

Abstract
Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) is triggered by constitutively activated BCR-ABL and SRC family tyrosine kinases.They account for the activations of multiple growth-signaling pathways, including Raf/MEK/ERK, Akt/mTOR and STAT5 pathways. The BCR-ABL tyrosine kinase inhibitor imatinib is the standard treatment for Ph+ leukemia and plays efficacious role in CML. However, imatinib has few inhibitory effects on SRC tyrosine kinase with response rate of Ph+ ALL lower and relapse more frequent and quicker compared with CML. Previous studies showed that oridonin inhibits proliferation and induces apoptosis in many tumor cells. However, the anticancer activity and mechanism of oridonin in Ph+ ALL is unknown. To investigate the anticancer activity of oridonin, we examined its role in constitutively activated Akt/mTOR, Raf/MEK/ERK, STAT5 and SRC pathway, mRNA level of bcr/abl gene, cell viability and apoptosis in Ph+ ALL SUP-B15 cells. Furthermore, we detected synergetic effect of oridonin plus imatinib. Our results showed that oridonin inhibiting activations of LYN (one of SRC family kinases) and ABL and their downstream Akt/mTOR, Raf/MEK/ERK and STAT5 pathways, downregulated Bcl-2 but upregulated Bax protein and then induced apoptosis in Ph+ ALL cells. Oridonin plus imatinib exerted synergetic effects by overcoming imatinib defect of upregulating Akt/mTOR and LYN signaling. Additionally, we examined the effect of oridonin on the signaling pathways in the primary specimens from Ph+ ALL patients. Our data showed that oridonin remarkably suppressed activations of Akt/mTOR, Raf/MEK and STAT5 pathway in these primary specimens and oridonin with imatinib exerted synergetic suppressive effects on mTOR, STAT5 and LYN signaling in one imatinib resistant patient specimen. Additional evaluation of oridonin as a potential therapeutic agent for Ph+ ALL seems warranted.
Keywords: Philadelphia chromosome-positive acute lymphoblastic leukemia, oridonin, imatinib, anti-leukemia effect, LYN, Akt/mTOR, RAF/MEK/ERK, STAT5
Introduction
The Philadelphia chromosome is produced by a reciprocal translocation t(9;22) between chromosomes 9 and 22, which results in the generation of a bcr-abl fusion gene. Bcr-abl fusion gene is the primary cause of Philadelphia chromosome–positive (Ph+) leukemia. Because of different breakpoint in the bcr locus, two alternate products of BCR-ABL fusion proteins, P210 or P190, can be translated. P210 is found predominantly in chronic myelogenous leukemia (CML), whereas the P190 form is mainly associated with Ph+ acute lymphoblastic leukemia (ALL).1,2 BCR-ABL fusion protein has much greater tyrosine kinase activity compared with ABL and results in the development of leukemia.3,4 The Philadelphia chromosome is present in about 5% of childhood ALL and 20–30% of adult ALL and the chances of occurrence of this chromosome increases with age, approaching 50% in patients older than 50 y.5-7 Ph+ ALLL has a very poor prognosis. In the pre-imatinib era, the treatment outcome of Ph+ ALL was dismal and five-year overall survival rates with chemotherapy alone are 10–20%.8,9 Allogeneic hematopoietic stem cell transplantation (allo-HSCT) was virtually the sole curative modality, while it was limited by the availability of a matched donor, the risk of treatment-related mortality and disease resistance or relapse in many cases.
Growth-signaling pathways play vital important roles in tumorigenesis, proliferation, anti-apoptosis and drug resistance. Akt/mTOR, RAF/MEK/ERK and JAK/STAT signaling pathways are such three pathways. Because of BCR-ABL tyrosine kinase activity, many growth-signaling pathways, including Akt/mTOR, RAF/MEK/ERK and JAK/STAT signaling pathways, which play vital roles in development of leukemia are activated in Ph+ leukemia.10-13 These pathways represent promising molecular targets of leukemia. Imatinib (STI 571, Gleevec) deregulates activity of BCR-ABL and it is widely used clinically for treating Ph+ leukemia.14,15 BCR-ABL alone is necessary and sufficient for the development of chronic myeloid leukemia, therefore, imatinib is a very effective therapy for chronic phase CML.16-18 Except BCR-ABL, other kinases are also involved in the development of Ph+ ALL, particularly SRC kinases,19,20 which are not blocked by imatinib. Thus, the response rate of imatinib alone is lower while, resistance and relapse is frequent in Ph+ ALL. Second-generation tyrosine kinase inhibitors, such as dasatinib and nilotinib, can overcome resistance of imatinib to some extent, as patients treated with them achieved complete remission quickly, with complete remission rates of approximately 90%; however, CR duration is short too.21-23 To further improve the clinical outcome and provide therapeutic options for Ph+ ALL patients, other investigational therapy should be developed.
Oridonin (Fig. 1A), an active diterpenoid compound isolated from Rabdosia Rubescens,24 has been traditionally used to treat various diseases.Oridonin serves various biological, pharmaceutical and physiological functions, such as anti-cancer, anti-bacteria and anti- inñammation activity.25,26 Studies showed that oridonin has inhibitory effects on activated signaling pathways in some cancer cells27-29 and is a promising anti-cancer agent which induces apoptosis in various cancer cells, including liver, prostate, breast, cervical, lung cancer and acute myelocytic leukemia, glioblastoma multiforme.30-33 However, whether oridonin can induce apoptosis by inhibiting constitutively activated growth-signaling pathways in Ph+ ALL cells remains obscure. Therefore, we set out to investigate the antileukemia activity of oridonin in Ph+ ALL. Here, we reported that oridonin inhibited proliferation and induced apoptosis in Ph+ ALL leukemia SUP-B15 cells by inhibiting activations of ABL and SRC kinases and their downstream Akt/mTOR, Raf/MEK/ERK and STAT5 pathways, upregulating the expression of Bax but downregulating the expression of Bcl-2. Furthermore, our results indicated that oridonin with imatinib exerted synergetic anti-leukemia effects by suppressing activated LYN and Akt/mTOR pathway. Otherwise, oridonin suppressed activations of Akt/mTOR, Raf/MEK, STAT5 and LYN pathway in Ph+ ALL patient samples.

Figure 1. Chemical structure of oridonin and cytotoxic effect of oridonin alone or in combination with imatinib on SUP-B15 cells. (A) chemical structure of oridonin.B, SUP-B15 cells were incubated with 0, 5, 7.5, 10, 12.5 μM oridonin for 1 d, 3 d and 5 d. At the end of incubation, MTT methods determined the cell survival rates. Cell viability is expressed as the percentage of cell survival compared with the control. (B) SUP-B15 cells were incubated with imatinib alone or in combination with 1 μM, 2 μM and 3 μM oridonin for 72 h. At the end of incubation, MTT methods were used to determine the cell survival rates and the IC50 of imatinib. (C) The cell viability treated with imatinib alone or in combination with indicated concentration of oridonin for 72 h. D,The IC50 of imatinib alone or in combination with indicated concentration of oridonin for 72 h. Data were from three independent experiments. *p < 0.05 was used to compare the effects of compounds.
Results
The anti-proliferative effects of oridonin, or its combination with imatinib on SUP-B15 cells
The results of MTT assay showed that oridonin inhibits cell growth in time- and dose- dependent manner in SUP-B15 cells (Fig. 1B). After 72 h of treatment, IC50 (the concentrations of oridonin which caused 50% inhibition of cell growth) was 7.8 ± 1.1886 μM. The IC50 of imatinib alone was 1.3 ± 0.14 μM and the cell viability was 60.73 ± 2.66% after 1 μM imatinib treatment. When combined with 1 μM, 2 μM, 3 μM oridonin, the IC50 of imatinib decreased to 0.57 ± 0.11 μM, 0.45 ± 0.13 μM, 0.34 ± 0.16 μM and the cell viability declined to 39.51 ± 4.45%, 37.35 ± 2.6% and 31.02 ± 3.21% respectively (Fig. 1C,D). The interaction index (I) for 50% inhibition of cell growth was 0.57, 0.6, 0.64 at the 1 μM, 2 μM, 3 μM oridonin, respectively. These results indicated there was a synergistic inhibitory effect of oridonin plus imatinib.
We obtained mononuclear cell preparations from four Ph+ ALL patients. Patient 1 was newly diagnosed de novo Ph+ ALL, patient 2 relapsed after chemotherapy and the other two patients 3 and 4 were progressing under imatinib therapy. The oridonin IC50 of the four patients was 8.71 μM, 9.19 μM, 9.56 μM and 10.18 μM, respectively.
Oridonin-mediated growth inhibition by induction of apoptosis in SUP-B15 cells
To determine whether oridonin-mediated growth inhibition is associated with apoptosis, cells were treated with 10 μM, 15 μM oridonin for 24 h and furthermore, to determine whether synergistic inhibitory effect of oridonin and imatinib is associated with apoptosis, cells were treated with 3 μM oridonin, 1 μM imatinib and oridonin plus imatinib for 24 h. The morphologic changes were observed using a light microscope. Hoechst 33258 staining was used to identify changes in the nuclei. The membrane of control cells (without drugs) was lubricious without losing integrity and the nuclei were round and homogeneously stained. 10 μM and 15 μM oridonin-treated cells changed significantly into the bluffing and lobulate appearances and marked condensation of the nuclei and apoptotic bodies (Fig. 2A and B). As shown in Figure 2A and B, in the treatment of 3 μM oridonin, 1 μM imatinib showed little morphologic changes, similar to control cells. However, oridonin plus imatinib-treated cells showed bluffing and lobulate appearances of membrane and marked blebbing of the nuclei and apoptotic bodies. To further confirm the oridonin-related apoptosis in SUP-B15 cells, the cells apoptosis rate was performed by FCM. The results revealed that apoptotic rate of SUP-B15 cells without drug was 5.1% and increased to 47.6% and 71.1% following treatment with 10 μM and 15 μM oridonin, respectively (Fig. 2C). Imatinib induced cytotoxic effects dilatorily and had no significant effects on apoptotic rate at 24h. However, treatment with 1μM imatinib plus 3μM oridonin resulted in the increase of apoptotic rate from 6.9% to 25.8% (Fig. 2C). These results suggested that oridonin suppresses the growth of SUP-B15 cells by inducing apoptosis and that oridonin exerts synergistic effect with imatinib on inducing apoptosis in Ph+ ALL cells.

Figure 2. Oridonin and imatinib induced apoptosis in SUP-B15 cells. (A) The SUP-B15 cells were treated with indicated oridonin, imatinib, or combination for 24 h, changes in cellular morphology were examined by a light microscope. The membrane of control cells was lubricious and integrity, treatment of 3 μM oridonin, 1 μM imatinib showed little morphologic changes, but oridonin plus imatinib-treated cells showed bluffing and lobulate appearances of membrane ( × 400). (B) The cells were treated with indicated oridonin, imatinib or in combination for 24 h, then stained with hoechst 33258 fluorescent. Changes in cellular nuclei were observed under a fluorescence microscope. The nuclei of control cells were round and were homogeneously stained. Treatment of 3 μM oridonin, 1 μM imatinib showed little morphologic changes, similar to control cells, while oridonin plus imatinib-treated cells showed marked blebbing of the nuclei and apoptotic bodies ( × 200). (C) The cells were treated with indicated oridonin, imatinib or in combination for 24 h and apoptosis was examined using the AnnexinV-FITC /PI apoptosis detection kit.
Oridonin inhibits activations of Abl and LYN kinases,and signaling pathways of Akt/mTOR, Raf/MEK /ERK, STAT5 and in SUP-B15 cells
To study the mechanisms of anti-leukemia activity by oridonin, we examined activation levels of the Abl and LYN kinases and their downstream growth-signaling pathways of Akt/mTOR, Raf/MEK/ERK and STAT5 in SUP-B15 cells. Western blot analysis showed that SUP-B15 cells without oridonin expressed constitutively the phosphorylated form of Abl(Tyr245), Akt(Ser473), mTOR (ser2448), p70S6K (Tyr389), 4EBP1 (Thr37/46) proteins, Raf (ser338), MEK1/2 (ser217/221), ERK 1/2 (Thr202/Tyr204) proteins, Stat5 and Lyn (Fig. 3A), unlike K562 cells, a Ph+ leukemia cell line established from a patient with chronic myeloid leukemia in blast crisis. After oridonin treatment, the levels of the phosphorylated forms of Abl (Tyr245), Akt (Ser473), mTOR(ser2448), p70S6K(Tyr389) and 4EBP1(Thr37/46) were downregulated in a time- and dose-dependent manner (Fig. 3B and C). Raf/MEK/ERK signaling, phosphorylation of Stat5 and LYN were also downregulated by oridonin in time- and dose-dependent manner, as shown in Figure 3D, E, F and G.

Figure 3. Oridonin inhibited constitutively activation of growth-signaling pathway in SUP-B15 cells. Western blot analysis with indicated anti-phosphotyrosine, phosphoserine and phosphothreonine antibodies showed Akt/mTOR and RAF/MEK/ERK signaling pathway, Abl, STAT5 and LYN protein kinase were constitutively active in SUP-B15 cells, compared with the expression in K562 cells (A). The cells were incubated with 7.5 μM oridonin for 6, 12, 24 h or with 5, 10, 15 μM doses of oridonin for 24 h. Cells were then harvested and total proteins were extracted. Equal amounts of protein from each sample were separated on SDS-PAGE and immunoblotted with indicated antibodies, GAPDH was used as a loading control. Oridonin inhibited constitutively activation of Abl, Akt/mTOR (B and C), Raf/MEK/ERK (D and E) and STAT5,LYN (F and G) signaling pathway in time- and dose-dependent manner. The results shown were representative of two independent experiments.
Oridonin inhibits imatinib-mediated activation of Akt/mTOR signaling in SUP-B15 cells
Imatinib is an important therapeutic agent to Ph+ leukemia. Due to Abl and SRC kinases and their downstream signaling, Akt/mTOR, Raf/MEK/ERK and Stat5 were constitutively activated in SUP-B15 cells. Thus we examined the effect of imatinib on these signaling pathways. The results showed that 1μM imatinib inhibited the phosphorylation of Abl, MEK, ERK (Fig. 4C) and Stat5 (Fig. 4E), but had no significant effects on LYN activation (Fig. 4E). Of interest, imatinib-treatment further upregulated activation of Akt/mTOR signaling (Fig. 4A). We wondered whether oridonin could inhibit the imatinib-mediated upregulation of Akt/mTOR signaling. Therefore, the effect of oridonin plus imatinib was examined. The results confirmed our speculation that oridonin inhibited the imatinib-mediated activation of Akt/mTOR signaling (Fig. 4B) and exerted siginificant inhibitory effect on phosphorylation of LYN kinase (Fig. 4F), but had no synergistic inhibitory effects on Abl, MEK, ERK (Fig. 4D) and Stat5 (Fig. 4F).

Figure 4. Effects of imatinib alone or in combination with oridonin on growth-signaling pathways in SUP-B15 cells. The SUP-B15 cells were incubated with 1μM imtinib for 6, 12, 24 h, or with 1 μM imatinib plus 3 μM oridonin for 24 h. They were then harvested and total proteins were extracted. Equal amounts of protein from each sample were separated on SDS-PAGE and immunoblotted with indicated antibodies, GAPDH was used as a loading control. Imatinib upregulated Akt/mTOR signaling pathway (A), inhibited constitutively activation of MEK, ERK signaling proteins (C) and STAT5 signaling (E) and had no significant effect on RAF and LYN signaling proteins (C,E). Oridonin overcame the upregulation of Akt/mTOR signaling pathway by imatinib. (B), Oridonin plus imatinib had no synergetic inhibitory effects on activation of MEK, ERK (D) and STAT5 (F) signaling proteins. The combination of oridonin and imatinib exerted significant inhibitory effect on activation of LYN tyrosine kinase (F). The results shown were representative of two independent experiments.
The effect of oridonin on the protein levels of Bcl-2 and Bax in SUP-B15 cells
Defect in apoptosis may contribute to leukemia progression and treatment resistance. Deregulated expression of anti-apoptotic or pro-apoptotic molecules may disrupt apoptosis signaling of leukemia cells. Bcl-2 family members are important regulators of apoptosis which include anti-apoptotic (Bcl-2, Bcl-XL and Mcl-1), pro-apoptotic (Bax and Bak) and the BH-3 domain-only (Bim, Bid and Bik) proteins.To explore the possible mechanism by which oridonin induced apoptosis in SUP-B15 cells, we analyzed the expression of Bcl-2 and Bax by western blot analysis. The results as depicted in Figure 5 A and B exhibited alteration in the expression of both Bcl-2 and Bax proteins. Oridonin treatment resulted in decrease of Bcl-2, whereas Bax level increased in time- and dose-dependent manner.
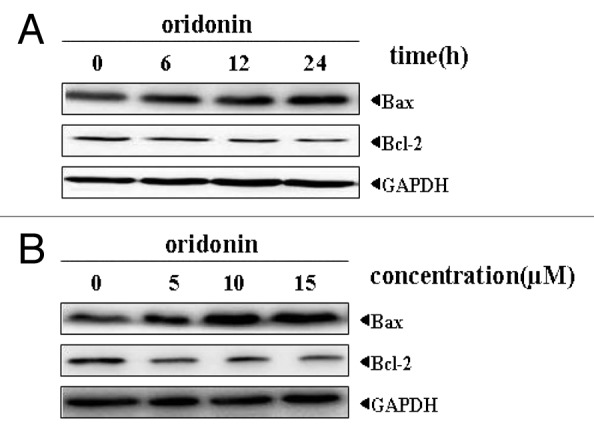
Figure 5. Oridonin modulated the expression of pro-apoptotic protein Bax and anti-apoptotic protein Bcl-2 in SUP-B15 cells. The cells were incubated with 7.5 μM oridonin for 6, 12, 24 h (A), or with 5, 10, 15 μM doses of oridonin for 24 h (B). Equal amounts of protein from each sample were separated on SDS-PAGE and immunoblotted with antibodies against Bcl2 and Bax. GAPDH was used as a loading control. The results shown were representative of two independent experiments. Oridonin upregulated the expression of pro-apoptotic protein Bax and downregulated the expression of anti-apoptotic protein Bcl-2 in SUP-B15 cells.
The effect of oridonin on mRNA levels of bcr-abl gene in SUP-B15 cells
Bcr-abl gene is the root cause of Ph+ leukemia, therefore, to investigate the anti-leukemia mechanism of oridonin, we evaluated the effects of oridonin on bcr-abl gene by performing semi-quantitative RT-PCR. As shown in Figure 6A and B, oridonin, imatinib or in combination treatment had no significant effects on mRNA levels of bcr-abl gene.

Figure 6. Analysis of mRNA expression of bcr-abl gene in SUP-B15 cells. The cells were treated with indicated oridonin, imatinib, or in combination for 24 h. Total RNA was isolated and RT-PCR was performed with β-actin primers as a control with the same amount of RNA. Oridonin, imatinib alone, or thier combination had no effects on the mRNA expression of bcr-abl gene in SUP-B15 cells. (A) Lane 1: control; Lane 2: oridonin 5 μM; Lane 3: oridonin 10 μM; Lane 4: oridonin 15μM; Lane 5: β-actin of conrtol; Lane 6: β-actin of oridonin 5μM; Lane 7: β-actin of oridonin 10μM. Lane 8: β-actin of oridonin 15 μM. (B) Lane 1: control; Lane 2: oridonin 3μM; Lane 3: imatinib 1μM; Lane 4: oridonin 3μM and imtinib 1μM; Lane 5: β-actin of control; Lane 6: β-actin of oridonin 3 μM; Lane 7: β-actin of imatinib 1μM; Lane 5: β-actin of oridonin 3 μM and imatinib 1 μM.
Oridonin suppresses activation of growth-signaling pathways in Ph+ ALL patient specimens
Different from SUP-B15 cells, we found ERK was scarcely activated in the four Ph+ ALL clinical specimens and we did not detect the effect of oridonin on it. Since we obtained only few specimens, these cells were incubated only with oridonin (10 μM) for 24h to measure changes in activation levels of signaling pathways. Synergetic effects of oridonin and imatinib were examined only in one imatinib resistant patient. As shown in Figure 7, oridonin markedly downregulated activation levels of Akt/mTOR, Raf/MEK, STAT5 and LYN signalings in the four patient specimens. In patient 4 who progressed under imatinib therapy, 5 μM imatinib had little inhibitory effects on activations of Akt/mTOR, Raf/MEK, STAT5 and LYN signalings, however, 5 μM imatinib plus 5 μM oridonin significantly inhibited the activation of mTOR, LYN and STAT5 signaling, but had no synergetic inhibitory effects on Raf/MEK.

Figure 7. Oridonin downregulates signaling pathways in Ph+ ALL patient primary cells. The mononuclear cells from Ph+ ALL patients were treated with 5 μM oridonin,10 μM oridonin, 5 μM imatinib, or 5 μM oridonin with 5 μM imatinib for 24 h. Total proteins were extracted and equal amounts of protein from each sample were separated on SDS-PAGE and immunoblotted with indicated antibodies. GAPDH was used as a loading control. Oridonin significantly suppressed Akt/mTOR, Raf/MEK, STAT5 and LYN signaling pathways in all four clinical specimens. In patient 4 who progressed under imatinib therapy, 5 μM imatinib had little inhibitory effects on Akt/mTOR, Raf/MEK, STAT5 and LYN signaling while, 5 μM imatinib with 5 μM oridonin significantly inhibited the activation of mTOR, LYN and STAT5 signalings, but had no synergetic inhibitory effects on Raf/MEK.
As a control, we examined the effects of oridonin on non-BCR/ABL positive leukemia cell line CEM cells (human acute T-lymphoblastic leukemia cell line) and normal human peripheral blood mononuclear cells. Our data suggested that the cytotoxic effects of oridonin were not cell or BCR/ABL kinase dependent. Oridonin also inhibited proliferation of CEM cells in a time- and dose-dependent manner (Fig. S1) and induced apotosis of CEM cells (Fig. S2A and B) via downregulating signaling m-TOR and Raf/ERK signaling pathways (Fig. S3). Oridonin had minimal cytotoxic effect on normal human peripheral blood mononuclear cells under these conditions (Fig. S4). We found that PBMCs from healthy donors were negative for ABL/AKT/mTOR, RAF/MEK/ERK and STAT5 signaling activation (Fig. S5); therefore we did not examined the effects on them.
Discussion
In this study, oridonin exhibited a strong anti-leukemia effect in Ph+ ALL in vitro. Our results showed that oridonin inhibited the spontaneous growth of SUP-B15 cells in time- and dose-dependent manner and exerted synergetic anti-leukemia effects in combination with imatinib. To further understand the mechanisms in the anti-leukemia activity of oridonin, we analyzed the effect of oridonin on apoptosis and activations of Abl and LYN kinases,and their downstream Akt/mTOR,Raf/MEK/ERK,and SATA5 signaling pathways which were constitutively activated in SUP-B15 leukemia cells. Our results showed that oridonin induced apoptosis by inhibiting simultaneously activations of Abl and LYN kinases and their downstream Akt/mTOR, Raf/MEK/ERK and SATA5 signaling pathways, downregulating the expression of anti-apoptosis protein Bcl-2 and upregulating the expression of pro-apoptosis protein Bax. Synergetic effects of oridonin and imatinib were derived from the inhibition of LYN/Akt/mTOR signaling pathway. In Ph+ ALL patient primary cells, oridonin also exhibited predominant suppressive effects on activations of Akt/mTOR, Raf/MEK, STAT5 and LYN signaling, upregulateed markedly the level of Bax and downregulated the level of Bcl2. Oridonin with imatinib exerted synergetic inhibitory effect on mTOR signaling, STAT5 and LYN signaling.
Unlike normal cells, tumor cells often express constitutively active growth-signaling pathways such as Akt/mTOR, RAF/MEK/ERK, JAK/STAT and NF-κB signaling pathways owing to genic mutations, rearrangements and chromosomal translocations. These growth-signaling pathways play vital important roles in tumorigenesis, proliferation, anti-apoptosis and drug resistance.35-38 More constitutively active growth-signaling pathways in leukemia indicate poorer the prognosis.39 Thus, these pathways may represent promising molecular targets of leukemia. BCR-ABL tyrosine kinase triggers Ph+ leukemia. Otherwise, the Src-family kinases (SFKs) are required for the development and progression of Ph+ ALL. Furthermore, SFKs cooperate with BCR-ABL resulting in the activation of certain downstream signaling pathways involved in Ph+ ALL transformation, including Akt/mTOR, Raf/MEK/ERK, STAT5 and NFκB signaling pathways.10,13,40-43
Although, BCR-ABL tyrosine kinase induces both Ph+ ALL and CML, activation status of downstream signaling pathways in them is different. Our studies showed that Akt/mTOR, Raf/MEK/ERK and STAT5 signaling were constitutively activated prominently and coequally in SUP-B15 cells. In K562 cells, the main activated pathway was Raf/MEK/ERK signaling. The activation levels of Akt/mTOR and STAT5 in K562 cells were markedly lower than in SUP-B15 cells. These may be one of the reasons that Ph+ ALL is more aggressive than CML and account for the poor prognosis of Ph+ ALL. Therefore, we speculate that agents inhibiting activation of Akt/mTOR, Raf/MEK/ERK or STAT5 would create anti-leukemia effects, especially since the inhibitors suppressing the three pathways simultaneously may prove more effective in SUP-B15 cells. Oridonin is an active diterpenoid compound isolated from Rabdosia Rubescens. Previous studies showed that oridonin had suppressive effect on Akt and RAF/ERK signaling pathways in some tumor cells. Song et al. showed oridonin induced apoptosis in human osteosarcoma cells through Akt and MAPKs signaling pathways.44 Dan et al. reported that oridonin induced the apoptosis of human epidermoid carcinoma A431 cells through blockage of the Ras/RAF/ERK signal pathway.45 Our study showed oridonin simultaneously inhibited activations of Akt/mTOR, Raf/MEK/ERK and STAT5 signaling pathways, downregulated Bcl-2 level and upregulated Bax expression and then induced apoptosis in SUP-B15 cells. Oridonin is a promising anti-leukemia agent for Ph+ ALL.
Both Ph+ ALL and CML are triggered by constitutively activated BCR-ABL tyrosine kinase while they clearly differ in their aggressiveness and response to target therapy. The BCR-ABL tyrosine kinase inhibitor imatinib mesylate is the standard treatment for CML; however, the response rate for Ph+ ALL is lower, response duration is shorter than that of CML and relapse is frequent when used as a single agent.46 Although the reason for the poor response of Ph+ ALL to imatinib therapy remains unclear, previous studies indicated that imatinib resistance exists even in imatinib-naive BCR-ABL positive cells. How do BCR-ABL positive cells escape from imatinib inhibition? In Ph+ leukemia, the activation level of STAT5 is a parameter that determines the sensitivity of BCR-ABL+ cells against tyrosine kinase inhibitors (TKIs) and it is associated with resistance against TKIs.47 Higher level of STAT5 is associated with lower level of TKI sensitivity. Our data showed that the activation level of STAT5 was higher in SUP-B15 than in K562. This may be one reason for lower response of Ph+ ALL to imatinib. Burchert et al. demonstrated that Akt/mTOR signaling was activated in response to IM-treatment in BCR-ABL+ cells.48 Furthermore, they indicated that IM-naive BCR-ABL+ cells employed the compensatory Akt/mTOR-signal activation to escape BCR-ABL inhibition of IM. Finally, the researchers found that imatinib-induced Akt/mTOR-signaling activation was involved in mediating early IM-resistance and they stated that inhibition activation of this signaling pathway prevented imatinib resistance development. Chu et al. reported that IM treatment of primary CML-cells unexpectedly did not inhibit the Akt-activity and that compensatory activation of the Ras/MAPK-signaling contributed to survival of BCR-ABL positive cells under IM selection pressure.49 Our studies showed that imatinib treatment notably inhibited the activation of Abl, MEK, ERK and STAT5, but further activated Akt/mTOR sigaling pathway in SUP-B15 cells. Otherwise, unlike CML, three SRC-family kinases, LYN, HCK and FGR, are required for the development of B-ALL and activation of these SRC kinases is independent of BCR-ABL kinase activity. Thus inactivation of BCR-ABL kinase activity alone is insufficient to control the disease.20 In SUP-B15 cells, imatinib treatment had no significant inhibitory effect on activity of LYN kinase. At the same time, our data showed imatinib induced cytotoxic effects in SUP-B15 cells dilatorily and had no significant effects on apoptosis at 24h; therefor these leukemia cells had adequate time to employ markedly activated Akt/mTOR survival pathway to escape imatinib treatment. These may particularly account for the poor response of imatinib-naive Ph + ALL cells and may be one of the mechanisms of imatinib primary resistance. According to our data, we supposed that Akt/mTOR signaling is not the intimate downstream of BCR-ABL kinase in SUP-B15 cells, as its activation is independent of BCR-ABL kinase activity. When we combined imatinib with mTOR inhibitor rapamycin, anti-leukemia efficacy of imatinib in SUP-B15 cells rose and unfortunately, the employment of rapamycin caused the upregulation of Akt because of the feedback which may be the base of new resistance (data not shown). Considering the striking inhibitory effects of oridonin on activation of Akt/mTOR signaling pathway and LYN kinase, we anticipated that oridonin might overcome the defect of imatinib and that synergetic anti-leukemia effects might be elicited when combining the two compounds. Consistent with our prediction, combination of oridonin and imatinib overcame the defect of imatinib alone and elicited inhibitory effect on activation of Akt/mTOR signaling and LYN signaling. No synergetic effects on Abl/Raf/MEK/ERK, STAT5 pathway and mRNA level of bcr-abl gene were observed. Therefore, we deduced that oridonin with imatinib exerts synergetic anti-leukemia effects on SUP-B15 cells by inhibiting activations of Akt/mTORsignaling pathway and LYN signaling. We are tempting to speculate that combination of imatinib and oridonin is a promising therapy strategy which may increase anti-leukemia potency of imatinib and retard development of imatinib resistance.
Taken together, oridonin simultaneously inhibits activations of Abl and LYN kinases and their downstream Akt/mTOR, Raf/MEK/ERK and STAT5 panthways which are constitutively activated in Ph+ ALL SUP-B15 cells and in Ph+ ALL patient specimens. It then induces apoptosis and inhibits proliferation. Moreover, oridonin with imatinib exert synergetic anti-leukemia effects by overcoming imatinib’s drawback of activation of Akt/mTOR signaling and its effects on LYN. Oridonin is a promising therapeutic agent for Ph+ ALL.
Materials and Methods
Cell culture, patient specimens and materials
Ph+ acute B lymphoblastic leukemia cell SUP-B15 expressing P190 fusion protein was purchased from American Type Culture Collection (ATCC, Rockville, MD). The cells were cultured in IMDM medium containing 10% FBs, 100 U/ml penicillin and 100 g/ml streptomycin and in 5% CO2 incubator at 37°C. Primary leukemia samples (blood or bone marrow) were obtained from four Ph+ ALL patients diagnosed at the Department of hematology at Sichuan University in West China, after acquiring written informed consent and the approval from the local ethics committees. The mononuclear cells were isolated by density centrifugation (Ficoll-Hypaque), re-suspended in cell culture media (RPMI-1640 with 10% fetal bovine serum, 100 U/ml penicillin and 100 g/ml streptomycin) and incubated at 37°C in a 5% CO2 incubator. The normal human peripheral blood mononuclear cells were isolated from blood of blood donor.
The rabbit monoclonal antibodies to phosphor-Abl(Tyr245), phospho-mTOR (ser2448), mTOR, phospho-Akt (ser473), Akt, phospho-P70S6 (Thr389), P70S6, phospho-4EBP1 (Thr37/46), 4EBP1, phospho-cRaf (ser338), phospho-MEK1/2 (ser217/221), MEK1/2, phospho-ERK1/2 (Thr202/Tyr204), GAPDH, Bax, Bcl-2 and goat anti-rabbit horseradish peroxidase (HRP) conjugate were purchased from the Cell Signaling Technology. The rabbit monoclonal antibody to phospho-Lyn (Tyr396) was purchased from Abcam. The rabbit monoclonal antibody phospho-STAT5a (Thr694) was purchased from Beijing Biosynthesis Biotechnology Co., Ltd.. AnnexinV-FITC/PI appotosis detection kit was purchased from KeyGen Biotech. Co., Ltd. 3-(4, 5-dimethylthiazol-2-yl)-2 and 5-diphenyl-2H-tetrazolium bromide (MTT) was purchased from Sigma Chemical Co. Oridonin was purchased from Suzhou Baozetang Bio-Pharm Co., Ltd. and was prepared at a 20-mM solution in dimethyl sulfoxide and diluted in cell culture medium before use. Imatinib from Novartis was dissolved in dimethyl sulfoxide and stored in -20°C with the concentration of 10mM. RPMI 1640, fetal bovine serum (FBS) and penicillin/streptomycin were obtained from Hyclo Company.
MTT assay
The cytotoxic effect of oridonin on SUP-B15 cells and mononuclear cells from Ph+ ALL patients was determined by MTT assay. Brieñy, the cells (4 × 104 cells/well in SUP-B15 cells and 5 × 105 cells/well in patient specimens) were incubated for 72 h at 37°C in triplicate in a 96-well plate in the presence or absence of indicated concentrations of oridonin in a ðnal volume of 100μL. Thereafter, 20μL MTT solution (5mg/mL in PBS) was added to each well. After 4-h incubation at 37°C, 100μL SDS-isobutanol-HCl solution was added and incubation was continued overnight at room temperature. Subsequently, the optical density (OD) was measured using μQuant MQX200 Microplate Spectrophotometer (Biotek) at a wavelength of 570 nm. The cell viability was displayed as a percentage: OD(oridonin)-OD(blank)/[OD (control)-OD(blank) ] × 100%. The percentage of cell growth inhibition was calculated as follows: Cell growth inhibition (%) = OD(control)- OD(oridonin)/[ OD(control)- OD(blank)] × 100. Synergistic cytotoxicity was determined by calculating the interaction index (I) according to the classic isobologram equation 34: I = (D)1/(Dx)1+(D)2/(Dx)2, where Dx is the concentration of one compound alone required to produce the effect (in this case 50% inhibition of cell growth) and (D)1 and (D)2 are the concentration of both compounds that produce the same effect. I = 1 indicates an additive effect, I < 1 indicates synergy, I > 1 means antagonism between two drugs.
Light microscopy
SUP-B15 cells (2 × 106) were seeded into 6-well culture plates and cultured with indicated concentration of oridonin, imatinib and oridonin plus imatinib for 24 h. The cellular morphologic changes were observed using a light microscope (Olympus).
Fluorescence microscopy
Apoptotic morphology was studied by staining the cells with Hoechst 33258 fluorescent. Cells (2 × 106) were seeded into a 6-well plate with indicated oridonin, imatinib and oridonin plus imatinib. After 24 h incubation, the cells were harvested and washed three times with PBS and stained with 20 mg/ml of Hoechst 33258 for 10 min. Thereafter, the cells were washed in PBS and observed under a fluorescence microscope (Olympus).
Flow cytometric analysis
Cell apoptosis of the treated cells was analyzed using a ñow cytometer. After the treatment with different concentrations of oridonin, imatinib and imatinib plus oridonin for 24 h, SUP-B15 cells were harvested by centrifugation at 300 × g for 5 min and then washed in phosphate-buffered saline (PBS) once. The cells were resuspended in 100 μL binding buffer. Fuorescein isothiocyanate (FITC)-conjugated annexin V was added to the samples and kept in the dark for 10 min, subsequently, PI was added and analyzed by ñow cytometry. For each determination, 10,000 cells were counted.
Western blot analysis
Whole-cell extracts (2 × 106 cells) were prepared from control, oridonin, imatinib and oridonin plus imatinib in RAPI lysis buffer (20 mM Tris, pH 7.4; 250 mM NaCl; 2 mM EDTA, pH 8.0; 0.1% Triton-X100; 0.01 mg/mL aprotinin; 0.005 mg/mL leupeptin; 0.4 mM PMSF; 4 mM NaVO4), protein extracts were quantitated loading on 6% to 15% sodium dodecyl sulfate-polyacrylamide gel. After electrophoresis, proteins were electro-transferred to nitrocellulose membrane. The membrane was incubated overnight with speciðc primary antibody at 4°C, then washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature and ðnally detected by enhanced chemiluminescence (ECL) detection system and ðlm (Bio-Rad Laboratories Inc.) according to manufacturer’s instruction. GAPDH was used as an endogenous control to standardize the amount of the sample proteins.
RT-PCR assay
SUP-15 cells were treated with indicated concentration of ordonin, imatinib and ordonin plus imatinib for 24 h. Total RNA was isolated from 2 × 106 cells according to the manufacturer’s instructions (Tiangen Biotech Co., Ltd.). 2 μg of RNA was converted to cDNA by Superscript reverse transcriptase (Takara Biotechnology Co., Ltd.). The relative expression of bcr-abl was analyzed using semi-quantitative RT-PCR with β-actin as an internal control. The RT-PCR reaction mixture contained 10 μl Taq mixture, 3 μl of cDNA, 1 μl of sense and anti-sense primer and 5 μl RNA-free water in a ðnal volume of 20 μl. The primer sequences for bcr-abl were (sense) 5′-CCG GAG TTT TGA GGA TTG CGG A3′; (anti-sense) 5′-TTG GAG TTC CAA CGA GCG GC3′. For β-actin the primer sequences were as follows (sense) 5′CCA AGG CCA ACC GCG AGA AGA TGA C3′; (anti-sense 5′AGG GTA CAT GGT GGT GCC GCC AGA C3′. The reaction was performed at 94°C for 2 min, 94°C for 30 cycles of 30 sec each, 56°C for 30s, 72°C for 35 s and a ðnal extension at 72°C for 10 min. The PCR products (10 μl) were analyzed by electrophoresis on 2% (w/v) agarose gel, photographed and quantiðed by densitometric scanning.
Statistical analysis
The IC50 and viability were analyzed with one-way ANOVA and independent sample t test. P values less than 0.05 were considered statistically significant and were derived from 2-sided statistical tests. All statistical analyses were performed using the software SPSS 13.0 for windows.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the grants from National Natural Science Foundation of China (No. 30770912), Foundation of the Science and Technology Department of Sichuan Province (No. 2008SZ0017) and National Science and Technology Pillar Program (No. 2008BAI61B01).
Supplemental Material
Supplemental Material may be found here:
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/21460
References
- 1.Laurent E, Talpaz M, Kantarjian H, Kurzrock R. The BCR gene and philadelphia chromosome-positive leukemogenesis. Cancer Res. 2001;61:2343–55. [PubMed] [Google Scholar]
- 2.Kurzrock R, Kantarjian HM, Druker BJ, Talpaz M. Philadelphia chromosome-positive leukemias: from basic mechanisms to molecular therapeutics. Ann Intern Med. 2003;138:819–30. doi: 10.7326/0003-4819-138-10-200305200-00010. [DOI] [PubMed] [Google Scholar]
- 3.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–30. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 4.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247:1079–82. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 5.Cobaleda C, Gutiérrez-Cianca N, Pérez-Losada J, Flores T, García-Sanz R, González M, et al. A primitive hematopoietic cell is the target for the leukemic transformation in human philadelphia-positive acute lymphoblastic leukemia. Blood. 2000;95:1007–13. [PubMed] [Google Scholar]
- 6.Faderl S, Jeha S, Kantarjian HM. The biology and therapy of adult acute lymphoblastic leukemia. Cancer. 2003;98:1337–54. doi: 10.1002/cncr.11664. [DOI] [PubMed] [Google Scholar]
- 7.Radich JP. Philadelphia chromosome-positive acute lymphocytic leukemia. Hematol Oncol Clin North Am. 2001;15:21–36. doi: 10.1016/S0889-8588(05)70198-2. [DOI] [PubMed] [Google Scholar]
- 8.Dombret H, Gabert J, Boiron JM, Rigal-Huguet F, Blaise D, Thomas X, et al. Groupe d’Etude et de Traitement de la Leucémie Aiguë Lymphoblastique de l’Adulte (GET-LALA Group) Outcome of treatment in adults with Philadelphia chromosome-positive acute lymphoblastic leukemia--results of the prospective multicenter LALA-94 trial. Blood. 2002;100:2357–66. doi: 10.1182/blood-2002-03-0704. [DOI] [PubMed] [Google Scholar]
- 9.Jones LK, Saha V. Philadelphia positive acute lymphoblastic leukaemia of childhood. Br J Haematol. 2005;130:489–500. doi: 10.1111/j.1365-2141.2005.05611.x. [DOI] [PubMed] [Google Scholar]
- 10.Hoover RR, Gerlach MJ, Koh EY, Daley GQ. Cooperative and redundant effects of STAT5 and Ras signaling in BCR/ABL transformed hematopoietic cells. Oncogene. 2001;20:5826–35. doi: 10.1038/sj.onc.1204549. [DOI] [PubMed] [Google Scholar]
- 11.Jin A, Kurosu T, Tsuji K, Mizuchi D, Arai A, Fujita H, et al. BCR/ABL and IL-3 activate Rap1 to stimulate the B-Raf/MEK/Erk and Akt signaling pathways and to regulate proliferation, apoptosis and adhesion. Oncogene. 2006;25:4332–40. doi: 10.1038/sj.onc.1209459. [DOI] [PubMed] [Google Scholar]
- 12.Nieborowska-Skorska M, Wasik MA, Slupianek A, Salomoni P, Kitamura T, Calabretta B, et al. Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J Exp Med. 1999;189:1229–42. doi: 10.1084/jem.189.8.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16:6151–61. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–53. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 15.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 16.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. IRIS Investigators Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–17. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 17.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 18.O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. IRIS Investigators Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 19.Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, et al. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36:453–61. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- 20.Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci U S A. 2006;103:16870–5. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fei F, Stoddart S, Müschen M, Kim YM, Groffen J, Heisterkamp N. Development of resistance to dasatinib in Bcr/Abl-positive acute lymphoblastic leukemia. Leukemia. 2010;24:813–20. doi: 10.1038/leu.2009.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ottmann O, Dombret H, Martinelli G, Simonsson B, Guilhot F, Larson RA, et al. Dasatinib induces rapid hematologic and cytogenetic responses in adult patients with Philadelphia chromosome positive acute lymphoblastic leukemia with resistance or intolerance to imatinib: interim results of a phase 2 study. Blood. 2007;110:2309–15. doi: 10.1182/blood-2007-02-073528. [DOI] [PubMed] [Google Scholar]
- 23.Redaelli S, Piazza R, Rostagno R, Magistroni V, Perini P, Marega M, et al. Activity of bosutinib, dasatinib and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J Clin Oncol. 2009;27:469–71. doi: 10.1200/JCO.2008.19.8853. [DOI] [PubMed] [Google Scholar]
- 24.Zhang JX, Han QB, Zhao AH, Sun HD. Diterpenoids from Isodon japonica. Fitoterapia. 2003;74:435–8. doi: 10.1016/S0367-326X(03)00107-2. [DOI] [PubMed] [Google Scholar]
- 25.Fujita E, Nagao Y, Node M, Kaneko K, Nakazawa S, Kuroda H. Antitumor activity of the Isodon diterpenoids: structural requirements for the activity. Experientia. 1976;32:203–6. doi: 10.1007/BF01937766. [DOI] [PubMed] [Google Scholar]
- 26.Fujita T, Takeda Y, Sun HD, Minami Y, Marunaka T, Takeda S, et al. Cytotoxic and antitumor activities of Rabdosia diterpenoids. Planta Med. 1988;54:414–7. doi: 10.1055/s-2006-962485. [DOI] [PubMed] [Google Scholar]
- 27.Cheng Y, Qiu F, Ye YC, Tashiro S, Onodera S, Ikejima T. Oridonin induces G2/M arrest and apoptosis via activating ERK-p53 apoptotic pathway and inhibiting PTK-Ras-Raf-JNK survival pathway in murine fibrosarcoma L929 cells. Arch Biochem Biophys. 2009;490:70–5. doi: 10.1016/j.abb.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Li D, Wu LJ, Tashiro S, Onodera S, Ikejima T. Oridonin inhibited the tyrosine kinase activity and induced apoptosis in human epidermoid carcinoma A431 cells. Biol Pharm Bull. 2007;30:254–60. doi: 10.1248/bpb.30.254. [DOI] [PubMed] [Google Scholar]
- 29.Li XT, Lin C, Li PY, Zhang TM. [Comparative study on the sensitivities of seven human cancer cell lines to rubescensine A] Yao Xue Xue Bao. 1985;20:243–6. [PubMed] [Google Scholar]
- 30.Chen S, Gao J, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. The cytostatic and cytotoxic effects of oridonin (Rubescenin), a diterpenoid from Rabdosia rubescens, on tumor cells of different lineage. Int J Oncol. 2005;26:579–88. [PubMed] [Google Scholar]
- 31.Ikezoe T, Chen SS, Tong XJ, Heber D, Taguchi H, Koeffler HP. Oridonin induces growth inhibition and apoptosis of a variety of human cancer cells. Int J Oncol. 2003;23:1187–93. [PubMed] [Google Scholar]
- 32.Zhang CL, Wu LJ, Zuo HJ, Tashiro S, Onodera S, Ikejima T. Cytochrome c release from oridonin-treated apoptotic A375-S2 cells is dependent on p53 and extracellular signal-regulated kinase activation. J Pharmacol Sci. 2004;96:155–63. doi: 10.1254/jphs.FPJ04008X. [DOI] [PubMed] [Google Scholar]
- 33.Zhou GB, Kang H, Wang L, Gao L, Liu P, Xie J, et al. Oridonin, a diterpenoid extracted from medicinal herbs, targets AML1-ETO fusion protein and shows potent antitumor activity with low adverse effects on t(8;21) leukemia in vitro and in vivo. Blood. 2007;109:3441–50. doi: 10.1182/blood-2006-06-032250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berenbaum MC. What is synergy? Pharmacol Rev. 1989;41:93–141. [PubMed] [Google Scholar]
- 35.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 36.Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Bäsecke J, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22:686–707. doi: 10.1038/leu.2008.26. [DOI] [PubMed] [Google Scholar]
- 37.Talapatra S, Thompson CB. Growth factor signaling in cell survival: implications for cancer treatment. J Pharmacol Exp Ther. 2001;298:873–8. [PubMed] [Google Scholar]
- 38.Teachey DT, Grupp SA, Brown VI. Mammalian target of rapamycin inhibitors and their potential role in therapy in leukaemia and other haematological malignancies. Br J Haematol. 2009;145:569–80. doi: 10.1111/j.1365-2141.2009.07657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kornblau SM, Womble M, Qiu YH, Jackson CE, Chen W, Konopleva M, et al. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006;108:2358–65. doi: 10.1182/blood-2006-02-003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med. 2010;2:98–110. doi: 10.1002/emmm.201000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klejman A, Schreiner SJ, Nieborowska-Skorska M, Slupianek A, Wilson M, Smithgall TE, et al. The Src family kinase Hck couples BCR/ABL to STAT5 activation in myeloid leukemia cells. EMBO J. 2002;21:5766–74. doi: 10.1093/emboj/cdf562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyn MA, 3rd, Wilson MB, Abdi FA, Fahey N, Schiavone AP, Wu J, et al. Src family kinases phosphorylate the Bcr-Abl SH3-SH2 region and modulate Bcr-Abl transforming activity. J Biol Chem. 2006;281:30907–16. doi: 10.1074/jbc.M605902200. [DOI] [PubMed] [Google Scholar]
- 43.Sawyers CL. Signal transduction pathways involved in BCR-ABL transformation. Baillieres Clin Haematol. 1997;10:223–31. doi: 10.1016/S0950-3536(97)80004-2. [DOI] [PubMed] [Google Scholar]
- 44.Jin S, Shen JN, Wang J, Huang G, Zhou JG. Oridonin induced apoptosis through Akt and MAPKs signaling pathways in human osteosarcoma cells. Cancer Biol Ther. 2007;6:261–8. doi: 10.4161/cbt.6.2.3621. [DOI] [PubMed] [Google Scholar]
- 45.Li D, Wu LJ, Tashiro S, Onodera S, Ikejima T. Oridonin-induced A431 cell apoptosis partially through blockage of the Ras/Raf/ERK signal pathway. J Pharmacol Sci. 2007;103:56–66. doi: 10.1254/jphs.FPJ06016X. [DOI] [PubMed] [Google Scholar]
- 46.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–42. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 47.Warsch W, Kollmann K, Eckelhart E, Fajmann S, Cerny-Reiterer S, Hölbl A, et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood. 2011;117:3409–20. doi: 10.1182/blood-2009-10-248211. [DOI] [PubMed] [Google Scholar]
- 48.Burchert A, Wang Y, Cai D, von Bubnoff N, Paschka P, Müller-Brüsselbach S, et al. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia. 2005;19:1774–82. doi: 10.1038/sj.leu.2403898. [DOI] [PubMed] [Google Scholar]
- 49.Chu S, Holtz M, Gupta M, Bhatia R. BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood. 2004;103:3167–74. doi: 10.1182/blood-2003-04-1271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.