

Abstract
Chronic ethanol consumption was previously shown to induce CYP2A5 in mice, and this induction of CYP2A5 by ethanol was CYP2E1 dependent. In this study, the mechanisms of CYP2E1-dependent ethanol induction of CYP2A5 were investigated. CYP2E1 was induced by chronic ethanol consumption to the same degree in wild-type (WT) mice and CYP2A5 knockout (Cyp2a5 –/–) mice, suggesting that unlike the CYP2E1-dependent ethanol induction of CYP2A5, ethanol induction of CYP2E1 is not CYP2A5 dependent. Microsomal ethanol oxidation was about 25% lower in Cyp2a5 –/– mice compared with that in WT mice, suggesting that CYP2A5 can oxidize ethanol although to a lesser extent than CYP2E1 does. CYP2A5 was induced by short-term ethanol consumption in human CYP2E1 transgenic knockin (Cyp2e1 –/– KI) mice but not in CYP2E1 knockout (Cyp2e1 –/–) mice. The redox-sensitive transcription factor nuclear factor-erythroid 2-related factor 2 (Nrf2) was also induced by acute ethanol in Cyp2e1 –/– KI mice but not in Cyp2e1 –/– mice. Ethanol induction of CYP2A5 in Nrf2 knockout (Nrf2 –/–) mice was lower compared with that in WT mice, whereas CYP2E1 induction by ethanol was comparable in WT and Nrf2 –/– mice. Antioxidants (N-acetyl-cysteine and vitamin C), which blocked oxidative stress induced by chronic ethanol in WT mice and acute ethanol in Cyp2e1 –/– KI mice, also blunted the induction of CYP2A5 and Nrf2 by ethanol but not the induction of CYP2E1 by ethanol. These results suggest that oxidative stress induced by ethanol via induction of CYP2E1 upregulates Nrf2 activity, which in turn regulates ethanol induction of CYP2A5. Results obtained from primary hepatocytes, mice gavaged with binge ethanol or fed chronic ethanol, show that Nrf2-regulated ethanol induction of CYP2A5 protects against ethanol-induced steatosis.
Key Words: CYP2A5, CYP2E1, Nrf2, ROS, ethanol, antioxidant.
The CYP2A subfamily includes rat CYP2A3, mouse CYP2A5, and human CYP2A6 (Su and Ding, 2004). Although rat CYP2A3 is not detectable in the liver (Su and Ding, 2004), human CYP2A6 and mouse CYP2A5 are expressed predominantly in the liver (Su et al., 2002). Mouse CYP2A5 and human CYP2A6 metabolize several important xenobiotics including nicotine, cotinine, testosterone, aflatoxin B1, and nitrosamines (Kobliakov et al., 1993; Su and Ding, 2004). Coumarin, a plant alkaloid, is hydroxylated specifically by coumarin 7-hydroxylase (COH) encoded by the mouse Cyp2a5 gene and human Cyp2a6 gene (Mäenpää et al., 1993). COH activity is considered as a specific marker for catalytic activities of CYP2A5 and CYP2A6. Increased expression of CYP2A5 occurs during viral, fulminant, or bacterial hepatitis and in certain tumors (De-Oliveira et al., 2006; Jounaidi et al., 1994). CYP2A5 is highly inducible in mouse liver by diverse chemicals and heavy metals, which are neither structurally similar nor generally considered to be CYP inducers (Abu-Bakar et al., 2004; Lamsa et al., 2010; Su and Ding, 2004). Similarly, CYP2E1 is also induced by diverse chemicals that are not structurally similar, e.g., ethanol, acetone, benzene, and pyrazole (Koop and Tierney, 1990).
Recently, we found that CYP2A5 was induced by chronic ethanol feeding and, interestingly, ethanol induction of CYP2A5 was CYP2E1 dependent (Lu et al., 2011). Ethanol has long been known to elevate CYP2E1 levels (Lieber and DeCarli, 1972), and CYP2E1 plays an important role in ethanol-induced oxidative stress (Cederbaum et al., 2009; Lu and Cederbaum, 2008). CYP2E1 and CYP2A5 can be induced by ethanol or pyrazole. Much interest in CYP2E1 has reflected its ability to activate hepatotoxins and to generate reactive oxygen species (ROS) during its catalytic cycle (Cheung et al., 2005; Koop and Tierney, 1990; Lieber, 1997; Lu and Cederbaum, 2008). Induction of CYP2A5 has been suggested to occur in response to hepatocellular damage and to endoplasmic reticulum stress (Gilmore and Kirby, 2004). On the other hand, CYP2A5 has been shown to be involved in regulating endogenous antioxidant bilirubin levels during stress conditions and that it may play a role in cytoprotection as its basal and inducible expressions are regulated by the nuclear factor-erythroid 2-related factor 2 (Nrf2) pathway (Abu-Bakar et al., 2004, 2005, 2007, 2011; Lamsa et al., 2010).
We previously found that chronic ethanol feeding to mice resulted in an increase in Nrf2, which was associated with an increase in CYP2E1 (Gong and Cederbaum, 2006). We hypothesize that the elevation of liver CYP2E1 by chronic ethanol feeding causes an elevation of ROS, which increases Nrf2 levels, and the elevated Nrf2 upregulates CYP2A5. In this study, we show that CYP2A5 induction by ethanol was accompanied by an initial elevation of CYP2E1 and Nrf2; antioxidants such as N-acetyl-cysteine (NAC) and vitamin C (Vc) blunted elevation of CYP2A5 and Nrf2, but ethanol induction of CYP2E1 was not affected; ethanol induction of CYP2A5 but not CYP2E1 was lowered by Nrf2 deficiency. These results suggest that CYP2E1-dependent ethanol induction of CYP2A5 was regulated through the redox-sensitive transcription factor Nrf2.
MATERIALS AND METHODS
Animals and ethanol treatment. SV/129-background CYP2E1 knockout (Cyp2e1 –/–) (Lee et al., 1996) and humanized CYP2E1 transgenic knockin mice (Cyp2e1 –/– KI) (Cheung et al., 2005) were kindly provided by Dr Frank J. Gonzalez (Laboratory of Metabolism, National Cancer Institute, Bethesda, MD). Colonies of these mice were established at Mount Sinai, and the offspring of these mating pairs were used in this study. The SV129 wild-type (WT) mice were purchased from Charles River Laboratory. The C57BL/J6 background CYP2A5 knockout (Cyp2a5 –/–) mouse colony was established at Mount Sinai by rederivation from male Cyp2a5 –/– mice (Zhou et al., 2010) (kindly provided by Dr Xinxin Ding, Wadsworth Center, New York State Department of Health, Albany, NY) and female C57BL/J6 WT mice (purchased from Charles River Laboratory). All mice were housed in temperature-controlled animal facilities with 12-h light/12-h dark cycles and were permitted consumption of tap water and Purina standard chow ad libitum until being fed the liquid diets. The mice received humane care, and experiments were carried out according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals and with approval of the Mount Sinai Animal Care and Use Committee. Liver tissues from WT and Nrf2 knockout (Nrf2 –/–) mice fed dextrose or ethanol were kindly provided by Drs J. Lamlé and A. Vogel (Lamlé et al., 2008).
All mice were initially fed the control liquid dextrose diet (Bio-Serv, Frenchtown, NJ) for 3 days to acclimate them to the liquid diet. Then, the mice were fed either the liquid ethanol diet (Bio-Serv) or the control liquid dextrose diet as described by Lieber and DeCarli (1972) for 3 weeks. The content of ethanol was gradually increased every 3–4 days from 10% (1.77% [vol/vol]) of total calories to 20% (3.54% [vol/vol]), 25% (4.42% [vol/vol]), 30% (5.31% [vol/vol]), and finally 35% of total calories (6.2% [vol/vol]). For experiments involving less than 2 weeks of feeding, the mice were directly subjected to the diet containing ethanol as 35% of total calories. The control mice were pair-fed the control dextrose diet on an isoenergetic basis. For 3-week antioxidant intervention experiments, during the 3 weeks of feeding with ethanol or dextrose liquid diets, NAC (75 mg/kg body weight, ip) and Vc (125 mg/kg body weight, ip) were injected daily into the ethanol-fed SV129 WT mice. For CYP2E1 inhibitor chlormethiazole (CMZ) intervention experiments, during 2 weeks of ethanol feeding, CMZ (50 mg/kg body weight, ip) was injected every other day into the ethanol-fed SV129WT mice (Lu et al., 2008). For 2-day antioxidant intervention experiments, NAC and Vc were first injected the day before ethanol diet feeding, and then NAC was injected at 150 mg/kg body weight, ip, once a day or Vc was injected at 125 mg/kg body weight, ip, every 12 h. The ethanol-fed mice had access to their rations ad libitum, and the conditions of WT, knockout, and humanized transgenic mice were comparable. The amount of food consumed by the Cyp2e1 –/–, Cyp2a5 –/–, and Cyp2e1 –/– KI mice and the SV129 and C57BL/J6 background WT mice was approximately the same.
The mice were sacrificed by cervical dislocation under anesthesia, and the livers were rapidly excised into fragments and washed with cold saline. The liver tissue aliquots were stored at –80°C for further assays. Liver homogenates were prepared in ice-cold 0.15M KCl and stored at –80°C in aliquots. Liver triglycerides (TG) were assayed using commercial available kits (POINTE Scientific Inc., Canton, MI). Liver samples were fixed in 10% formalin solution and embedded in paraffin. Liver sections (5-µm thick) were stained with hematoxylin and eosin for pathological evaluation.
For the binge ethanol model, the mice were gavaged with ethanol once at 6 g/kg body weight (0.2 ml of 38% ethanol (v/v) per 10 g body weight), and the mice were sacrificed 3, 6, 9, and 14 h after the gavage.
Preparation of hepatic microsomes. Hepatic microsomes were prepared by placing liver aliquots in 0.15M KCl and homogenization in a Polytron homogenizer for 10 strokes. The homogenate was centrifuged at 9000 g for 20 min, and then the resulting supernatant fraction was centrifuged further at 105,000 g for 60 min. The resulting pellets (microsomes) were resuspended in 50mM sodium phosphate buffer (pH 7.4). All procedures were carried out on ice.
Isolation of primary hepatocytes. The hepatocytes were isolated as previously described (Lu et al., 2004). The cells were seeded in collagen-coated plates. After overnight preculture, the cells were treated with 100mM ethanol for 36 h. The cells were observed under an inverted microscope, photos of the cells were taken, and then the cells were subjected to Oil Red O staining. Some of the cells were collected for Western blotting analysis or TG assay.
Ethanol oxidation. The production of acetaldehyde from ethanol by microsomes was assayed as previously described (Cederbaum and Cohen, 1984). The ethanol oxidation buffer containing 100mM potassium phosphate buffer, pH 7.4, 25mM ethanol, and 1mM NADPH was supplemented with 1mM sodium azide to inhibit catalase activity and with 0.1mM deferoxamine to inhibit microsomal hydroxyl radical formation because both catalase and hydroxyl radical contribute to ethanol oxidation (Cederbaum, 1991). Microsomal protein (50 µg) was added to 0.1 ml of the reaction buffer and was incubated for 15 min at 37°C. Reactions were carried out in center-well flasks containing 0.15 ml of 15mM semicarbazide HCl in 180mM potassium phosphate, pH 7.4, in the center well. Reactions were terminated by the addition of trichloroacetic acid (TCA) to a final concentration of 4.5% (w/v). The sealed flasks were incubated overnight at room temperature to allow diffusion of acetaldehyde into the center well. The absorbance of the aldehyde-semicarbazone complex was determined at 224 nm.
Cytochrome P450 2E1 and 2A5 activity. CYP2E1 activity was measured by the rate of oxidation of 1mM p-nitrophenol to p-nitrocatechol by 100 µg of microsomal protein for 15 min at 37°C (Lu and Cederbaum, 2006a). CYP2A5 activity was measured by assessing COH activity with 100µM coumarin as substrate plus 100 µg of microsomal protein and incubation for 15 min at 37°C (Lu and Cederbaum, 2006a).
Measurement of reduced glutathione levels. Liver homogenate was mixed with TCA to a final concentration of 5%, and the mixture was incubated at 4°C for 30 min to extract glutathione (GSH). The TCA extracts (10 µl) were added to 200 µl of methanol containing 1 mg/ml o-phthaldehyde and then were incubated for 15 min at 37°C in the dark. Fluorescence was measured at 350/420 nm (excitation/emission). The concentration of GSH was determined from a GSH standard curve (Lu and Cederbaum, 2006b).
Determination of thiobarbituric acid reactive substances. In brief, hepatic homogenates supplemented with butylated hydroxytoluene to prevent further oxidation during assay were incubated with 0.2 ml of TCA [15% (wt/vol)]-TBA [0.375% (wt/vol)]-HCl (0.25 N) solution in a boiling water bath for 10 min. After centrifugation at 524 × g for 5 min, the resulting supernatant was used to determine the formation of thiobarbituric acid reactive substances (TBARS) by evaluating absorbance at 535 nm. TEP (1,1,3,3-tetraethoxypropane) treated as above served as a standard (Lu et al., 2005).
Western blotting. Hepatic proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. After 1 h of blocking with 2% fat-free milk, membranes were incubated overnight with anti-CYP2E1 IgG (a gift from Dr Jerome Lasker, Hackensack Biomedical Research Institute, Hackensack, NJ), anti-CYP2A5 IgG (a gift from Dr Risto Juvonen, Department of Pharmacology and Toxicology, University of Kuopio, Kuopio, Finland), and antibodies against Nrf2, lamin, Calnexin, heme oxygenase 1 (HO-1), or β-actin (Santa Cruz) followed by 1-h incubation with peroxidase secondary anti-rabbit, anti-chick, and anti-goat antibodies (Millipore), respectively. Chemiluminescence was detected by Image Reader LAS-4000 (Fujifilm) after adding PierceECL Western Blotting Substrate (Thermo Scientific, Rockford, IL). All specific bands were quantified with the Automated Digitizing System (ImageJ gel programs, version 1.34S; National Institutes of Health, Bethesda, MD).
Transcriptional activity of Nrf2. Nuclear proteins were isolated from frozen liver tissues using the NE-PER nuclear protein extraction kit (Thermo Scientific) according to the manufacturer’s protocol. Nrf2 DNA binding activity was assessed using a TransAM Transcription Factor ELISA kit (Active Motif, catalog no. 50296). This kit contains 96-well plates coated with oligonucleotides containing the antioxidant sensitive response element (ARE) consensus binding site (5′-GTCACAGTGACTCAGCAGAATCTG-3). Nuclear protein (2.5 µg) was added to each well. Nrf2 bound to the imbedded ARE oligonucleotides on the 96-well plates was detected colorimetrically by incubating the plates with a primary antibody against Nrf2 followed by incubating with a secondary antibody conjugated to horseradish peroxidase. Nuclear extracts from COS-7 cells transfected with Nrf2 were used as a positive control for Nrf2 binding activity.
Statistics. Results are expressed as means ± SEMs. Statistical evaluation was carried out by one-way ANOVA followed by the Student-Newman-Keuls post hoc test. p < 0.05 was considered as statistically significant.
RESULTS
Induction of CYP2A5 by Chronic Ethanol Feeding in WT and Cyp2e1–/– Mice
After 3 weeks of ethanol administration, activities of CYP2E1 and CYP2A5 were increased by ethanol in SV129 WT mice compared with those in the dextrose-fed controls by threefold, but no increases in the activities of CYP2E1 or CYP2A5 were observed in the Cyp2e1 –/– mice (Fig. 1A). Protein levels of CYP2E1 and CYP2A5 were increased by ethanol in the WT mice by two- and threefold, respectively, but protein levels of CYP2E1 and CYP2A5 were not elevated in the Cyp2e1 –/– mice (Fig. 1B). To further evaluate the role of CYP2E1 in chronic ethanol induction of CYP2A5, the CYP2E1 inhibitor CMZ was applied to inhibit CYP2E1 activity. As shown in Fig. 1C, after 2 weeks of ethanol feeding, CYP2E1 and CYP2A5 activities were increased about 3- and 2.5-fold, respectively; the ethanol-induced CYP2E1 and CYP2A5 activities were decreased by CMZ injection to increases of about 2- and 1.5-fold over the dextrose-fed mice. Thus, CMZ blunted the ethanol elevation of CYP2E1 and CYP2A5.
Fig. 1.

Induction of CYP2E1 and CYP2A5 by chronic ethanol feeding in WT, Cyp2e1 –/–, and Cyp2a5 –/– mice. After 3 weeks of ethanol feeding, induction of CYP2E1 and CYP2A5 was measured as described in Materials and Methods section. (A) Activities of CYP2E1 and CYP2A5 in WT and Cyp2e1 –/– mice. (B) Western blotting analyses for CYP2E1 and CYP2A5 in WT and Cyp2e1 –/– mice. *p < 0.05, compared with Cont group; #p < 0.05, compared with WT EtOH group. Cont, Control; EtOH, Ethanol. (C) Activities of CYP2E1 and CYP2A5 were inhibited by CMZ. *p < 0.05, compared with Cont group; #p < 0.05, compared to EtOH group. (D) Activities of CYP2E1 and CYP2A5 in WT and Cyp2a5 –/– mice. *p < 0.05, compared with Cont group; #p < 0.05, compared with WT EtOH group. (E) Western blotting analyses for CYP2E1 and CYP2A5 in WT and Cyp2a5 –/– mice. *p < 0.05, compared with Cont group. (F) Ethanol oxidation in microsomes isolated from WT and Cyp2a5 –/– mice fed dextrose or ethanol liquid diet. *p < 0.05, compared with Cont group; #p < 0.05, compared with WT EtOH group.
Induction of CYP2E1 by Chronic Ethanol Feeding in WT and Cyp2a5–/– Mice
Because ethanol induction of CYP2A5 is CYP2E1 dependent, as a comparison, we evaluated the reverse condition, i.e., whether ethanol induction of CYP2E1 is CYP2A5 dependent. We observed that CYP2E1 induction by chronic ethanol feeding occurs in Cyp2a5 –/– mice. Activity of CYP2E1 was increased by ethanol in C57BL/6J WT mice compared with the activity of CYP2E1 in the dextrose-fed controls by threefold (Fig. 1D). The activity of CYP2E1 was comparably increased by chronic ethanol feeding in the Cyp2a5 –/– mice and WT mice (Fig. 1D). Protein levels of CYP2E1 were increased by ethanol comparably by 2.4- and 2.2-fold in WT and Cyp2a5 –/– mice, respectively (Fig. 1E). CYP2A5, as expected, was induced in the WT mice but not in the Cyp2a5 –/– mice (Fig. 1E). These results indicated that CYP2E1 induction by ethanol is independent of CYP2A5 although ethanol induction of CYP2A5 is CYP2E1 dependent.
CYP2E1 induction by ethanol contributes to microsomal ethanol oxidation (Lieber and DeCarli, 1972). To see whether CYP2A5 induction by ethanol is able to metabolize ethanol, microsomes isolated from WT and Cyp2a5 –/– mice fed dextrose or ethanol liquid diets were incubated with ethanol. Microsomal ethanol oxidation was increased about eightfold by ethanol feeding in the WT mice. Microsomal ethanol oxidation was 25% lower in the Cyp2a5 –/– mice compared with that in the WT mice (Fig. 1F). The lower ethanol induction in the Cyp2a5 –/– mice might be attributed to the absence of CYP2A5 because ethanol induction of CYP2E1 was comparable in WT and Cyp2a5 –/– mice (Figs. 1D and E). These results suggest that CYP2A5 can oxidize ethanol to acetaldehyde; however, its catalytic activity with ethanol is lower than that by CYP2E1.
Induction of CYP2A5 by Short-Term Ethanol Feeding in Cyp2e1–/– KI Mice
We previously found that after 1 week of ethanol feeding, levels of CYP2A5 did not change in WT mice although levels of CYP2E1 were increased by twofold (Lu et al., 2011). However, in Cyp2e1 –/– KI mice, which have higher levels of CYP2E1 than WT mice (Lu et al., 2010), activities of CYP2A5 and CYP2E1 were increased by 1 week of ethanol feeding (data not shown). To set up an acute model for ethanol induction of CYP2A5 and CYP2E1, Cyp2e1 –/– KI mice were fed ethanol for 1–3 days, and results were compared with the results of Cyp2e1 –/– mice. CYP2E1 was induced in the Cyp2e1 –/– KI mice after 1 day of ethanol feeding, and levels remained increased after 2 and 3 days of feeding (Fig. 2A). CYP2A5 was slightly induced after 1 day ofethanol feeding but was clearly increased after 2 and 3 daysof ethanol feeding in the Cyp2e1 –/– KI mice (Fig. 2A). Activity of CYP2E1 was increased 1.5-fold in the Cyp2e1 –/– KI mice after 1 and 2 days of feeding and further increased by 3-fold after 3 days of feeding (Fig. 2B). Activity of CYP2A5 was not increased after 1 day of ethanol feeding but did increase threefold after 2 days of feeding and further increased by fivefold after 3 days of feeding (Fig. 2C). Thus, the induction of CYP2E1 by short-term ethanol feeding in the Cyp2e1 –/– KI mice precedes the induction of CYP2A5. However, in Cyp2e1 –/– mice, CYP2E1 and CYP2A5 were not induced by ethanol feeding (Fig. 2A–C), suggesting that like chronic ethanol induction of CYP2A5, CYP2A5 induction by short-term ethanol feeding in the Cyp2e1 –/– KI mice is also CYP2E1 dependent.
Fig. 2.

Time course of induction of CYP2E1, CYP2A5, and Nrf2 by short-term ethanol feeding in Cyp2e1 –/– KI mice. Cyp2e1 –/– and Cyp2e1 –/– KI mice were fed an ethanol diet for 1, 2, and 3 days. Western blotting analyses for hepatic CYP2E1, CYP2A5, and Nrf2 (A), activities of CYP2E1 (B) and CYP2A5 (C), Western blotting analyses for hepatic nuclear Nrf2 (D), and nuclear Nrf2 binding activities (E) were measured as described in Materials and Methods section. *p < 0.05, compared with 0 day (control group); #p < 0.05, compared with Cyp2e1 –/– KI mice.
Induction of Nrf2 by Short-Term Ethanol Feeding in Cyp2e1–/– KI Mice
CYP2A6, the human orthologue of CYP2A5, is regulated by the redox-sensitive transcription factor Nrf2 (Yokota et al., 2011). CYP2A5 induction by heavy metals such as lead chloride, methylmercury chloride, and cadmium chloride is regulated by Nrf2 (Lamsa et al., 2010). Does ethanol induction of CYP2A5 involve Nrf2? In Cyp2e1 –/– KI mice, Nrf2 was increased in the liver after 1 day of ethanol feeding and remained elevated after 2 and 3 days of feeding (Fig. 2A). Interestingly, the ethanol-induced increase in Nrf2 precedes the ethanol induction of CYP2A5. Because the shortest time point assayed after administration of ethanol was 1 day, more definitive time course experiments are needed to evaluate whether CYP2E1 induction occurs prior to Nrf2 pathway activation, e.g., assay hours after ethanol administration rather than days. Nrf2 induction was not observed in Cyp2e1 –/– mice through 3 days of ethanol feeding (Fig. 2A). Translocation of Nrf2 to the nucleus was determined by assaying Nrf2 levels in liver nuclear extracts. As shown in Fig. 2D, Nrf2 in the nuclear extracts from the Cyp2e1 –/– KI mice was increased after 1 day of ethanol feeding and further increased after2 and 3 days of feeding. Nrf2 was not increased in the nuclear extracts from the Cyp2e1 –/– mice after ethanol feeding (Fig. 2D). Nrf2 DNA binding activity assay showed that Nrf2 binding activity in the Cyp2e1 –/– KI mice was increased after1 day of ethanol feeding and remained increased after 2 days of feeding, followed by a decline after 3 days of feeding (Fig. 2E).Ethanol feeding did not increase Nrf2 DNA binding activity in the Cyp2e1 –/– mice (Fig. 2E). These results suggest that CYP2E1 promotes Nrf2 signaling pathway activationby ethanol.
Induction of Nrf2 Activity by Short-Term Ethanol Feeding in Cyp2e1–/– KI Mice Is Blocked by Antioxidants NAC and Vc
Ethanol can elevate CYP2E1 levels (Lieber, 1997; Lieber and DeCarli, 1972), and CYP2E1 plays an important role in ethanol-induced oxidative stress (Cederbaum et al., 2009; Lu and Cederbaum, 2008). CYP2E1-mediated oxidative stress upregulates Nrf2 (Gong and Cederbaum, 2006). If CYP2E1-dependent induction of Nrf2 is via CYP2E1-mediated ROS, then antioxidants should block the induction of Nrf2. After 2 days of ethanol feeding to the Cyp2e1 –/– KI mice, hepatic TBARS was increased by twofold; treatment with NAC or Vc completely blocked the increase in TBARS (Fig. 3A). After 2 days of ethanol feeding, hepatic GSH levels were decreased by 80%; treatment with NAC or Vc completely restored hepatic GSH levels (Fig. 3A). These results suggest that 2 days of ethanol feeding induced oxidative stress in Cyp2e1 –/– KI mice, and the selected doses of NAC and Vc effectively blocked this ethanol-induced oxidative stress. Two days of ethanol feeding caused an increase in the level of Nrf2 in the liver nuclear fraction, and this increase was lowered by the treatments with NAC or Vc (Fig. 3B). Nrf2 DNA binding activity was increased by the ethanol feeding, and this increase was inhibited by NAC and Vc (Fig. 3C). These results suggest that Nrf induction by short-term ethanol feeding in Cyp2e1 –/– KI mice is mediated by CYP2E1-mediated oxidative stress.
Fig. 3.
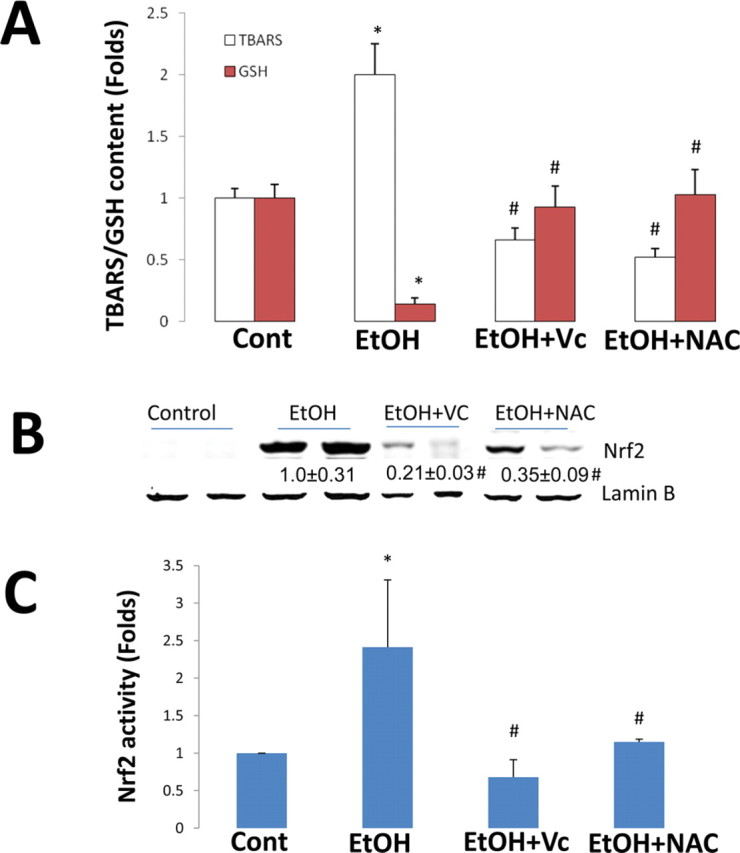
. Induction of oxidative stress and Nrf2 by short-term ethanol feeding in Cyp2e1 –/– KI mice was blocked by antioxidants. Cyp2e1 –/– KI mice were fed ethanol for 2 days, and NAC and Vc were injected the day before initiating ethanol feeding; NAC was injected at 150 mg/kg body weight, ip, once a day; Vc was injected at 125 mg/kg body weight, ip, every 12 h. Hepatic TBARS and GSH (A), Western blotting analyses for nuclear Nrf2 (B), and ELISA for Nrf2 DNA binding activity (C) were measured as described in Materials and Methods section. *p < 0.05, compared with Cont group; #p < 0.05, compared with EtOH group. Cont, Control; EtOH, Ethanol.
Induction of CYP2A5 by Short-Term Ethanol Feeding in Cyp2e1–/– KI Mice Is Also Blocked by NAC and Vc
If CYP2E1-dependent induction of Nrf2 via increased ROS production plays a role in ethanol induction of CYP2A5, then antioxidants should block the induction of CYP2A5 but should not block the induction of CYP2E1. After 2 days of ethanol feeding in Cyp2e1 –/– KI mice, activity of CYP2A5 was increased by threefold; induction of CYP2A5 was inhibited by the treatments with NAC and Vc by 70 and 60%, respectively (Fig. 4A). Activity of CYP2E1 was increased by threefold, but treatment with NAC and Vc did not inhibit ethanol induction of CYP2E1 (Fig. 4A). The increased protein levels of CYP2E1 after 2 days of ethanol feeding were also not affected by treatment with NAC and Vc, but the ethanol-induced increase in CYP2A5 protein was blunted by treatment with NAC and Vc (Fig. 4B).
Fig. 4.
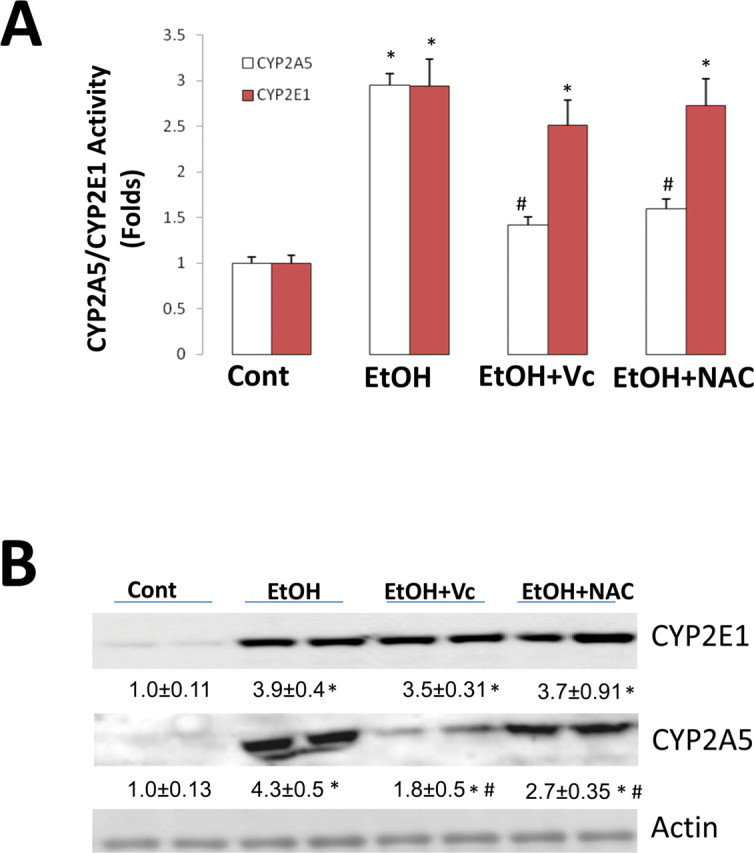
Induction of CYP2A5 but not CYP2E1 by short-term ethanol feeding in Cyp2e1 –/– KI mice was blocked by antioxidants. Cyp2e1 –/– KI mice were fed ethanol for 2 days, and NAC and Vc were injected the day before initiating ethanol feeding; NAC was injected at 150 mg/kg body weight, ip, once a day; Vc was injected at 125 mg/kg body weight, ip, every 12 h. Hepatic microsomal CYP2A5 and CYP2E1 activities (A) and Western blotting analyses for CYP2A5 and CYP2E1 (B) were measured as described in Materials and Methods section. *p < 0.05, compared with Cont group; #p < 0.05, compared with EtOH group. Cont, Control; EtOH, Ethanol.
Induction of Oxidative Stress, Nrf2, and CYP2A5 by Chronic Ethanol Feeding in WT Mice Is Also Blocked by Antioxidants NAC and Vc
To see whether chronic ethanol induction of CYP2A5, like the induction by short-term ethanol feeding in Cyp2e1 –/– KI mice, is also dependent on elevated oxidative stress, WT mice were fed the ethanol liquid diet for 3 weeks, and NAC and Vc were injected daily. After 3 weeks of ethanol feeding, CYP2A5 and CYP2E1 activities were increased by threefold (Fig. 5A). Injection of the antioxidants almost completely decreased ethanol-induced CYP2A5 activity but had no effect on ethanol-induced CYP2E1 activity (Fig. 5A). Protein levels of CYP2A5 and CYP2E1 were induced by chronic ethanol, and injection of antioxidants blocked the ethanol-induced increase in CYP2A5 but not CYP2E1 protein (Fig. 5B). Similar to the acute model, chronic ethanol feeding to WT mice caused increases in liver TBARS and decreases in hepatic GSH (Fig. 5C), indicating that chronic ethanol induced hepatic oxidative stress. The antioxidants decreased this ethanol-induced oxidative stress (Fig. 5C). The chronic ethanol feeding also elevated nuclear levels of Nrf2, and this ethanol induction of Nrf2 was also blunted by the antioxidants (Fig. 5B).
Fig. 5.
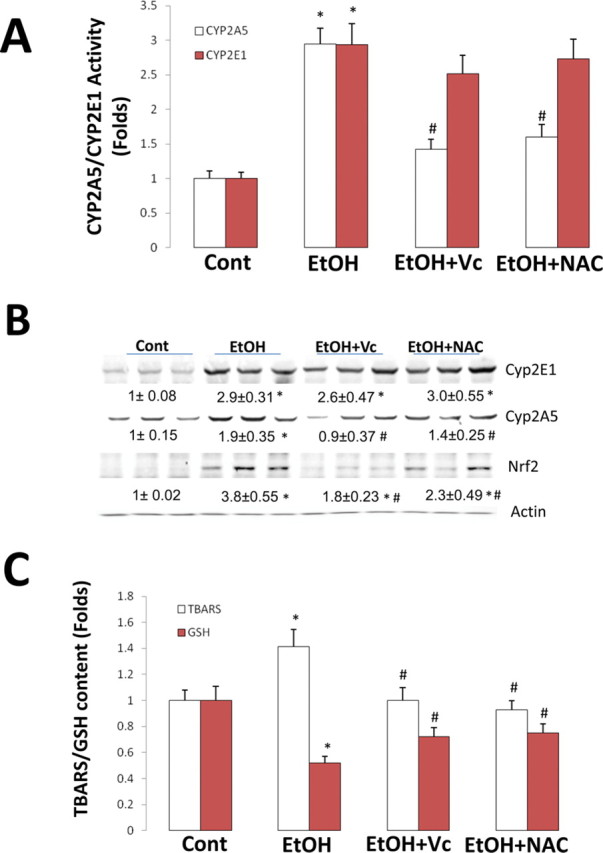
Induction of CYP2A5 but not CYP2E1 by chronic ethanol feeding in WT mice was blocked by antioxidants. WT mice were fed an ethanol diet for 3 weeks; NAC (75 mg/kg body weight) and Vc (125 mg/kg body weight) were injected, ip, once a day. Hepatic microsomal CYP2A5 and CYP2E1 activities (A), Western blotting analyses for CYP2A5, CYP2E1, and Nrf2 (B), and hepatic TBARS and GSH (C) were measured as described in Materials and Methods section. *p < 0.05, compared with Cont group; #p < 0.05, compared with EtOH group. Cont, Control; EtOH, Ethanol.
Induction of CYP2A5 by Chronic Ethanol Feeding in Nrf2–/– Mice Is Lower Compared With That in WT Mice
It was reported by the Vogel group that CYP2E1 levels were comparable between chronic ethanol-fed WT and Nrf2–/– mice; CYP2A5 levels were not measured in this study (Lamlé et al., 2008). To further evaluate the role of Nrf2 in ethanol induction of CYP2A5, levels and activities of CYP2A5 and CYP2E1 in livers from WT and Nrf2 –/– mice were assayed. Expression of CYP2A5 was increased by 2.5-fold in WT mice fed ethanol, but CYP2A5 expression was increased by only 60% in the ethanol-fed Nrf2 –/– mice (Fig. 6A). CYP2A5 activity was increased ninefold by ethanol in WT mice but only fourfold in Nrf2 –/– mice (Fig. 6B). CYP2E1 induction by ethanol was comparable (3.4- vs. 3.7-fold) in Nrf2 –/– mice and WT mice (Fig. 6A), and CYP2E1 activities were slightly but not significantly lower in ethanol-fed Nrf2 –/– mice compared with the activities in the WT mice ( Fig. 6C). These results suggest that Nrf2, in part, regulates ethanol induction of CYP2A5 but not CYP2E1.
Fig. 6.

Induction of CYP2A5 and CYP2E1 by chronic ethanol feeding in WT and Nrf2 –/– mice. WT and Nrf2 –/– mice were fed an ethanol diet for 3 weeks, Western blotting analyses for CYP2E1 and CYP2A5 (A) and activities of CYP2A5 (B) and CYP2E1 (C) were measured as described in Materials and Methods section. *p < 0.05, compared with Cont group; #p < 0.05, compared with WT EtOH group. Cont, Control; EtOH, Ethanol.
Nrf2-Mediated CYP2A5 Induction and Liver Injury
Is there any physiological or pathophysiological significance to the induction of CYP2A5 by ethanol? Nrf2 usually regulates expression of cytoprotective or antioxidant genes such as HO-1 in catalyzing the detoxification of oxidants. Does Nrf2-regulated ethanol induction of CYP2A5 potentiate or protect against alcohol-induced liver injury? The results obtained from WT and Cyp2a5 –/– mice fed ethanol for 3 weeks (Figs. 7A and B) or from primary hepatocytes isolated from WT and Cyp2a5 –/– mice treated with ethanol (Fig. 7C–E), or from WT and Cyp2a5 –/– mice gavaged with binge ethanol (Figs. 7F and G) all show that ethanol-induced steatosis was higher in Cyp2a5 –/– mice or hepatocytes compared with that in WT mice or hepatocytes. This suggests that CYP2A5 protects against alcohol-induced steatosis. One possible mechanism for this might involve Nrf2 upregulation of protective factors such as HO-1. Binge alcohol inhibits HO-1 expression (Gerjevic et al., 2011). Does CYP2A5 prevent or lower alcohol-induced steatosis via upregulation of HO-1? HO-1 protein levels were indeed low after ethanol treatment in hepatocytes from WT and Cyp2a5 –/– mice (Figs. 8A and B). HO-1 was inhibited by binge alcohol (Figs. 8C and D). These results suggest that HO-1 inhibition is associated with binge alcohol-induced steatosis, especially in Cyp2a5 –/– mice. The relationship between HO-1 (and possibly other protective factors) and CYP2A5 requires further study.
Fig. 7.
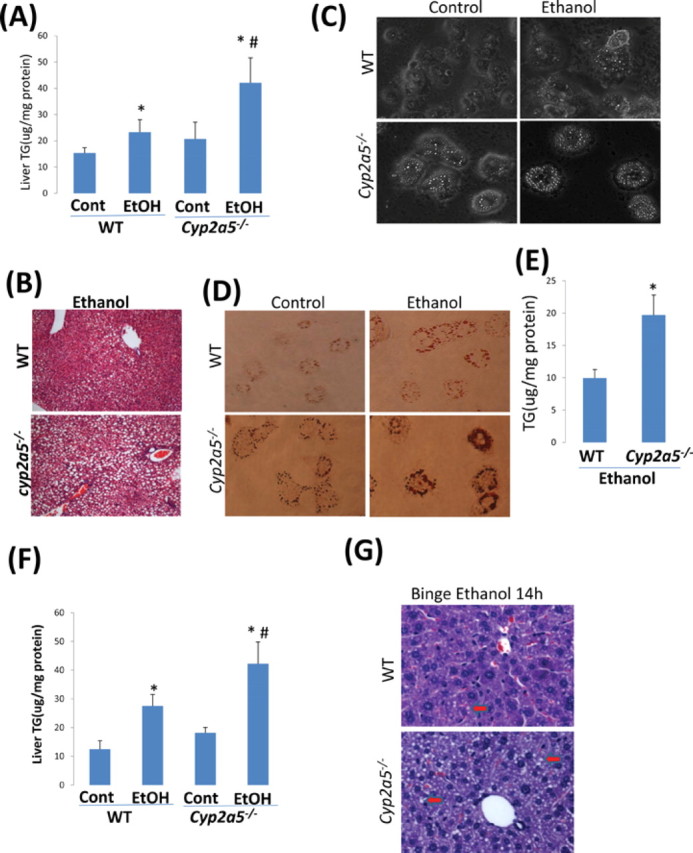
Ethanol-induced steatosis in Cyp2a5
–/– mice was more severe compared with that in WT mice. (A, B) WT and Cyp2a5
–/– mice were fed ethanol or dextrose liquid Lieber-DeCarli diets for 3 weeks. (A) Hepatic TG levels were increased more by ethanol in Cyp2a5
–/– mice than in WT mice. *p < 0.05, compared with Cont group; #p < 0.05, compared with WT EtOH group. Cont, Control; EtOH, Ethanol. (B) Hematoxylin and eosin (H&E) staining shows more lipid droplets in liver sections from ethanol-fed Cyp2a5
–/– mice than those from ethanol-fed WT mice. (C, D, E) Primary hepatocytes isolated from WT and Cyp2a5
–/– mice were treated with 0–100mM ethanol for 36 h and then were stained with Oil Red O or lysed for TG assay. (C) More lipid droplets were observed in hepatocytes from Cyp2a5
–/– mice than those from WT mice. (D) Oil Red O staining was elevated in hepatocytes from Cyp2a5
–/– mice than those from WT mice. (E) Higher TG levels were found in ethanol-treated hepatocytes from Cyp2a5
–/– mice than those from WT mice. *p < 0.05, compared with WT group. (F, G) WT and Cyp2a5
–/– mice were treated by gavage with one dose of ethanol (6 g/kg body weight for 14 h). (F) Hepatic TG levels were increased more by ethanol in Cyp2a5
–/– mice than in WT mice. *p < 0.05, compared with Cont group; #p < 0.05, compared with WT EtOH group. Cont, Control; EtOH, Ethanol. (G) H&E staining shows more lipid droplets ( ) in liver sections from Cyp2a5
–/– mice than those from WT mice.
) in liver sections from Cyp2a5
–/– mice than those from WT mice.
Fig. 8.

Ethanol treatment decreases HO-1 protein levels. (A) Western blotting analysis showing that HO-1 protein levels are decreased after ethanol treatment in WT and Cyp2a5 –/– hepatocytes. (B) Quantification of Western blotting in (A). *p < 0.05, compared with Cont group. (C) Time course for the decline in HO-1 protein level in livers after binge ethanol treatment in WT mice. (D) Quantification of Western blotting in C. *p < 0.05, compared with 0 h.
DISCUSSION
Recent studies have implicated a role for cellular redox status and possible activation of stress-related transcription factors in activation of CYP2A5 (Abu-Bakar et al., 2007; Gilmore and Kirby, 2004; Lamsa et al., 2010; Su and Ding, 2004). Treatment of hepatocytes with menadione, a redox cycling agent, elevated CYP2A5 expression (Gilmore and Kirby, 2004). Pretreatment of hepatocytes with antioxidants such as NAC or vitamin E blunted pyrazole-mediated increases in CYP2A5 mRNA levels (Gilmore and Kirby, 2004). CYP2A5 constitutive expression and induction by heavy metals such as cadmium or lead or mercury was dependent on the redox-sensitive transcription factor Nrf2 in liver (Abu-Bakar et al., 2004; Abu-Bakar et al., 2007; Lamsa et al., 2010).
We have previously shown that chronic ethanol feeding elevated CYP2A5 protein and catalytic activity by a mechanism in which CYP2E1 plays a central role (Lu et al., 2011). These studies were extended in the current report to a short-term model of ethanol feeding: 2–3 days of ethanol intake was sufficient to elevate CYP2A5 protein and activity in Cyp2e1 –/– KI mice but not in Cyp2e1 –/– mice. The induction of CYP2A5 (2–3 days) occurs after induction of CYP2E1 (1 day). Ethanol-mediated increases in ROS production are important for the induction of CYP2A5 as two antioxidants blunted the short-term and the chronic ethanol induction of CYP2A5 but not the induction of CYP2E1. Ethanol increases oxidative stress in the WT and Cyp2e1 –/– KI mice but not in the Cyp2e1 –/– mice (Lu et al., 2010), suggesting that ethanol-induced oxidative stress is CYP2E1 dependent. Nrf2 appears to play a central role in the ethanol induction of CYP2A5 but not in the induction of CYP2E1 as the increase in CYP2A5 was less in Nrf2 –/– mice compared with that in WT mice, whereas the increase in CYP2E1 was the same. Short-term and chronic ethanol feeding elevated ROS production and nuclear levels of Nrf2 and DNA binding activity of Nrf2. NAC and Vc lowered ROS, decreased the activation of Nrf2, and subsequently blunted the elevation of CYP2A5 by ethanol feeding. We hypothesize that ethanol induction of CYP2E1 followed by increases in production of ROS and subsequent activation and translocation of Nrf2 to the nucleus are important steps in the mechanism by which ethanol induces CYP2A5 (Fig. 9). CYP2A6, the human orthologue of CYP2A5, is regulated by the redox-sensitive transcription factor Nrf2 (Yokota et al., 2011). Levels of CYP2E1 and 2A6 were elevated in livers of patients with alcoholic and nonalcoholic liver diseases (Niemela et al., 2000). Whether human CYP2A6 in patients with alcoholic liver disease is regulated by CYP2E1-ROS-Nrf2 pathways requires further study.
Fig. 9.

Scheme of ethanol induction of CYP2A5. Ethanol causes proliferation of SER and upregulates levels of CYP2E1. This leads to increased production of ROS. CYP2E1-mediated ROS activates the redox-sensitive transcription factor Nrf2, which in turn upregulates CYP2A5 expression. For details, please refer to the text.
Recently it was found that Nrf2 can regulate CYP2A5 induction, which contains an ARE in its promoter (Lamsa et al., 2010).Pyrazole-induced CYP2A5 was lower in Nrf2 –/– mice, compared with that in WT mice, suggesting that CYP2A5 induction by pyrazole was mediated by Nrf2 (Lu et al., 2008). CYP2A5 induction by heavy metals including CdCl2 is also dependent on Nrf2 (Abu-Bakar et al., 2004, 2005, 2007; Lamsa et al., 2010). Ethanol induces CYP2A5, at least in part, at the level of transcription because ethanol increased Cyp2a5 mRNA levels (Lu et al., 2011). We found that CYP2A5 induction by ethanol in Nrf2 knockout mice was lower compared with that in WT mice (Fig. 6), suggesting that CYP2A5 induction by ethanol is regulated, in part, by Nrf2. In addition to Nrf2, several other transcription factors have been implicated in the basal and inducible expression of the Cyp2a5 gene (Arpiainen et al., 2005, 2007, 2008; Cai et al., 2002). It is possible that one or some of those transcription factors may take part in regulating ethanol induction of CYP2A5, especially because CYP2A5 was still induced by ethanol in Nrf2 knockout mice although the extent of induction was lower compared with that in WT mice (Fig. 6).
Nrf2 usually regulates expression of cytoprotective or antioxidant genes such as HO-1, glutamylcysteine synthetase, glutathione S-transferases, and NAD(P)H:quinone oxidoreductase in catalyzing the detoxification of reactive electrophiles and oxidants that contribute to the formation of mutations and ultimately cancers; thus, Nrf2 plays important roles in protection against oxidative damage or chemoprevention of cancer (Martín-Montalvo et al., 2011). Nrf2 has been considered as a determinant of susceptibility to carcinogenesis (Slocum and Kensler, 2011). On the other hand, CYP2A5/6 may metabolize nicotine (Benowitz et al., 2006; Zhou et al., 2010), which is of special interest because most alcoholics are also smokers (Wall et al., 2007). Nicotine is inactivated to cotinine primarily by CYP2A5/6, and ethanol induction of CYP2A5/6 may decrease the half time of blood nicotine and thereby change smoking behaviors (Siu et al., 2006). In addition, CYP2A5/6 also metabolizes and activates carcinogens such as aflatoxin B1, N-nitrosodiethylamine, and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) (Su and Ding, 2004), and NNK is a major carcinogen in tobacco smoke (Miyazaki et al., 2005). Therefore, ethanol induction of CYP2A5/6 via Nrf2 might also increase the risks of tumor formation. Nrf2 may have different effects on CYP2A5/6-mediated carcinogen activation and chemical carcinogenesis in alcohol drinkers compared with that in nondrinkers.
Nrf2 has been demonstrated to effectively prevent hepatotoxicity in mice induced by acetaminophen (Enomoto et al., 2001) and pyrazole (Lu et al., 2008a). Ethanol-induced liver injury was more severe in Nrf2 –/– mice compared with the injury in WT mice, suggesting that Nrf2 also protects against alcoholic liver disease (Lamlé et al., 2008). However, ethanol induction of CYP2A5 was lower in Nrf2 –/– mice compared with the induction in WT mice (Fig. 6); alcohol-induced steatosis was higher in Cyp2a5 –/– mice (Fig. 7), suggesting that induction of CYP2A5 may mediate antioxidant defense and cytoprotection in liver. Lower HO-1 levels are in association with the higher fat accumulation in the WT and Cyp2a5 –/– mice. Bilirubin is a HO-1 metabolite. Bilirubin is an effective antioxidant at concentrations ranging from ~0.01 to 10µM, but it is cytotoxic at concentrations > 20µM. Recently, CYP2A5 was identified as a hepatic bilirubin oxidase in oxidative metabolism of cellular bilirubin (Abu-Bakar et al., 2005). Thus, in addition to HO-1, CYP2A5 could control the intracellular concentration of bilirubin to a level with effective antioxidant properties instead of cytotoxic properties ( Abu-Bakar et al., 2011). Therefore, ethanol induction of CYP2A5 under Nrf2 regulation may have a homeostatic function, i.e., alcohol-induced steatosis was more severe when CYP2A5 was absent (Fig. 7). Further studies on the metabolic effects induced by ethanol induction of CYP2A5 and its pathophysiological significance are continuing in our lab.
FUNDING
United States Public Health Service grants (RO1 AA-017425, AA-018790, and P20-AA017067) from the National Institute on Alcohol Abuse and Alcoholism and ABMRF/The Foundation for Alcohol Research.
ACKNOWLEDGMENTS
We thank Dr Frank J. Gonzalez for Cyp2e1 –/– and Cyp2e1 –/– KI mice, Dr Xinxin Ding for Cyp2a5 –/– mice, Dr Jerome Lasker for CYP2E1 antibody, Dr Risto Juvonen for CYP2A5 antibody, and Drs Arndt Vogel and Jutta Lamlé (Department of Hepatology, Gastroenterology and Endocrinology, Hannover, Germany) for liver samples from WT and Nrf2 –/– mice fed with ethanol.
REFERENCES
- Abu-Bakar A., Arthur D. M., Aganovic S., Ng J. C., Lang M. A. 2011. Inducible bilirubin oxidase: A novel function for the mouse cytochrome P450 2A5 Toxicol. Appl. Pharmacol 257 14–22 [DOI] [PubMed] [Google Scholar]
- Abu-Bakar A., Lamsa V., Arpainen S., Moore M. R., Lang M. A., Hakkola J. 2007. Regulation of CYP2A5 gene by the transcription factor nuclear factor (erythroid-derived 2)-like 2 Drug Metab. Disp 35 787–794 [DOI] [PubMed] [Google Scholar]
- Abu-Bakar A., Moore M. R., Lang M. A. 2005. Evidence for induced microsomal bilirubin degradation by cytochrome P450 2A5 Biochem. Pharmacol 70 1527–1535 [DOI] [PubMed] [Google Scholar]
- Abu-Bakar A., Satarug S., Marks G. C., Lang M. A., Moore M. R. 2004. Acute cadmium chloride administration induces hepatic and renal CYP2A5 mRNA, protein and activity in the mouse: Involvement of transcription factor NRF2 Toxicol. Lett 148 199–210 [DOI] [PubMed] [Google Scholar]
- Arpiainen S., Järvenpää S. M., Manninen A., Viitala P., Lang M. A., Pelkonen O, Hakkola J. 2008. Coactivator PGC-1alpha regulates the fasting inducible xenobiotic-metabolizing enzyme CYP2A5 in mouse primary hepatocytes Toxicol. Appl. Pharmacol 232 135–141 [DOI] [PubMed] [Google Scholar]
- Arpiainen S., Lämsä V., Pelkonen O., Yim S. H., Gonzalez F. J., Hakkola J. 2007. Aryl hydrocarbon receptor nuclear translocator and upstream stimulatory factor regulate cytochrome P450 2a5 transcription through a common E-box site J. Mol. Biol 369 640–652 [DOI] [PubMed] [Google Scholar]
- Arpiainen S., Raffalli-Mathieu F., Lang M. A., Pelkonen O., Hakkola J. 2005. Regulation of the Cyp2a5 gene involves an aryl hydrocarbon receptor-dependent pathway Mol. Pharmacol 67 1325–1333 [DOI] [PubMed] [Google Scholar]
- Benowitz N. L., Swan G. E., Jacob P., Lessov-Schlaggar C. N., Tyndale R. F. 2006. CYP2A6 genotype and the metabolism and disposition kinetics of nicotine Clin. Pharmacol. Ther 80 457–467 [DOI] [PubMed] [Google Scholar]
- Cai Y., Konishi T., Han G., Campwala K. H., French S. W., Wan Y. J. Y. 2002. The role of hepatocyte RXR alpha in xenobiotic-sensing nuclear receptor-mediated pathways Eur. J. Pharm. Sci 15 89–96 [DOI] [PubMed] [Google Scholar]
- Cederbaum A. I. 1991. Microsomal generation of reactive oxygen species and their possible role in alcohol hepatotoxicity Alcohol Alcohol.Suppl 1 291–296. [PubMed] [Google Scholar]
- Cederbaum A. I., Cohen G. 1984. Microsomal oxidant radical production and ethanol oxidation. Methods Enzymol 105 516–522 [DOI] [PubMed] [Google Scholar]
- Cederbaum A. I., Lu Y., Wu D. 2009. Role of oxidative stress in alcohol-induced liver injury Arch. Toxicol 83 519–548 [DOI] [PubMed] [Google Scholar]
- Cheung C., Yu A. M., Ward J. M., Krausz K. W., Akiyama T. E., Feigenbaum L., Gonzalez F. J. 2005. The CYP2E1-humanized transgenic mouse: Role of CYP2E1 in acetaminophen hepatotoxicity Drug Metab. Disp 33 449–457 [DOI] [PubMed] [Google Scholar]
- De-Oliveira A. C., Da-Matta A. C., Paumgartten F. J. 2006. Plasmodium Berghei: Infection induces CYP2A5 and 2E1 while depressing other CYP isoforms in the mouse liver Exp. Parasitol 113 256–261 [DOI] [PubMed] [Google Scholar]
- Enomoto A., Itoh K., Nagayoshi E., Haruta J., Kimura T., O’Connor T., Harada T., Yamamoto M. 2001. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes Toxicol. Sci 59 169–177 [DOI] [PubMed] [Google Scholar]
- Gerjevic L. N., Lu S., Chaky J. P., Harrison-Findik D. D. 2011. Regulation of heme oxygenase expression by alcohol, hypoxia and oxidative stress World J. Biol. Chem 2 252–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore W. J., Kirby G. M. 2004. Endoplasmic reticulum stress due to altered cellular redox status positively regulates murine hepatic CYP2A5 expression J. Pharmacol. Exp. Ther 308 600–608 [DOI] [PubMed] [Google Scholar]
- Gong P., Cederbaum A. I. 2006. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1 Hepatology 43 144–153 [DOI] [PubMed] [Google Scholar]
- Jounaidi Y., Bonfils C., Perin F., Negishi M., Lange R. 1994. Overexpression of a cytochrome P450 of the 2a family (CYP2a5) in chemically induced hepatomas from female mice Eur. J. Biochem 219 791–798 [DOI] [PubMed] [Google Scholar]
- Kobliakov V., Kulikova L., Samoilov D., Lang M. A. 1993. High expression of cytochrome P4502a5 (coumarin 7-hydroxylase) in mouse hepatomas Mol. Carcinog 7 276–280 [DOI] [PubMed] [Google Scholar]
- Koop D. R., Tierney D. J. 1990. Multiple mechanisms in the regulation of ethanol-inducible cytochrome P450IIE1 Bioessays 12 429–435 [DOI] [PubMed] [Google Scholar]
- Lamlé J., Marhenke S., Borlak J., von Wasielewski R., Eriksson C. J., Geffers R., Manns M. P., Yamamoto M., Vogel A. 2008. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury Gastroenterology 134 1159–1168 [DOI] [PubMed] [Google Scholar]
- Lamsa V., Levonen A. L., Leinonen H., Yla-Herttuala S., Yamamoto M., Hakkola J. 2010. Cytochrome P4502A5 constitutive expression and induction by heavy metals is dependent on redox-sensitive transcription factor Nrf2 in liver Chem. Res. Toxicol 23 977–985 [DOI] [PubMed] [Google Scholar]
- Lee S. S., Buter J. T., Pineau T., Fernandez-Salguero P., Gonzalez F. J. 1996. Role of CYP2E1 in the hepatotoxicity of acetaminophen J. Biol. Chem 271 12063–12067 [DOI] [PubMed] [Google Scholar]
- Lieber C. S. 1997. Cytochrome P-4502E1: Its physiological and pathological role Physiol. Rev 77 517–544 [DOI] [PubMed] [Google Scholar]
- Lieber C. S., DeCarli L. M. 1972. The role of the hepatic microsomal ethanol oxidizing system (MEOS) for ethanol metabolism in vivo J. Pharmacol. Exp. Ther 181 279–287 [PubMed] [Google Scholar]
- Lu Y., Cederbaum A. I. 2006a. Enhancement by pyrazole of lipopolysaccharide-induced liver injury in mice: Role of cytochrome P4502E1 and 2A5 Hepatology 44 263–274 [DOI] [PubMed] [Google Scholar]
- Lu Y., Cederbaum A. I. 2006b. Cisplatin-induced hepatotoxicity is enhanced by elevated expression of cytochrome P450 2E1 Toxicol. Sci 89 515–523 [DOI] [PubMed] [Google Scholar]
- Lu Y., Cederbaum A. I. 2008. CYP2E1 and oxidative liver injury by alcohol Free Rad. Biol. Med 44 723–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Gong P., Cederbaum A. I. 2008. Pyrazole induced oxidative liver injury independent of CYP2E1/2A5 induction due to Nrf2 deficiency Toxicology 252 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Kawashima A., Horii I., Zhong L. 2004. Effects of BSO and L-cysteine on drug-induced cytotoxicity in primary cell cultures: Drug-, cell type-, and species-specific difference Drug Chem. Toxicol 27 269–280 [DOI] [PubMed] [Google Scholar]
- Lu Y., Wang X., Cederbaum A. I. 2005. Lipopolysaccharide-induced liver injury in rats treated with the CYP2E1 inducer pyrazole Am. J. Physiol. Gastrointest. Liver Physiol 289 G308–G319 [DOI] [PubMed] [Google Scholar]
- Lu Y., Wu D., Wang X., Ward S. C., Cederbaum A. I. 2010. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice Free Radic. Biol. Med 49 1406–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Zhuge J., Wu D., Cederbaum A. I. 2011. Ethanol induction of CYP2A5: Permissive role for CYP2E1 Drug Metab. Dispos 39 330–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Zhuge J., Wang X., Bai J., Cederbaum A. I. 2008. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice Hepatology 47 1483–1494 [DOI] [PubMed] [Google Scholar]
- Mäenpää J., Sigusch H., Raunio H., Syngelmä T., Vuorela P., Pelkonen O. 1993. Differential inhibition of coumarin 7-hydroxylase activity in mouse and human liver microsomes Biochem. Pharmacol 45 1035–1042 [DOI] [PubMed] [Google Scholar]
- Martín-Montalvo A., Villalba J. M., Navas P., de Cabo R. 2011. NRF2, cancer and calorie restriction Oncogene 30 505–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M., Yamazaki H., Takeuchi H., Saoo K., Yokohira M., Masumura K., Nohmi T., Funae Y., Imaida K., Kamataki T. 2005. Mechanisms of chemopreventive effects of 8-methoxypsoralen against 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced mouse lung adenomas Carcinogenesis 26 1947–1955 [DOI] [PubMed] [Google Scholar]
- Niemela O., Parkkila S., Juvonen R. O., Viitala K., Gelboin H. V., Passanen M. 2000. Cytochromes P4502A6, 2E1 and 3A and production of protein aldehyde adducts in the liver of patients with alcoholic and non-alcoholic liver diseases J. Hepatol 33 893–901 [DOI] [PubMed] [Google Scholar]
- Siu E. C., Wildenauer D. B., Tyndale R. F. 2006. Nicotine self-administration in mice is associated with rates of nicotine inactivation by CYP2A5 Psychopharmacology (Berl) 184 401–408 [DOI] [PubMed] [Google Scholar]
- Slocum S. L., Kensler T. W. 2011. Nrf2: Control of sensitivity to carcinogens Arch. Toxicol 85 273–284 [DOI] [PubMed] [Google Scholar]
- Su T., Ding X. 2004. Regulation of the cytochrome P4502A genes Toxicol. Appl. Pharmacol 199 285–294 [DOI] [PubMed] [Google Scholar]
- Su T., Zhang Q., Swiatek P., Ding X. 2002. Expression of the rat CYP2A3 gene in transgenic mice Drug Metab. Dispos 30 548–552 [DOI] [PubMed] [Google Scholar]
- Wall T. L., Schoedel K., Ring H. Z., Luczak S. E., Katsuyoshi D. M., Tyndale R. F. 2007. Differences in pharmacogenetics of nicotine and alcohol metabolism: Review and recommendations for future research Nicotine Tobacco Res 9(Suppl. 3), S459–S474 [DOI] [PubMed] [Google Scholar]
- Yokota S., Higashi E., Fukami T., Yokoi T., Nakajima M. 2011. Human CYP2A6 is regulated by nuclear factor-erythroid 2 related factor 2 Biochem. Pharmacol 81 289–294 [DOI] [PubMed] [Google Scholar]
- Zhou X., Zhuo X., Xie F., Kluetzman K., Shu Y. Z., Humphreys W. G., Ding X. 2010. Role of CYP2A5 in the clearance of nicotine and cotinine: Insights from studies on a CYP2a5-null mouse model J. Pharmacol. Exp. Ther 332 578–587 [DOI] [PMC free article] [PubMed] [Google Scholar]