQ5 cycler optics module (Bio-Rad, Hertfordshire, U.K.). Commercially available TaqMan Gene Expression Assays and human endogenous controls were purchased from Applied Biosystems (U.K.) and supplied as premixed primers and FAM/MGB probe. To perform one-step real-time PCR, OGG1 target gene (Hs00213454_m1*) and HPRT reference gene (4333768) probes were used in conjunction with QuantiFast Probe RT-PCR kit (Qiagen). Approximately 0.2 µg RNA was used for cDNA synthesis and PCR in a reaction volume of 20 µl, containing 10 µl of 2X QuantiFast Probe RT-PCR Master Mix, 0.4 µl QuantiFast RT Mix, and 1 µl of TaqMan probe mix (primers and probe at final concentrations of 900nM and 250nM, respectively).The following PCR reaction protocol was used: initially cDNA production occurred for one cycle at 50°C for 10 min followed by 95°C for 5 min. Subsequently, PCR was initiated for 40 cycles of 95°C for 10 s and 61°C for 30 s. Reactions were performed in triplicate, and the level of gene expression was reported as the ratio between the mRNA level of the target gene and the HPRT reference gene using the relative standard curve method.
Western Blot Analysis
Following exposure to the test pro-oxidants for 0-, 2-, 4-, and 24-h time points, total cellular protein was isolated from AHH-1 cells. Cells were pelleted by centrifugation, washed in ice-cold PBS, and then lysed in RIPA buffer (Sigma), supplemented with protease inhibitor cocktail (Sigma) at 4°C for 5 min. Cells were further lysed by agitation and centrifugation, and the cell pellet was discarded. Protein extracts (30 ng) of AHH1 cells were resolved on 10% SDS-PAGE at 120 V, transferred onto polyvinylidene difluoride membranes (Bio-Rad), and blocked for 1 h at room temperature with 5% bovine serum albumin in TBS-T (20mM Tris [pH 7.6], 125mM NaCl, 0.1% (v/v) Tween20). Membranes were incubated with monoclonal mouse anti-OGG1 (Sigma), polyclonal rabbit anti-beta tubulin (AbCam, Cambridge, U.K.), monoclonal rabbit anti-ATM (Cell Signaling Technology, MA), polyclonal rabbit anti-BRCA1 (Cell Signaling Technology), or monoclonal rabbit anti-beta actin (Cell Signaling Technology) antibodies overnight at 4°C. Following washing (4 X 5 min in TBS-T), membranes were incubated with appropriate horseradish peroxidase conjugated secondary antibodies (AbCam). Protein bands were detected using the Immun-Star WesternC chemiluminescence kit (Bio-Rad). Membranes were visualized using the Bio-Rad Chemidoc XRS, and average band density analysis was performed using Quantity One version 4.6.3 (Bio-Rad).
Human DNA Repair PCR Arrays
To assess the role of a wider range of DNA repair enzymes in the observed nonlinear damage responses to pro-oxidants, gene expression PCR arrays tailored for DNA repair were utilized. AHH-1 cells were treated with specific doses above and below confirmed threshold inflection points (IPs), which were 5µM and 25µM H2O2, 0.2mM and 0.8mM KBrO3, and 0.5mM and 3.5mM menadione, for 4 h, and the RNA was extracted. RNA (1.6 µg) from each sample was reverse transcribed into cDNA using the RT² First Strand Kit (Qiagen). Gene expression was performed utilizing Human DNA Repair RT² Profiler PCR Arrays (PAHS-042A) and 2X SABiosciences RT² qPCR Master Mix (Qiagen) and MyiQ real-time PCR Platform (Bio-Rad) according to the manufacturer’s instructions. Gene expression was normalized using five housekeeping genes within the array and quantified using the Ct method by accessing the PCR Array Data Analysis Web Portal (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php).
Gas Chromatography/Mass Spectrometry Determination of Oxidatively Induced DNA Damage
Gas chromatography/mass spectrometry (GC/MS) with isotope dilution was used to determine the absolute levels of five different oxidatively modified bases: 8-oxoG, TG, 5- hydroxy-5-methylhydantoin (5-OH-5-MeHyd), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FAPyG), and 4,6- diamino-5-formamidopyrimidine (FAPyA). AHH-1 cells were seeded for 24 h and treated with 0, 5, and 25µM/ml H2O2 for 4 h. Following washing, DNA was extracted from the treated cells using the DNeasy Blood & Tissue Kit (Qiagen). The DNA was precipitated, and GC/MS analyses were performed as previously described (Dizdaroglu, 1985; Jaruga et al., 2008).
Alteration of Cellular GSH Levels
In order to modulate GSH levels prior to measuring pro-oxidant-induced MN, GSH was either supplemented or depleted. AHH-1 (10 ml) suspensions of cells at 1 X 105/ml were seeded for 24 h at 37°C, 5% CO2. Cells were dosed with 2mM NAC to promote GSH levels and increase cellular antioxidant status or with 0.5mM BSO, an inhibitor of GSH synthesis, for 24 h prior to pro-oxidant treatment and CBMN assay (as previously described). The levels of GSH in treated cells were estimated utilizing the Glutathione Assay kit (Sigma) according to the manufacturer’s instructions.
Statistical Analysis
For the genotoxicity data and DNA adduct data, a one-way ANOVA, followed by a Dunnett’s post hoc test, was used to determine if any of the treatment doses were significantly different from the zero dose, except when cells were pretreated with BSO or NAC, where a t-test was employed. The hockey stick modeling of apparent thresholds was carried out using software, kindly provided by Lutz and Lutz (2009) and implemented in the R package. A t-test was performed to establish any differences between relative protein and mRNA expression of OGG1 after pro-oxidant treatments at different time points analyzed.Independent sample t-test was utilized to analyze differences in normalized protein levels of ATM and BRCA1 after treatment with H2O2 (0–25µM) or KBrO3 (0–0.8mM).
RESULTS
The CBMN and HPRT forward mutation assays were used to assess the induction of chromosomal aberration (MN formation) and frequency of point mutations, respectively, after exposure of AHH-1 lymphoblastoid cells to low concentrations of the oxidizing agents. The range of concentrations used was determined following initial dose–setting experiments, analyzing genotoxicity and assessing cytotoxicity utilizing relative population doubling calculations in satellite cultures (data not shown). To ensure that any observed genetic damage was not due to cytotoxicity-related mechanisms, only those concentrations leading to more than 50% cell viability were analyzed (data not shown). Investigations into the MOA behind these threshold responses involved analysis of levels of DNA repair enzymes involved in the repair of oxidative damage, assessment of the levels of oxidatively modified bases in exposed DNA, and analysis of the effect of antioxidant status on the dose-response curves to chromosome damage. To ensure that cellular exposure to ROS was occurring at low doses of pro-oxidants, oxidation of the nonfluorescent probe 2′,7′-dichlorofluorescein-diacetate (DCFH-DA) to the fluorochrome 2,7-dichlorofluorescein (DCF) was used as an index to quantify the overall level of intracellular ROS produced by 0–100µM H2O2 (Supplementary fig. S1).
Gene Mutation Induction
The HPRT forward mutation assay was used here to i nvestigate the mutagenic activity of H2O2, KBrO3, and menadione. All three pro-oxidants demonstrated a range of low doses with minimal levels of mutation induction in AHH-1 human lymphoblastoid cells, compared with solvent control data (Fig. 1). The first statistically significant increase in mutation frequency above background levels (lowest observed effect level [LOEL]) was 18µM for H2O2 (p = 0.004), 0.5mM for KBrO3 (p = 0.004), and 2.9µM for menadione (p = 0.001) utilizing one-way ANOVA, followed by a Dunnett’s post hoc test. Further analysis of these results applying a “hockey-stick model” developed by Lutz and Lutz (2009) rejected linearity of each dose-response curve while supporting a threshold model (p < 0.05)and showed that H2O2 had an IP (or threshold dose) at 7.3µM with lower confidence interval (CI) of 2.04µM; KBrO3, an IP of 0.18mM with lower CI of 0.08mM; and menadione, an IP of 1.89µM with lower CI of 0.76mM (Fig. 1) (Lutz and Lutz, 2009).
Fig. 1.
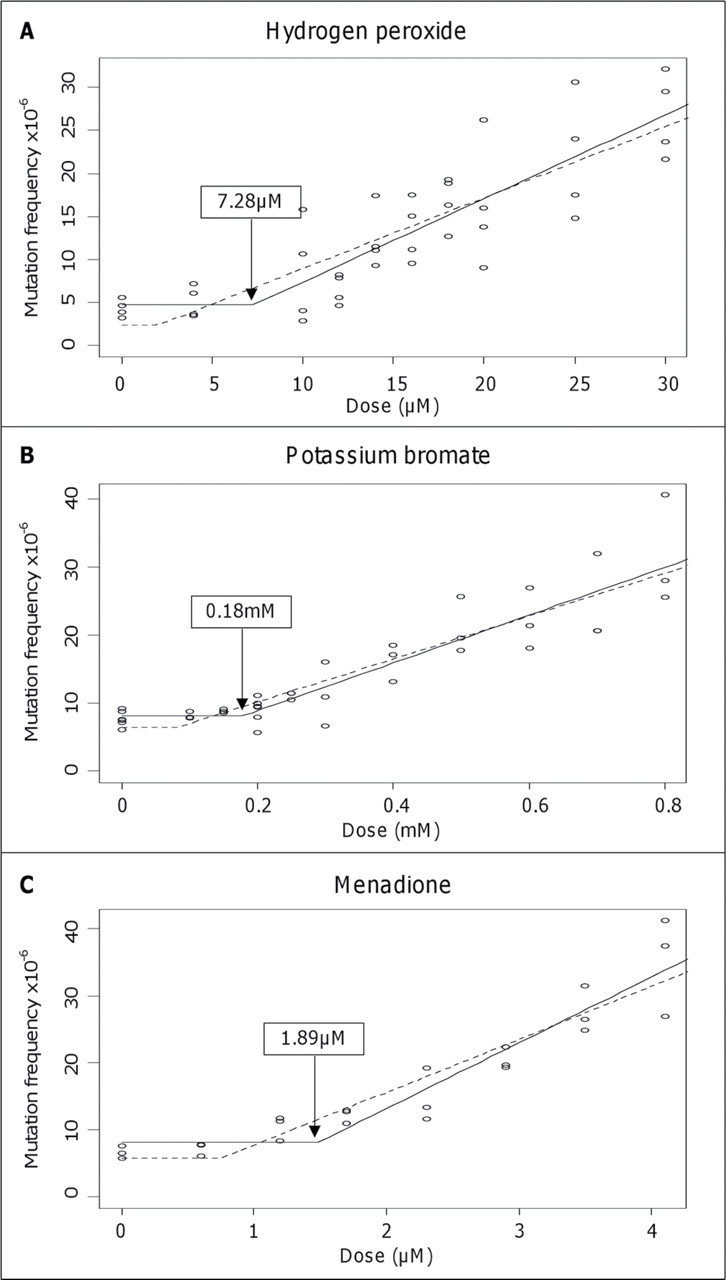
Gene mutation frequency in response to pro-oxidants. Dose- response relationships of hydrogen peroxide (H2O2), potassium bromate (KBrO3), and menadione in the AHH-1 cell line with respect to HPRT gene mutation frequency. Hockey-stick statistical modeling analysis has been applied to each data set to calculate the IP, probability for nonlinearity (p), and Y- intercept. (A) H2O2, threshold, IP = 7.28µM, lower IP/CI = 2.04µM, Y = 1.95, and p= 0.038. (B) KBrO3, threshold, IP = 0.18mM, lower IP/CI = 0.08mM, Y = 6.11, and p = 0.004. (C) Menadione, threshold, IP = 1.89µM, lower IP/C = 0.76µM, Y= 5.05, and p= 0.004.
Chromosomal Damage Induction
The dose-response relationships, with respect to the induction of chromosome aberrations, obtained after pro-oxidant treatments with H2O2, KBrO3, and menadione are illustrated in Figure 2. LOEL concentrations of 25µM (p = 0.03), 0.6mM (p = 0.007), and 2.9µM (p = 0.036) were identified for exogenous treatment with H2O2, KBrO3, and menadione, respectively. Subsequent increases in concentration above each LOEL produced a progressive rise in cellular DNA damage, as detected by a higher incidence of MN. Hockey-stick statistical modeling of the data using the Lutz approach rejected a linear fit to the dose-response curves and confirmed an IP (threshold value) of 10.88µM with lower CI of 5.93µM and 0.35mM with lower CI of 0.22mM, for H2O2 and KBrO3, respectively. The dose-response curve for menadione, despite indicating a nonlinear response for the induction of genotoxic damage, favored a linear model and did not achieve significance when hockey-stick modeling was performed on the data.
Fig. 2.
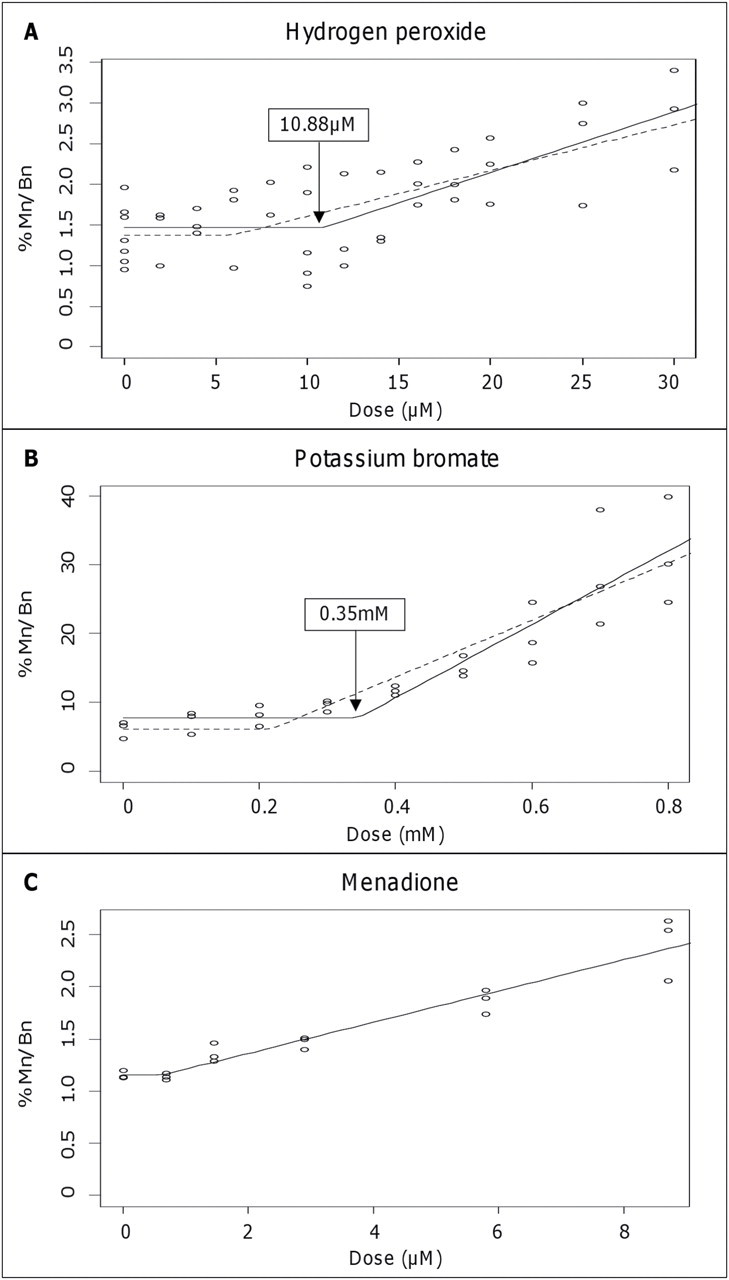
Chromosomal damage in response to pro-oxidants. Dose-response relationships of hydrogen peroxide (H2O2), potassium bromate (KBrO3), and menadione in the AHH-1 cell line with respect to micronucleus frequency. Hockey-stick statistical modeling analysis has been applied to each data set to calculate the IP, probability for nonlinearity (p), and Y-intercept. (A) H2O2, threshold, IP = 10.88µM, lower IP/CI = 5.93µM, Y = 1.32, and p = 0.023. (B) KBrO3, threshold, IP = 0.35mM, lower IP/CI = 0.22mM, Y = 1.05, and p = 0.002. (C) Menadione, linear, and p = 0.35. Mn/Bn%:Percentage of binucleated cells containing one or more MN.
BER Glycosylase Cellular Levels
In order to examine the protective mechanisms behind the genotoxic tolerance to low doses of pro-oxidants shown here, we focused firstly on the role of DNA repair. Previous work on thresholded responses for the alkylating agents has demonstrated upregulation of the DNA repair enzymes MPG and MGMT by EMS and MMS, respectively, at doses below the threshold (Doak et al., 2008; Zair et al., 2011). Because 8-oxoG is a major DNA lesion formed as a consequence of ROS exposure and 8-oxoG in DNA is primarily repaired by the DNA glycosylase OGG1, the mRNA and protein levels of this enzyme were measured in response to specific pro-oxidant treatment (in this case, H2O2). OGG1 expression levels in treated and untreated AHH-1 lymphoblastoid cells, measured by real-time RT-PCR, are represented in Figure 3A. No significant modulation in OGG1 expression in response to H2O2 treatment (0–50µM) was observed at any of the time points (0–24 h) studied. OGG1 protein levels in AHH-1 cells following oxidative insult were also analyzed by Western blotting and normalized to beta- tubulin expression (Fig. 3B). Similar to mRNA levels, treatment with H2O2 (0–50µM) had no detectable effect on the amount of OGG1 protein recovered from nuclear extracts at any of the treatment periods (0–24 h) monitored; comparable results were observed for treatment with 1mM KBrO3 (results not shown).
Fig. 3.

Effect of hydrogen peroxide on OGG1 levels. (A) Effect of hydrogen peroxide (H2O2) on OGG1 expression in AHH-1 cells.Cells were treated with 0–50µM of H2O2, and total RNA was extracted between 0 and 24 h. Levels of OGG1 mRNA were assessed by real-time RT PCR. Values were normalized to levels of the constitutively expressed housekeeping gene, HPRT,and represent the mean (SD) fold change from control levels at each time point. Each data point represents three independent measurements. (B) Western blot of OGG1 protein and loading control B-tubulin in AHH-1 cells following treatment with 25µM H2O2 for 0, 2, 4, and 24 h; L: Ladder.
Human DNA Repair PCR Arrays
To investigate the effects of pro-oxidant treatment on a wider range of DNA repair genes within this interesting thresholded region, treated cells were analyzed using DNA repair-directed PCR gene expression arrays. Because only H2O2 and KBrO3 showed thresholds for both point mutation and chromosome damage, these studies concentrated on these two pro-oxidants. The PCR expression arrays focus on a selected panel of 84 genes involved in the base excision, nucleotide excision, mismatch, double-strand break, and other DNA repair processes (Supplementary fig. S2). Numerous fold changes in expression, compared with untreated control cells, were observed above and below the threshold doses (Table 1). ATM, for example (an important gene involved in the signaling response to DNA damage), showed unchanged expression levels at concentrations below the threshold doses but was observed to be 2.13-fold (H2O2) and 4.0-fold (KBrO3) downregulated above the threshold dose for chromosome damage induction, suggestive of a relatively higher degree of damage recognition below the threshold. The cancer susceptibility gene BRCA1, involved in the repair of DNA double-strand breaks (DSBs), was upregulated by 2.39- and 2.04-fold (H2O2 and KBrO3, respectively) at concentrations below but not above the thresholds for chromosomal damage in both chemicals. H2O2-treated cells also exhibited a 2.69-fold increase in expression of APEX1 (APEX nuclease [multifunctional DNA repair enzyme] 1; GenBank ID:328) at a concentration below, but not above, the threshold dose, indicative of a role for BER in minimizing chromosome damage at low H2O2 doses. Further, the BER DNA glycosylase MUTYH (mutY homolog [Escherichia coli]; GenBank ID:4595), which catalyzes the removal of adenine bases from the DNA backbone at sites where adenine is inappropriately paired with guanine, cytosine, or 8-oxoG, was downregulated by 2.17-fold after exposure to 0.8mM KBrO3.
TABLE 1.
Alterations in DNA Repair Gene Expression Above and Below the Threshold for Chromosome Damage Induction
| Gene symbol | Fold change in gene expression | |||
|---|---|---|---|---|
| 5µM H2O2 | 25µM H2O2 | 0.2mM KBrO3 | 0.8mM KBrO3 | |
| APEX1 | 2.69 | 1.14 | n/a | 2.57 |
| ATM | 0.52 | –0.47 | 1.08 | –0.25 |
| BRCA1 | 2.39 | 1.71 | 2.04 | 1.89 |
| ERCC4 | 0.55 | 0.74 | 0.66 | –0.5 |
| FEN1 | –0.46 | 0.83 | 0.61 | –0.44 |
| LIG3 | 0.53 | 0.7 | 0.62 | –0.47 |
| MUTYH | 0.54 | 0.74 | 0.62 | –0.46 |
| PNKP | –0.41 | 0.71 | 0.53 | –0.34 |
| TOP3B | 0.51 | 0.79 | 0.55 | –0.43 |
Note. n/a, not applicable.
Relative Levels of DNA Damage Response Proteins
To further investigate the gene expression changes observed during the PCR gene array experiments and to ascertain whether such expression changes were consistently observed at the protein level, the amount of ATM and BRCA1 protein recovered from nuclear extracts after pro-oxidant treatment was determined in AHH-1 cells. Cells were dosed with H2O2 and KBrO3 at concentrations above and below the thresholds for chromosome damage induction, and relative protein levels were deduced by Western blotting via the examination of band intensities of ATM or BRCA1 normalized to beta-actin loading controls (Fig. 4). Unlike the alterations in ATM and BRCA1 mRNA expression observed following treatment with concentrations of H2O2 above (25µM) or below (5µM) the threshold, no significant threshold-dependent differences in the levels of these DNA repair proteins were detected (Fig. 4A). Treatment with 0.2mM KBrO3 (below the threshold) appeared to induce an increase in protein levels of both ATM and BRCA1 (Fig. 4B), which then decreased at 0.8mM KBrO3 (above the threshold) to levels comparable to untreated samples; however, these differences were not deemed significant by independent sample t-test.
Fig. 4.
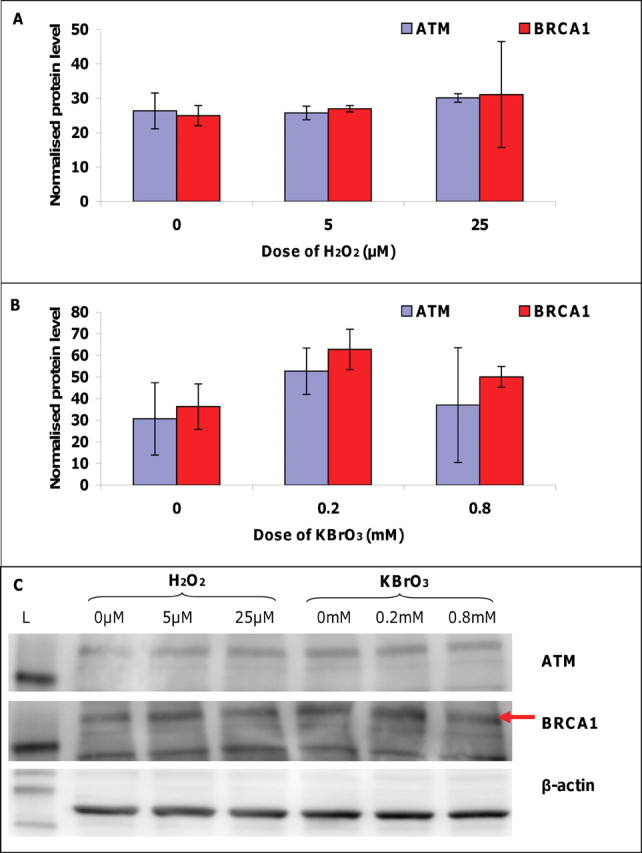
Effects of pro-oxidants on protein levels of ATM and BRCA1. (A) Effect of hydrogen peroxide (H2O2) on ATM and BRCA1 protein levels recovered from AHH-1 cells treated for 4 h with concentrations above (25µM) and below (5µM) the genotoxic threshold for DNA damage induction. (B) Effect of potassium bromate (KBrO3) on ATM and BRCA1 protein levels recovered form AHH-1 cells treated for 4 h with concentrations above (0.2mM) and below (0.8mM) the genotoxic threshold for DNA damage induction. Normalized protein level: Relative protein concentration calculated using membrane band intensities normalized to beta-actin loading control band intensities. Bars: Standard deviation.
GC/MS Determination of Oxidatively Induced DNA Damage
Four out of the five modified bases were detected in the AHH-1 DNA extracts (Fig. 5). The FAPyA lesion was not detectable in either the control or the H2O2-treated samples, most likely due to the low amount (~30 µg) of extracted DNA/ sample available for MS analysis. Of the four detected lesions, the levels of the 8-oxoG, FAPyG, and 5-OH-5-MeHyd lesions in the H2O2-treated samples were not significantly different from the background levels in the control samples. The TG lesion, however, was found to be significantly (p = 0.0259) increased in the 25µM H2O2-treated sample (Fig. 5D), in comparison with the TG background level in the control samples. The level of TG in the 5µM H2O2-treated sample was not significantly different from the TG levels in the controls. The accumulation of TG in the 25µM H2O2-treated sample could potentially have biological relevance as TG is a cytotoxic lesion that is a known block to DNA polymerase.
Fig. 5.

Determination of oxidatively induced DNA damage in hydrogen peroxide-treated AHH-1 cells.(A) Accumulation of 8-oxoG in relation to exposure dose, (B) accumulation of FAPyG in relation to exposure dose, (C) accumulation of 5-OH-5-MeHyd in relation to exposure dose, and (D) accumulation of TG in relation to exposure dose. *indicates statistically significant result (p < 0.05) compared with that of the control samples using one-way ANOVA followed by Dunnett’s multiple comparison test. All data points represent the mean of 4 independent measurements. Uncertainties are standard deviations.
Impact of GSH on Pro-oxidant-Induced DNA Damage
GSH is the principal nonprotein thiol involved in the antioxidant cellular defense, playing a critical role in protecting cells from oxidative damage and the toxicity of xenobiotic electrophiles, as well as maintaining redox homeostasis (Forman et al., 2009). To elucidate the function of antioxidants in the observed genotoxic threshold effects described above, intracellular levels of GSH were modified, and the shapes of the resultant dose-response curves for H2O2, with respect to chromosomal damage, were assessed. AHH-1 cells were pretreated with BSO (0.5mM), a specific GSH synthesis inhibitor, or NAC (2.0mM), a GSH precursor, for 24 h, and the effects on H2O2-induced DNA damage were determined by the CBMN assay (Fig. 6). BSO pretreatment altered the shape of the H2O2 dose-response curve significantly from a thresholded curve to a linear dose-response, which saturates at higher doses. BSO pretreatment reduced the first dose to induce a statistically significant increase in genetic damage from 18µM to 8µM (p = 0.0008), which indicates that GSH depletion by BSO (confirmed utilizing a GSH assay) reduces protection against oxidatively induced DNA damage, at low-dose treatments. Furthermore, statistically significant increases in MN frequency were observed in BSO pretreated cells at 10µM (p < 0.001)and 12µM (p <0.01) H2O2 compared with cells without pretreatment. In contrast, NAC pretreated cells had significantly lower levels of MN induction at 18µM (p < 0.001)and 25µM (p <0.01) H2O2 compared with H2O2 cells without pretreatment, suggestive of a protective effect of enhanced GSH status.
Fig. 6.

Pro-oxidant dose response following alteration of cellular GSH antioxidant levels. Effect of BSO (0.5mM) or NAC (2.0mM) pretreatment on the frequency of hydrogen peroxide (H2O2)-induced MN in AHH-1 cells. *At 8µM H2O2, first significant increase in MN frequency above the control in BSO pretreated cells (p = 0.0008) as determined by t-test. &: At 10µM H2O2, BSO pretreated cells demonstrated a statistically significant increase in percentage MN compared with non-pretreated and NAC pretreated cells (p <0.001 and p < 0.05, respectively); **At 12µM H2O2, BSO pretreated cells showed a statistically significant elevation in MN compared with non-pretreated cells (p < 0.01); $: At 18µM H2O2, first statistically significant increase in MN frequency above the control in non-pretreated cells (p < 0.0001); also, non-pretreated cells exhibited statistically significant higher levels of MN to NAC pretreated cells (p < 0.001); ***At 25µM H2O2, non-pretreated cells had statistically significant higher levels of MN than NAC pretreated cells (p < 0.01). Mn/Bn%:Percentage of binucleated cells containing one or more MN.
DISCUSSION
Elevated cellular levels of ROS can imbalance homeostasis and create oxidative stress, and chronic exposure to this stress can cause permanent genomic changes. Accumulation of oxidative lesions has been associated with ageing and a host of human diseases including cancer, chronic inflammation, atherosclerosis, and neurodegenerative diseases such as Alzheimer’s disease (Sedelnikova et al., 2010). The use of oxidative DNA damage measurements in populations may have important implications for human health risk assessment, distinguish relevant environmental exposures, and predict the frequency of disease (Hatt et al., 2008; Loft et al., 2008).
Traditionally, characterization of DNA-reactive agents has involved the assumption of a linear relationship between genotoxin exposure and induction of mutagenic modifications (Henderson et al., 2000). This view, however, does not account for homeostatic mechanisms that potentially counteract DNA damage induced after genotoxic exposure. Knowledge of the dose-response relationship of a given chemical is paramount; if a threshold exists, safe levels can be calculated; if there is a linear dose-response, there are no safe exposure levels. This can have economic implications, impacting the availability and use of certain compounds.
To investigate the biological significance of low-dose exposures, a robust analysis of mutation and chromosomal damage induction was performed with the DNA-reactive compounds H2O2, KBrO3, and menadione. All three pro-oxidants displayed a range of low doses with no statistically significant increase above background levels of DNA damage induction. Objective analysis of the data employing hockey stick statistical modeling demonstrated inflection (threshold) points for each of the dose relationships, except for chromosomal damage induction in menadione, which conformed to a linear model. Menadione may thus be considered a nonthresholded compound overall. The linear relationship of damage induction observed after menadione exposure may be explained by the reported production of a high frequency of DNA strand scission by the compound mediated by HO•, which may be too extensive for the repair machinery to correct, even at low doses (Nutter et al., 1992). Although DNA strand breaks occur after exposure to H2O2 and KBrO3, other oxidative lesions such as 8-oxoG and TG occur preferentially (Cadet et al., 2010; Kawanishi and Murata, 2006).
Thresholds were reported at lower concentrations (1.5- to 2-fold) for point mutations than for MN induction, which may be explained by the higher probability of a single oxidative lesion formation yielding a base substitution, compared with a DSB arising from multiple clustered lesions. Indeed, abortive BER processing of radical damage can form DSBs when the position of lesions is closely opposed on the two strands (Wallace, 2002).
Several mechanisms may be responsible for contributing to genotoxic thresholds in response to ROS; however, DNA repair is likely to be the primary mode of defense. Repair pathways may well successfully remove newly formed adducts at low doses, and if the rate of lesion repair is faster than its rate of formation, a no observed–effect limit (NOEL) will result. We have previously noted that thresholds for alkylating agents (EMS and MMS) are accompanied by increases in the expression of DNA repair genes MGMT and MPG (Doak et al., 2008; Zair et al., 2011).
A well-studied biomarker of oxidative damage, and a key repairable DNA lesion induced by the pro-oxidants studied, is 8-oxoG. 8-oxoG is a premutagenic DNA lesion due to its propensity to mispair with adenine, generate errors in replication, and G:C to T:A transversions. It is a substrate for the BER pathway, initiated by the OGG1 enzyme (Boiteux and Radicella, 2000; Nishimura, 2002). To investigate the potential of OGG1 as a thresholded MOA, gene and protein expression of OGG1 was examined. No modulation in OGG1 levels was observed in response to oxidative stress, which is in agreement with other studies (Mistry and Herbert, 2003; Saitoh et al., 2001). Furthermore, OGG1 has been described as a housekeeping gene with a constant level of expression throughout the cell cycle (Dhenaut et al., 2000). Basal, not inducible, expression of OGG1, therefore, may play a role in the maintenance of homeostasis in the presence of low levels of pro-oxidants.
A lack of OGG1 induction may reflect the redundancy that exists between BER glycosylases and indeed between other pathways to repair oxidative lesions. Further investigation into the responses of other DNA repair genes to pro-oxidant stress was needed, and to fulfill this requirement, the gene expression profile of 84 key DNA repair enzymes was compared at doses above and below the thresholds of chromosome damage induction observed for H2O2 and KBrO3 (S2). For most of the nine genes with altered expression, there was no clear pattern for the two chemicals, and therefore, it is difficult to propose a direct mechanistic link in which a threshold level of exposure determines a switch in the expression of the DNA damage response program. Despite this, two repair genes showed interesting results: ATM, a central component of the DSB repair pathway in mammalian cells, was downregulated above the threshold doses for damage induction and BRCA1, a nuclear phosphoprotein that plays a role in maintaining genomic stability, was upregulated below the threshold doses for damage induction in both H2O2 and KBrO3.
Analysis of protein levels of these DNA damage response genes following exposure to pro-oxidants, however, did not substantiate the PCR array findings, and no significant alterations in the protein levels of ATM or BRCA1 were observed above or below the genotoxic thresholds. Thus, although DSB may occur under oxidative stress conditions, such as when ROS-induced DNA damage interferes with either DNA replication or transcription, for example, during the processing of bulky DNA adducts such as FAPyG produced from ring opening of guanine upon exposure to HO•; it is not involved in the MOA of genotoxic thresholds observed for pro-oxidants. In agreement with this conclusion, in this study, FAPyG lesions were observed at similar levels at doses above and below the H2O2 threshold and thus appeared to be repaired effectively or were not formed upon exposure to low doses. In contrast, TG levels were significantly higher in 25µM, but not in 5µM, H2O2-treated samples than in untreated controls.
TG is the most common thymine lesion found after treatment with oxidizing agents and exerts significant distortion on the duplex DNA molecule, blocking replication. However, there are instances whereby DNA polymerases can by-pass TG and a low level of misincorporation of guanine opposite TG occurs, giving rise to mutation (Wallace, 2002). The presence of higher levels of TG above the threshold suggests a role for this lesion and BER in the observed genotoxic thresholds. The BER glycosylases NEIL1 (nei endonuclease VIII-like 1; GenBank ID:79661) and NTHL1 (nth endonuclease III-like 1; GenBank ID: 4913) may successfully remove TG at low doses, but above the threshold the glycosylases are “saturated”; lesions start to escape repair, becoming fixed permanent defects, and subsequent increases in dose result in a more linear increase in damage. Alternatively, formation of TG may be reduced below the threshold due to lower exposure to ROS.
DNA repair represents but one tier of protection against oxidatively generated DNA damage present in multicellular organisms. Antioxidants may also contribute to the above described thresholds. Alteration of the antioxidant status of cells via the manipulation of GSH levels transformed the shape of the dose-response curve of H2O2-induced chromosomal damage. For example, inhibition of GSH by BSO altered the shape of the curve from a nonlinear threshold to a more linear response and reduced the lowest dose to induce a statistically significant increase in genetic damage from 25µM to 8µM H2O2. Furthermore, boosting GSH levels with NAC shifted the dose response to the right, with treated cells showing significantly lower levels of MN induction at 18µM and 25µM H2O2 compared with H2O2 cells without pretreatment. Such effects s uggest a causal role for GSH in the genotoxic thresholds of pro-oxidants, competing with DNA to accept electrons from ROS, removing their oxidative capacity and the potential to create mutagenic lesions. Altering the redox status in vitro by increasing the levels of antioxidants has beneficial, protective effects against pro-oxidant agents.
Although it is difficult to extrapolate from the in vitro data described here to an in vivo setting, the existence of a NOEL implies at least a pragmatic threshold for carcinogenicity of H2O2 and KBrO3. Genotoxic tolerance to low levels of pro-oxidant chemicals appears to be due, in part, to basal BER DNA repair plus the protective capacity of antioxidants against DNA damage. The abundance of repair pathways and significant redundancy achieved by overlapping substrates in maintaining REDOX homeostasis suggest that the persistence of oxidative DNA damage is extremely detrimental to cells. Theoretically, as genetic alterations do not arise at very low doses, the risk of carcinogenesis (and also several degenerative chronic diseases) is unlikely to occur after exposure to concentrations below the LOEL, as no biologically significant effects were observed experimentally. This outcome has implications on the numerous uses of pro-oxidant chemicals, including the uses as cosmetic bleaches, as cancer treatment agents, and in food production. Furthermore, this study strengthens the evidence of the existence of thresholds for direct-acting genotoxins.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci. oxfordjournals.org/.
Funding
Unilever.
ACKNOWLEDGMENTS
Certain commercial equipment, instruments, and materials are identified in this article to specify an experimental procedure as completely as possible. The i dentification of particular equipment or materials does not imply a recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the materials, instruments, or equipment are necessarily the best available for the purpose.
REFERENCES
- Ballmaier D., Epe B. (1995). Oxidative DNA damage induced by potassium bromate under cell-free conditions and in mammalian cells Carcinogenesis 16 335–342 [DOI] [PubMed] [Google Scholar]
- Ballmaier D., Epe B. (2006). DNA damage by bromate: Mechanism and consequences Toxicology 221 166–171 [DOI] [PubMed] [Google Scholar]
- Basu A. K., Loechler E. L., Leadon S. A., Essigmann J. M. (1989). G enetic effects of thymine glycol: Site-specific mutagenesis and molecular modeling studies Proc. Natl. Acad. Sci. U.S.A. 86 7677–7681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiteux S., Radicella J. P. (2000). The human OGG1 gene: Structure, functions, and its implication in the process of carcinogenesis Arch. Biochem. Biophys. 377 1–8 [DOI] [PubMed] [Google Scholar]
- Cadet J., Douki T., Ravanat J. L. (2010). Oxidatively generated base damage to cellular DNA Free Radic. Biol. Med. 49 9–21 [DOI] [PubMed] [Google Scholar]
- Chung S. H., Chung S. M., Lee J. Y., Kim S. R., Park K. S., Chung J. H. (1999). The biological significance of non-enzymatic reaction of menadione with plasma thiols: Enhancement of menadione-induced cytotoxicity to platelets by the presence of blood plasma FEBS Lett. 449 235–240 [DOI] [PubMed] [Google Scholar]
- Dhenaut A., Boiteux S., Radicella J. P. (2000). Characterization of the hOGG1 promoter and its expression during the cell cycle Mutat. Res. 461 109–118 [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M. (1985). Application of capillary gas chromatography-mass spectrometry to chemical characterization of radiation-induced base damage of DNA: Implications for assessing DNA repair processes Anal. Biochem. 144 593–603 [DOI] [PubMed] [Google Scholar]
- Doak S. H., Brusehafer K., Dudley E., Quick E., Johnson G., Newton R. P., Jenkins G. J. (2008). No-observed effect levels are associated with up-regulation of MGMT following MMS exposure Mutat. Res. 648 9–14 [DOI] [PubMed] [Google Scholar]
- Doak S. H., Jenkins G. J., Johnson G. E., Quick E., Parry E. M., Parry J. M. (2007). Mechanistic influences for mutation induction curves after exposure to DNA-reactive carcinogens Cancer Res. 67 3904–3911 [DOI] [PubMed] [Google Scholar]
- Elhajouji A., Lukamowicz M., Cammerer Z., Kirsch-Volders M. (2011). Potential thresholds for genotoxic effects by micronucleus scoring Mutagenesis 26 199–204 [DOI] [PubMed] [Google Scholar]
- Evans M. D., Dizdaroglu M., Cooke M. S. (2004). Oxidative DNA damage and disease: Induction, repair and significance Mutat. Res. 567 1–61 [DOI] [PubMed] [Google Scholar]
- Fenech M. (2007). Cytokinesis-block micronucleus cytome assay Nat. Protoc. 2 1084–1104 [DOI] [PubMed] [Google Scholar]
- Forman H. J., Zhang H., Rinna A. (2009). Glutathione: Overview of its protective roles, measurement, and biosynthesis Mol. Aspects Med. 30 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Great Britain. Committee on Mutagenicity of Chemicals in Food. 2000). Guidance on a strategy for testing of chemicals for mutagenicity, Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment.
- Guest R. D., Parry J. M. (1999). P53 integrity in the genetically engineered mammalian cell lines AHH-1 and MCL-5 Mutat. Res. 423 39–46 [DOI] [PubMed] [Google Scholar]
- Hartman A. R., Ford J. M. (2003). BRCA1 and p53: Compensatory roles in DNA repair J. Mol. Med. 81 700–707 [DOI] [PubMed] [Google Scholar]
- Hatt L., Loft S., Risom L., Moller P., Sorensen M., Raaschou-Nielsen O., Overvad K., Tjonneland A., Vogel U. (2008). OGG1 expression and OGG1 Ser326Cys polymorphism and risk of lung cancer in a prospective study Mutat. Res. 639 45–54 [DOI] [PubMed] [Google Scholar]
- Henderson L., Albertini S., Aardema M. (2000). Thresholds in genotoxicity responses Mutat. Res. 464 123–128 [DOI] [PubMed] [Google Scholar]
- Jaruga P., Kirkali G., Dizdaroglu M. (2008). Measurement of formamidopyrimidines in DNA Free Radic. Biol. Med. 45 1601–1609 [DOI] [PubMed] [Google Scholar]
- Jenkins G. J., Doak S. H., Johnson G. E., Quick E., Waters E. M., Parry J. M. (2005). Do dose response thresholds exist for genotoxic alkylating agents? Mutagenesis 20 389–398 [DOI] [PubMed] [Google Scholar]
- Jeong M. S., Lee C. M., Jeong W. J., Kim S. J., Lee K. Y. (2010). Significant damage of the skin and hair following hair bleaching J. Dermatol. 37 882–887 [DOI] [PubMed] [Google Scholar]
- Kawanishi S., Murata M. (2006). Mechanism of DNA damage induced by bromate differs from general types of oxidative stress Toxicology 221 172–178 [DOI] [PubMed] [Google Scholar]
- Loft S., Hogh Danielsen P., Mikkelsen L., Risom L., Forchhammer L., Moller P. (2008). Biomarkers of oxidative damage to DNA and repair Biochem. Soc. Trans. 36 1071–1076 [DOI] [PubMed] [Google Scholar]
- Lutz W. K., Lutz R. W. (2009). Statistical model to estimate a threshold dose and its confidence limits for the analysis of sublinear dose- response relationships, exemplified for mutagenicity data Mutat. Res. 678 118–122 [DOI] [PubMed] [Google Scholar]
- Mistry P., Herbert K. E. (2003). Modulation of hOGG1 DNA repair enzyme in human cultured cells in response to pro-oxidant and antioxidant challenge Free Radic. Biol. Med. 35 397–405 [DOI] [PubMed] [Google Scholar]
- Naik S., Tredwin C. J., Scully C. (2006). Hydrogen peroxide tooth-whitening (bleaching): Review of safety in relation to possible carcinogenesis Oral Oncol. 42 668–674 [DOI] [PubMed] [Google Scholar]
- Nishimura S. (2002). Involvement of mammalian OGG1(MMH) in excision of the 8-hydroxyguanine residue in DNA Free Radic. Biol. Med. 32 813–821 [DOI] [PubMed] [Google Scholar]
- Nutter L. M., Ngo E. O., Fisher G. R., Gutierrez P. L. (1992). DNA strand scission and free radical production in menadione-treated cells. Correlation with cytotoxicity and role of NADPH quinone acceptor oxidoreductase J. Biol. Chem. 267 2474–2479 [PubMed] [Google Scholar]
- Platel A., Nesslany F., Gervais V., Marzin D. (2009). Study of oxidative DNA damage in TK6 human lymphoblastoid cells by use of the in vitro micronucleus test: Determination of no-observed-effect levels Mutat. Res. 678 30–37 [DOI] [PubMed] [Google Scholar]
- Powell C. L., Swenberg J. A., Rusyn I. (2005). Expression of base excision DNA repair genes as a biomarker of oxidative DNA damage Cancer Lett. 229 1–11 [DOI] [PubMed] [Google Scholar]
- Pryor W. A. (1986). Oxy-radicals and related species: Their formation, lifetimes, and reactions Annu. Rev. Physiol. 48 657–667 [DOI] [PubMed] [Google Scholar]
- Saitoh T., Shinmura K., Yamaguchi S., Tani M., Seki S., Murakami H., Nojima Y., Yokota J. (2001). Enhancement of OGG1 protein AP lyase activity by increase of APEX protein Mutat. Res. 486 31–40 [DOI] [PubMed] [Google Scholar]
- Sedelnikova O. A., Redon C. E., Dickey J. S., Nakamura A. J., Georgakilas A. G., Bonner W. M. (2010). Role of oxidatively induced DNA lesions in human pathogenesis Mutat. Res. 704 152–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov D. A., Cheung V. G. (2008). ATM gene mutations result in both recessive and dominant expression phenotypes of genes and microRNAs Am. J. Hum. Genet. 83 243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace S. S. (2002). Biological consequences of free radical-damaged DNA bases Free Radic. Biol. Med. 33 1–14 [DOI] [PubMed] [Google Scholar]
- Zair Z. M., Jenkins G. J., Doak S. H., Singh R., Brown K., Johnson G. E. (2011). N-methylpurine DNA glycosylase plays a pivotal role in the threshold response of ethyl methanesulfonate-induced chromosome damage Toxicol. Sci. 119 346–358 [DOI] [PubMed] [Google Scholar]