

Abstract
Deregulation of imprinted genes is an important molecular mechanism contributing to the development of cancer in humans. However, knowledge about imprinting defects in human hepatocellular carcinoma (HCC), the third leading cause of cancer mortality worldwide, is still limited. Therefore, a systematic meta-analysis of the expression of 223 imprinted loci in human HCC was initiated. This screen revealed that the DLK1-MEG3 locus is frequently deregulated in HCC. Deregulation of DLK1 and MEG3 expression accompanied by extensive aberrations in DNA methylation could be confirmed experimentally in an independent series of human HCC (n = 40) in more than 80% of cases. Loss of methylation at the DLK1-MEG3 locus correlates linearly with global loss of DNA methylation in HCC (r2 = 0.63, p<0.0001). Inhibition of DNMT1 in HCC cells using siRNA led to a reduction in MEG3-DMR methylation and concomitant increase in MEG3 RNA expression. Allele-specific expression analysis identified loss of imprinting in 10 out of 31 informative samples (32%), rendering it one of the most frequent molecular defects in human HCC. In 2 cases unequivocal gain of bi-allelic expression accompanied by substantial loss of methylation at the IG-DMR could be demonstrated. In 8 cases the tumour cells displayed allelic switching by mono-allelic expression of the normally imprinted allele. Allelic switching was accompanied by gains or losses of DNA methylation primarily at IG-DMR1. Analysis of 10 hepatocellular adenomas (HCA) and 5 cases of focal nodular hyperplasia (FNH) confirmed that this epigenetic instability is specifically associated with the process of malignant transformation and not linked to increased proliferation per se. This widespread imprint instability in human HCC has to be considered in order to minimize unwanted side-effects of therapeutic approaches targeting the DNA methylation machinery. It might also serve in the future as predictive biomarker and for monitoring response to epigenetic therapy.
Introduction
The development of cancer in humans is not only caused by genetic lesions (mutations, deletions, translocations etc.) [1], but also by epigenetic aberrations [2]. One epigenetic phenomenon whose deregulation contributes to the development and progression of cancer in humans is imprinting, the parent-of-origin specific expression of genes. In the human genome about 200 genes are imprinted [3], displaying preferential expression of one allele or even strict mono-allelic expression.
A causal role of imprinting aberrations in human carcinogenesis is suggested by several human disorders, e.g., complete parthenogenesis in ovarian teratomas [4] and androgenic conception in hydatidiform moles [5]. Contribution of imprinting defects in cancer is best exemplified in patients with Beckwith-Wiedemann syndrome (BWS) [6], [7]. Deregulation of imprinted genes in the 11p15.5 imprinting locus caused by mutations, epimutations, or uniparental inheritance affects proliferation control in BWS patients predisposing them with a 600 fold increase in cancer risk, especially for embryonic tumours such as Wilms' tumour or hepatoblastoma [8].
Since some genes demonstrate developmental stage-specific or tissue specific imprinting [9], the study of imprinting can be complicated and the comparison of results from different studies might be misleading. The use of proper controls for the identification of imprint alterations is of uppermost importance. These experimental challenges (see also [10]) might be the reason why much less is known about imprinting defects in human cancer compared to aberrant promoter hypermethylation of tumour suppressor genes, despite the fact the imprinting defects in tumour cells have actually been described several years before the identification of tumour suppressor gene hypermethylation [11], [12].
This holds true especially for human hepatocellular carcinoma (HCC), the third leading cause of cancer mortality worldwide [13]. Only 11 publications could be identified in PubMed (from 1991 to June 2012) addressing specifically the allele-specific DNA methylation and/or expression of imprinted loci in primary human HCC specimens (excluding all animal and cell line studies) [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24]. Since the majority of these studies (7 of 11) concentrate on a single locus, i.e., IGF2/H19, we started a systematic in silico analysis of the expression of all known imprinted loci in human HCC in order to identify imprinted loci deregulated in human HCC. After validation of candidates in our own cohort, we concentrated on the analysis of the DLK1-MEG3 imprinting locus on chromosome 14q32 which is frequently deregulated in several paediatric tumours [25] and reported to have tumour suppressor activities [26], [27].
At the start of the project (end of 2010) only a single publication about MEG3 expression in human HCC could be identified (reporting no alteration in MEG3 expression in 10 HCC samples, [15]). Recently, another study analysing MEG3 expression in a small series of human HCC was published [14] which is analysed in detail in the “Discussion” section.
After screening published expression data sets for deregulated imprinted loci in human HCC we could show that the expression of the DLK1/MEG3 locus is deregulated in more than 80% of human HCC accompanied by extensive aberrations in DNA methylation.
Results
Identification of imprinted loci deregulated in human HCC
Using expression profiles deposited in the database Oncomine [28] 223 imprinted loci of the human genome were screened for deregulated expression in human HCC. The comprehensive list of imprinted loci was retrieved from the databases “Geneimprint” (http://www.geneimprint.org/) and “A Catalogue of Parent-of-Origin Effects” (www.otago.ac.nz/IGC). Within Oncomine a set of 16 expression profiles comprising altogether 953 primary human HCC specimens were identified and evaluated (Table S1). From these datasets, we identified 26 imprinted genes as down-regulated and 12 genes as up-regulated in primary human liver tumour samples and/or HCC cell lines (see Table S2). Subsequent analyses focussed on the non-coding RNA MEG3, because it showed the most frequent deregulation of expression in primary HCC according to this meta-analysis and also in an initial screen performed by ourselves (see below). Since MEG3 is part of the DLK1-MEG3 imprinting locus (see Figure 1), the expression and regulation of DLK1 was also analysed in this study.
Figure 1. Schematic representation of the DLK1-MEG3 imprinted locus on chromosome 14q32.

Maternally expressed genes are depicted in black and paternally expressed genes in grey. The intergenic differentially methylated region (IG-DMR) is located approximately 14 kb upstream to the first exon of MEG3. Studies in patients with UPD chromosome 14 revealed two regions with differential methylation at IG-DMR [30]. Therefore two pyrosequencing assays were developed at this region (IG-DMR1 and 2). In addition, three pyrosequencing assays were designed for one CpG island and two CTCF binding sites within the MEG3-DMR which was found to display parent-of-origin specific methylation (MEG3-DMR1, 2, and 3). Horizontal bar represents scale of 100 kbp.
Deregulation of DLK1 and MEG3 expression in human HCC
The expression of MEG3 and DLK1 was analysed in a series of 34 primary human HCC specimens and the corresponding adjacent liver tissue samples using quantitative real-time PCR. This revealed frequent and extensive deregulation in MEG3 RNA and DLK1 mRNA expression (Figure 2): 20 HCC samples display a MEG3 down-regulation (59%), whereas 11 samples show an increase in expression (32%). DLK1 mRNA is increased in 18 (53%) and reduced in 15 cases (44%).
Figure 2. Expression and DNA methylation of MEG3 RNA and DLK1 mRNA in primary human HCC.

MEG3 RNA (A) and DLK1 mRNA (B) expression was measured in a series of 34 paired HCC using quantitative RT-PCR. For every sample, the RNA and mRNA levels were normalized to the mean expression of GUSB and GAPDH. Expression in primary HCC samples was then normalized to the corresponding adjacent non-cancerous liver tissues. Relative expression is presented as fold change in a logarithmic scale. Samples were classified as “up-regulated” if the lower limit of the 98% confidence interval was larger than 1 and “down-regulated” if the upper limit was smaller than 1. DNA methylation levels were measured using high-resolution quantitative pyrosequencing and are displayed below the expression data as hypermethylated (black box), hypomethylated (light grey) or normomethylated (white box). For definition of thresholds see Materials and Methods. The computed methylation levels are the average of two independent pyrosequencing runs. The pyrosequencing assays for IG-DMR1, IG-DMR2, and MEG3-DMR1 contain 5 CG sites, for MEG3-DMR2 9 CG sites, for MEG3-DMR3 6 CG sites. A positive association of high methylation with reduced MEG3 expression (panel A), left part) and reduced methylation and low DLK1 expression (panel B), left part) is clearly visible.
DNA methylation patterns at the DLK1-MEG3 imprinting locus in human HCC
Since the DLK1-MEG3 locus displays imprinting and mono-allelic expression [29], [30], [31], the loss or gain of DNA methylation as a cause of deregulated expression was studied. In a panel of established HCC cell lines frequent and extensive gain or loss of DNA methylation at this locus could be demonstrated using newly established pyrosequencing assays (see Figure S1). In line with these finding also primary HCC specimens display frequent and extensive alterations in DNA methylation patterns (Fig. 2 and Figure S2). If all differentially methylated regions (DMRs) under study are considered together, 33 out of 40 samples display aberrations in DNA methylation (82.5%, Fig. 2)
If the HCC samples are sorted according to their methylation status (i.e., hypomethylated, hypermethylated, and normomethylated, for threshold definition see “Materials and Methods”) the negative correlation between the methylation at the DLK1/MEG3-DMRs and the expression of the MEG3 RNA becomes obvious (Figure 3 A), Fisher's exact test: p = 0.018). By contrast methylation at the DLK1/MEG3-DMRs is positively correlated with DLK1 mRNA expression level (Figure 3 B), Fisher's exact test: p = 0.006). This relation between an increase in DNA methylation and reduction in MEG3 expression as well as a decrease in DNA methylation and a decrease in DLK1 expression is also obvious from Figure 2.
Figure 3. Relationship between DNA methylation and gene expression at the DLK1-MEG3 locus.

Primary HCC samples are defined as “hypermethylated” and “hypomethylated” using mean ±2×SD of the methylation levels of the adjacent non-cancerous liver tissues. Relative MEG3 and DLK1 expression is then compared among hyper-, normo-, and hypo-methylated subgroups. The horizontal lines inside the box represents the median of relative MEG3 expression and the upper and lower edges of each box represent the 75th and 25th percentile respectively, while the bars denote the highest and lowest relative expression measured. * = 0.001<p<0.05, ** = 0.0001<p<0.001.
Inhibition of DNA methylation restores MEG3 expression in HCC cells
To elucidate the effect of DNA methylation on the expression of MEG3-RNA in more detail we employed inhibition of DNA methyltransferase 1 (DNMT1) and subsequent quantification of MEG3-RNA expression and methylation of the DLK1-MEG3 locus. Figure 4 A) demonstrate that the DNMT1 knockdown in HLE cells is able to induce re-expression of MEG3 accompanied by significant loss of DNA methylation at IG- and MEG3-DMR (Fig. 4 C), p<0.0001). The effectiveness of the siRNA-mediated knock-down of DNMT1 mRNA and protein in the HCC cell line HLE is shown in Figure S3.
Figure 4. Restoration of MEG3-RNA and DLK1 mRNA expression after DNMT1 knock down and reduction of methylation at MEG3 imprinting loci.

(A) Quantitative MEG3 RNA and (B) DLK1 mRNA expression levels in HLE cells after DNMT1 knockdown after normalization to GUSB and GAPDH levels (p = 0.029). (C) Reduction of DNA methylation at DLK1/MEG3 imprinting locus after DNMT1 knock down (DLK1p: DLK1 promoter). Methylation analysis was performed using quantitative pyrosequencing. Paired t-test showed significant decrease of methylation after DNMT1 knockdown for all six regions. Values shown for qRT-PCR and DNA methylation analysis are means from two independent DNMT1-knockdown experiments (50 nM and 100 nM) and two different negative control siRNAs. * = 0.001<p<0.05, *** = p<0.0001.
Deregulated DLK1-MEG3 methylation and global methylation level in human HCC
In order to study the relationship between the methylation at the DLK1-MEG3 locus and the global DNA methylation level in these tumour specimens, the latter was measured using the methylation level of the repetitive sequence LINE-1 as surrogate marker for global DNA methylation [32].
Average LINE-1 DNA methylation was significantly lower in primary HCC samples (46%±12.6%) compared to the corresponding adjacent non-cancerous liver tissue (56.7%±8.8%), (see Figure 5), p = 0.0002). The mean global methylation level of healthy liver specimens (n = 5) was nearly identical to the mean value of the liver tissues adjacent to a tumour (57.1%).
Figure 5. Global DNA methylation level in primary human liver specimens.
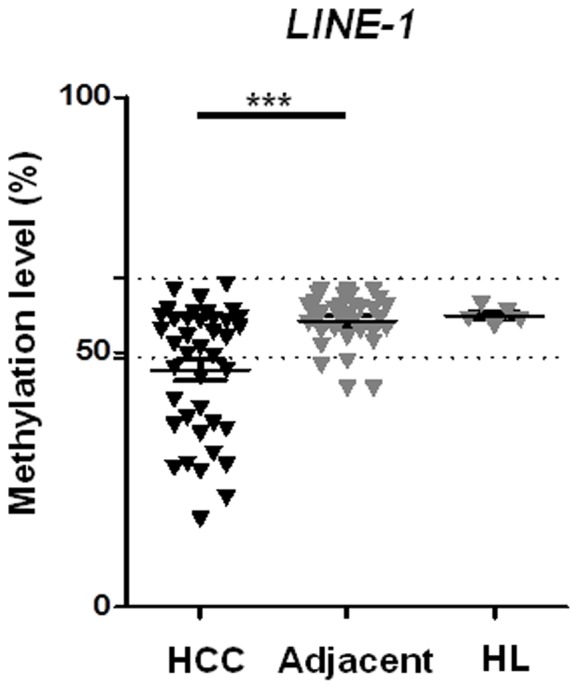
The global DNA methylation level in primary HCC samples, the corresponding adjacent non-cancerous liver tissues, as well as in unrelated healthy liver tissue was determined employing quantitative pyrosequencing of LINE1 sequences as described [40]. Methylation levels at LINE-1 locus were significantly lower in primary HCC samples compared to the corresponding adjacent non-cancerous liver tissues. Dashed lines represent cut off values defining hyper- or hypomethylation (mean+/−2× std).
The quantitative methylation levels at the IG-DMRs and the MEG3-DMRs in human specimens are overall linearly correlated with the global methylation level within these specimens (r2 = 0.24–0.63, p = 0.0012 and smaller, see figure S4). However, in several instances gain of methylation occurs in the context of global loss of methylation (e.g., #9 and #19 in Fig. 2).
DNA methylation at the DLK1-MEG3 imprinting locus in benign liver tumours
Addressing the question whether the above described aberrations in imprinting are specific for bona fide malignant proliferations and not associated with proliferation of hepatocytes per se DNA methylation patterns at the DLK1-MEG3 locus were analysed in hepatocellular adenoma (HCA, n = 10) and in focal nodular hyperplasia (FNH, n = 5) using quantitative pyrosequencing. HCA show a very limited propensity for progressing to overt malignancy [33] whereas FNH represents a clearly benign liver cell proliferation [34]. Neither showed any alteration in DNA methylation at the DLK1-MEG3 imprinting locus (figure S5). Also the global methylation level (as measured by LINE-1 methylation value) was not altered in HCA and FNH compared to adjacent liver tissue and compared to unrelated healthy liver (n = 5, figure S6).
Deregulated DLK1-MEG3 methylation leads to bi-allelic expression or allelic switching in human HCC
In order to analyse the impact of aberrant DNA methylation on the imprinting status of the DLK1-MEG3 locus, allele-specific expression analyses were performed. For this purpose SNPs located within exon 3 and exon 6 of the MEG3 gene as well as one SNP within exon 5 of the DLK1 gene as reported by Misyoshi et al. [31] and Kagami et al. [29] were analysed for the identification of informative specimens. Sequence analysis using genomic DNA from 40 primary HCC tumours revealed 16 specimens informative for MEG3 and 24 specimens informative for DLK1 (in total 31 informative cases, see Table S4 for details).
Quantitative SNP analysis of cDNA preparations from heterozygous samples employing pyrosequencing showed unequivocally bi-allelic MEG3 expression in one case and bi-allelic DLK1 expression in another case (Figure 6 A) and B)). These results were confirmed by Sanger sequencing (see Figure S7, panel A). The corresponding adjacent liver tissue displays mono-allelic expression in both cases. In addition to this well described loss of imprinting, 8 heterozygous samples displayed allelic switching (2 for MEG3, 6 for DLK1, see Figure 6 C) and D) as well as figure S8). Since in all 8 cases the genomic DNA from the tumour is clearly heterozygous (with the expected 50∶50 ratio, see Figure S8) this switch in the expressed allele cannot be due to relaxation of imprinting (resulting in bi-allelic expression) and subsequent loss of the originally imprinted allele but is caused by switching of the expressed allele. Therefore, altogether 32% (10 out of 31) of informative HCC specimens from our cohort display imprinting aberrations in the DLK1-MEG3 locus.
Figure 6. Allele-specific expression analysis of the DLK1-MEG3 locus in human HCC.

Quantitative SNP analysis was performed using pyrosequencing at a C/G polymorphism in exon 3, at SNP Rs8013873 for MEG3 and SNP Rs1802710 for DLK1. In tumour specimens showing genomic DNA polymorphisms, further SNP analysis was performed with the cDNAs from the same sample as well as with genomic and cDNA from the corresponding adjacent non-cancerous liver tissues. (A) Pyrosequencing analysis of the cDNA at SNP Rs8013873 shows mono-allelic MEG3 expression in HCC specimen #8 and bi-allelic expression in HCC specimen #21. (B) Pyrosequencing analysis of the cDNA at SNP Rs1802710 shows mono-allelic DLK1 expression in HCC specimen #33 and bi-allelic expression in HCC specimen #20. (C) MEG3 allelic switching is shown in case #25. Informative polymorphisms are found in the genomic DNAs of both tumour and the adjacent non-cancerous tissues and T/T expression in cDNA tumour but C/C expression in cDNA adjacent liver tissues. (D) DLK1 allelic switching is shown in case #30 with informative polymorphisms in the genomic DNAs of both tumour and the adjacent non-cancerous tissues and C/C expression in tumour cDNA but T/T expression in cDNA from the adjacent liver tissues. The confirmation of all genotyping results by Sanger sequencing is shown in Figure S5.
Changes in DNA methylation patterns underlying allelic switching at the DLK1-MEG3 locus in human HCC
The two samples displaying unequivocal gain of bi-allelic expression (#20, #21) indicating loss of imprinting control show substantial loss of methylation in the IG-DMR as analysed by bisulfite sequencing. Despite the absence of informative SNPs the loss of methylation in sample #20 and #21 is evident (Fig. 7 A). For comparison the methylation patterns of two samples (#4 and #9) displaying unperturbed imprinting control with retention of mono-allelic expression in the tumour cells from the same allele as in the adjacent liver tissue were analysed (Fig. 7 B). No changes in DNA methylation are discernible. The samples displaying allelic switching (#8, #15, #16, #17, #25, #26 #30, #31) indicating unstable imprinting control show gain or loss of DNA methylation at the IG-DMR1 to an intermediate extent (Fig. 7 C). Gain or loss of methylation at IG-DMR1 is the most recurrent alteration associated with allelic switching (see figure S9).
Figure 7. DNA methylation analysis of the IG-DMR1 of the DLK1-MEG3 locus in human HCC.

A) Gain of bi-allelic expression is accompanied by substantial loss of methylation. B) Tumour samples displaying retention of the original imprinting pattern by strict mono-allelic expression of the same allele as in the adjacent non-cancerous liver tissue show only minor variations in DNA methylation. C) Tumour samples demonstrating allelic switching (i.e., mono-allelic expression from the normally silenced imprinted allele) show variable changes of DNA methylation patterns to an intermediate degree. The numbers in the lower left corner of each bisulfite sequencing panel are the methylation values for the region according to pyrosequencing. “G” and “A” denote the sequence at SNP Rs12437020, which allows in heterozygous samples the differentiation of the two alleles. For the calculation of the statistical significance of the changes in DNA methylation the methylation level for each clone was calculated by computing the fraction of methylated CpG sites. From these values a mean methylation level of the tumor and adjacent liver tissue sample, respectively, was calculated. These two mean values were compared using Mann-Whitney-U-test and the p-values are depicted below the schematic representation of the bisulfite sequencing results.
Relationship of imprinting deregulation to clinical course
Despite the limited number of informative patient samples available the relationship between the above described deregulation of the DLK1-MEG3 imprinting locus and the histopathological characteristics as well as the clinical course were analysed.
The two patient showing gain of bi-allelic expression both died within very short time after diagnosis (after 1 and 8.5 months, respectively). Global loss of DNA methylation (Figure 5) correlates significantly with tumour size (>5 cm, p = 0.029), an indicator of advanced disease. However, future large collaborative studies are necessary in order to figure out in more detail the relationship between loss of imprinting and unfavourable course of disease in human HCC.
Discussion
Despite the fact that a relationship between imprinting defects and the development of cancer in humans is known for more than 20 years [35] this is still an underdeveloped area of research in comparison to e.g., the role of mutated oncogenes in human tumours. This is especially true for human HCC, the third leading cause of cancer mortality worldwide. Therefore, we initiated a systematic meta-analysis of the expression of all known imprinted loci in human HCC. Our study identifies loss of imprinting at the DLK1-MEG3 locus accompanied by deregulated expression as a common epigenetic aberration in human HCC indicating widespread epigenetic instability in this human malignancy.
Allelic switching turned out to be a frequent phenomenon in human HCC affecting 8 out of 31 informative cases (26%). This loss of proper imprinting control remains undetected in most studies, because its detection requires the analysis of the corresponding normal tissue from the same patient, as performed in a pioneering study by Mai et al. [36] These authors found switched mono-allelic expression of the p73 gene in 2 out of 12 renal cell carcinomas. However, in many studies dealing with loss of imprinting in solid tumours the imprinting status of a given gene is analysed in unaffected tissue samples from unrelated individuals and only those tumour samples which display bi-allelic expression are scored as “showing loss of imprinting”. This procedure clearly misses an unknown number of tumour samples displaying mono-allelic expression in the tumour cells but from the opposite allele compared to the adjacent normal tissue from the same patient. For myeloproliferative diseases, myelodysplastic syndrome, and overt leukaemia it is technically very difficult to obtain 100% pure cell fractions representing normal healthy haematopoiesis from the same patient. Therefore, all studies about loss of imprinting in these haematological malignancies screened by us used a series of unrelated healthy donors of bone marrow or peripheral blood for the assessment of mono-allelic expression excluding the detection of allelic switching for technical reasons. In addition, control groups with insufficient statistical power lead to incorrect conclusions about the gain or loss of imprinting [37].
For these reasons, the phenomenon of allelic switching which is an indicator of epigenetic instability is not adequately appreciated in the field and underrepresented in the literature. A study of the p73 gene in lung cancer suggests that allelic switching is a cell type- and disease-specific phenomenon, because in contrast to the situation in renal cell carcinoma no allelic switching could be found, only gain of bi-allelic expression in 5 out of 21 cases. [38]
The absence of any aberration in DNA methylation in the DLK1-MEG3 locus in altogether 15 benign liver tumours (figure S5) shows that in hepatocytes epigenetic instability is associated with the process of malignant transformation and not enhanced proliferation per se. In future studies the occurrence of these imprinting defects have to be analysed in dysplastic nodules of the liver. However, these lesions are quite rare, often very small, and cannot always be easily distinguished from well-differentiated HCC [39].
The data shown in Figure 2 show that not only loss of methylation but also substantial gain of methylation of DMRs can be observed in primary tumour specimens. Since this occurs in the context of global loss of methylation (see Figure 2 and 5) this is in line with previous publications showing that global loss of methylation and gain of methylation at specific sites are two independent processes in human HCC [40].
Several published studies and our own re-analysis of publicly available data sets confirmed allelic imbalances at 14q32 as a rare alteration in human HCC [41], [42]. Also all samples displaying allelic switching are heterozygous with the expected 50∶50 ratio as measured by quantitative pyrosequencing (see Figure S8). Therefore, allelic imbalance is very likely not the molecular mechanisms explaining the occurrence of allelic-switching. The most consistent feature in 7 out of 8 cases displaying allelic switching is an aberrant DNA methylation at IG-DMR1 (loss of methylation in 4 cases, gain in 3 cases). However, since strong decrease in methylation of IG-DMR1 is associated with gain of bi-allelic expression in case #20 and #21, additional molecular mechanisms have to be invoked.
The up-regulation of DLK1 mRNA expression after reduction of methylation in HLE cells after DNMT1 knock down (Fig. 4 B) seems to contradict the association between an increase in methylation of the DMRs and DLK1 up-regulation observed in primary specimens (Fig. 3). This discrepancy is most probably due to the fact that the DLK1 promoter is heavily methylated in HLE cells but rarely methylated in primary human HCC specimens (figure S2 F). Therefore, the activating effect of the demethylation of the DLK1 promoter (see Figure 4 C), right panel) obviously overrides the repressing effect of demethylation of IG-DMR1 and -2 in HLE cells, thereby uncoupling the correlation between IG-DMR methylation and DLK1 mRNA expression in these cells.
At the start of this project only a single publication could be identified dealing with DLK1-MEG3 locus in primary human HCC samples. However, Huang et al. [15] concentrate in their study of the DLK1-MEG3 locus in human HCC nearly exclusively on DLK1. While studying DLK1 expression using RT-PCR and immunohistochemistry in two independent and fairly large cohorts (82 and 88 cases, respectively) they measured the MEG3 RNA expression only in a small subset of samples (n = 10) and did not find any alteration. All additional experiments addressing bi-allelic expression and alterations of DNA methylation patterns deal only with DLK1 in very small groups: They found mono-allelic DLK1 expression in 3 informative cases, concluding that loss of imprinting does not play a role, and analysed the methylation of the DLK1 promoter and the IG-DMR in 6 cases (2 showing loss of methylation, 2 showing gain of methylation).
Braconi et al. [14] addressed the involvement of altered DNA methylation patterns at the DLK1-MEG3 locus indirectly by treating cells with the DNMT1 inhibitor aza-cytidine (resulting in MEG3 up-regulation in the HCC lines HepG2 and Huh-7 but not PLC/PRF-5) or knocking down DNMT1 and DNMT3b using an siRNA approach (resulting in a 1.5-fold induction of expression in HepG2 cells). In addition, they analysed the methylation of the MEG3-DMR in 11 HCC specimens using the MSP protocol published by Benetatos et al. [43] providing only very limited primary data for independent evaluation. Using the very same primer set published by Benetatos et al. we tried to correlate the quantitative pyrosequencing data with this MSP approach in our much larger cohort. In Figure S10 it is documented that it is not possible to detect the gains and losses of methylation identified by pyrosequencing in our study using the MSP protocol used by Benetatos et al. [43] and Braconi et al. [14].
The recently published study by Luk et al. about the DLK1-DIO3 locus in HCC [44] concentrates nearly exclusively on the expression of microRNAs contained within this genomic region. Analyses of the DNA methylation patterns and/or the allelic-specific expression were not performed. Only the overexpression of DLK1 mRNA in a small subset of samples is reported, without any reference to the expression of MEG3.
In contrast to our results shown in Figure 3 Khoury et al. could not show an inverse correlation of MEG3 and DLK1 expression in human acute myeloid leukaemia. [45] This shows again the cell-type specificity of imprint patterns and their deregulation in human malignancies.
In conclusion, this study demonstrates frequent epigenetic deregulation of the imprinted DLK1/MEG3 locus in human HCC leading to aberrant expression of MEG3 RNA and DLK1 mRNA. Altogether, 32% of informative samples displayed epigenetic instability rendering it one of the most frequent molecular defects in human HCC. This widespread occurrence of instability of DNA methylation patterns has to be considered for any therapeutic approach interfering with the establishment or maintenance of DNA methylation patterns (e.g. by DNMT inhibitors). On the other hand, it might serve as a useful predictive biomarker for epigenetic therapy selecting those patients most probably to respond with minimal unwanted side-effects and for monitoring response to therapy. In addition, we identified allelic switching, i.e., strict mono-allelic expression in tumour cells from the opposite allele compared to the corresponding normal tissue, as a frequent but under most circumstances overlooked indicator of epigenetic instability, which is much more prevalent than currently perceived.
Materials and Methods
Patient samples and cell lines
All primary patient samples were retrieved from the archive of the Institute of Pathology, Hanover Medical School (Germany) and analysed anonymously. The local Ethics committee (“Ethik-Kommission der Medizinischen Hochschule Hannover”, head: Prof. Dr. Tröger) exempted this study from review because all specimens under study were retrieved anonymously and retrospectively (left-over samples from diagnostic procedures) and waived the need for consent due to the fact the samples received were anonymous. Tumour cell content was determined to be greater than 70% using representative H&E sections.
Seven HCC cell lines (HLE, HLF, Huh7, HepG2, Hep3B, SNU182, and SNU387) and two immortalized hepatocyte lines (THLE2 and THLE3) were purchased from American Tissue Culture Collection (Rockville, MD, USA) and cultivated according to the supplier's recommendation.
DNA and RNA isolation
DNA was isolated by digestion with proteinase K (Merck, Darmstadt, Germany) followed by phenol/chloroform extraction from a total of 40 HCC, 10 HCA, 5 FNH, and 5 healthy liver fresh-frozen specimens (Table S3). Adjacent liver tissue from the same patient was available for 34 HCC, 8 HCA, and 2 FNH, respectively.
Total RNA from fresh frozen biopsies was extracted using TRIZOL™ reagent (Invitrogen, Darmstadt, Germany) following the protocol supplied by the manufacturer.
In silico screening of deregulated imprinted genes in HCC
To identify deregulation of imprinted genes in HCC, a total of 223 human imprinted genes listed at Geneimprint website (http://www.geneimprint.com/) and contained within ‘A Catalogue of Parent Origin Effects’ of the Otago University Dunedine, New Zealand (http://igc.otago.ac.nz/home.html) were screened using the microarray database Oncomine (https://www.oncomine.org/resource/login.html). Latest check of availability of all three web pages: 13th September 2012. Table S1 contains all studies used for meta-analysis.
Bisulfit conversion and methylation analysis
Bisulfit treatment of genomic DNAs was performed as described before [46]. Approximately 25 ng of converted DNA was amplified and used for pyrosequencing analysis as described [47]. All primers for PCR and pyrosequencing are available at Table S5. The methylation levels were computed by calculating the mean methylation level of all individual CpG sites covered by the corresponding pyrosequencing assay. At least two independent measurements were performed. Tumour samples were classified as “hypermethylated”, “hypomethylated”, and “normomethylated” if their methylation values were above, below, or within, respectively, the following range: mean+/−2× std of all adjacent liver tissue samples.
For allele-specific methylation analysis converted DNA was amplified and the PCR products were inserted into plasmid vector using TOPO-TA™ Cloning Kit (Invitrogen, Darmstadt, Germany). Individual clones of the PCR product were sequenced with GenomeLab™ Genetic Analysis System (Beckman Coulter, Brea, CA) using vector primers and GenomeLab™ DTCS Quick Start Kit (Beckman Coulter, Krefeld, Germany). For every sample at least 8 clones were sequenced.
Reverse transcription and quantitative Real Time PCR (qRT-PCR)
Reverse transcription (RT) was performed in total volume of 20 µl using 1 µg purified RNA with High Capacity cDNA Reverse Transcription Kit (Invitrogen, Darmstadt, Germany) according to the manufacture's protocol. For MEG3 and DLK1 expression analysis qRT-PCR was performed in the ABI Prism 7500 Sequence Detection System using TaqMan Gene Expression Assays™ (Invitrogen, Darmstadt, Germany) following the manufacture's protocol and βGUS and GAPDH as reference genes.
Allele-specific gene expression analysis
To evaluate allele-specific expression of MEG3 and DLK1 in primary HCC samples and HCC cell lines, polymorphisms at exonic regions showing parent-specific expression in MEG3 (Rs8013873) [31] and DLK1 (Rs1802710) [29] were analysed using pyrosequencing technology as described [48]. In heterozygous samples, further SNP analysis of the corresponding cDNA was carried out. Primers for PCR and pyrosequencing for SNP analysis are available in Table S4.
DNMT1 knockdown
DNMT1 knockdown in HLE cells was performed using pre-designed pools of four siRNA targeting DNMT1 (ON-TARGET plus SMARTpool, Dharmacon/Thermo Scientific, London, UK) following the manufacturer's protocol. In brief, 2×104 cells in 500 µl complete medium were seeded together with 100 µl of a previously prepared mixture containing 50 or 100 nM siRNA/well, Lipofectamine™ RNAiMAX (Invitrogen, Darmstadt, Germany), and Opti-MEM (Gibco-Invitrogen, Darmstadt, Germany) assembled sequentially as per manufacture's recommendation in individual wells of a 24-well plate. Medium containing transfection reagent was replaced with fresh medium after 24 h and repeated transfection was performed after 48 h from this point onward. After three times sequential transfections, cells were harvested for DNA, RNA, and protein extractions. Two scramble siRNAs (AllStars Negative Control siRNA, Qiagen, Hilden, Germany and Riboxx® control-N1, Riboxx, Dresden, Germany) were also included in the experiments as negative controls.
Western Blot analysis
Following protein concentration analysis with Bradford (Pierce, Bonn, Germany), 15 µg of total proteins from cultured cell lines after siRNA treatments were subjected to electrophoresis in 10% pre-cast SDS-polyacrylamide gels (Bio-RAD, München, Germany) and then transferred onto Hybrid-P polyvinylidene difluoride (PVDF) membrane (Amersham Biosciences, Freiburg, Germany). After blocking with 5% skim milk in phosphate buffer saline-tween, the membranes were incubated with primary and followed by secondary antibodies. The signals were subsequently detected using enhanced chemilumescent (ECL) (Pierce, Bonn, Germany). Antibodies used were mouse monoclonal anti-DNMT1 antibody (IMG-261A clone 60B1220.1, Imgenex, San Diego, CA), mouse monoclonal anti-β-actin antibody (ab6276 clone AC-15, Abcam, UK), and anti-mouse secondary antibody HRP (R1253HRP, Acris, Herford, Germany).
Statistical analysis
Statistical differences were calculated using the Mann-Whitney-U test. All calculations were performed using the software package GraphPad Prism (version 5.01 for Windows, La Jolla, CA, USA). p<0.05 were considered statistically significant.
Supporting Information
List of microarray datasets retrieved from Oncomine.
(DOC)
List of significantly up- or down-regulated imprinted loci in human HCC.
(DOC)
Patient data.
(DOC)
List of all primers used in this study.
(DOC)
DNA methylation levels at IG-DMR (1 and 2) and MEG3-DMR (1, 2 and 3) in a panel of human HCC cell lines (HLE, HLF, Huh7, HepG2, Hep3B, SNU182, SNU387) and immortalized hepatocytes (THLE2, THLE3).
(TIF)
DNA methylation of the IG-DMR 1 and 2 and MEG3-DMR 1, 2, and 3 as well as DLK1 promoter in primary human HCC. Display of all individual quantitative measurements.
(TIF)
Reduction of DNMT1 protein (A) and mRNA (B) after siRNA-mediated knock down.
(TIF)
Correlation between DLK1/MEG3 DMRs and global methylation (measured as LINE-1 methylation values).
(TIF)
DNA methylation analysis of DLK1/MEG3 imprinting locus in benign liver tumours. IG-DMR 1 (A), IG-DMR 2 (B), MEG3-DMR 1(C), MEG3-DMR 2 (D), MEG3-DMR 3 (E), and DLK1 promoter (F).
(TIF)
LINE-1 methylation in HCA (n = 10) and FNH (n = 5) and in corresponding adjacent liver tissue as well as in unrelated healthy liver tissues (n = 5).
(TIF)
Sanger sequencing of cDNA confirming gain of bi-allelic expression (A) and allelic switching (B) in tumour samples.
(TIF)
Quantitative SNP analysis of the genomic and cDNAs from the tumour specimens displaying allelic switching for MEG3 (A) and DLK1 (B–F).
(TIF)
Bisulfite sequencing of MEG3 DMR1 – 3 for the 8 tumours displaying allelic switching.
(TIF)
Methylation analysis using MSP with primers described by Benetatos et al. [43] , and also used by Braconi et al. [14] .
(DOC)
Acknowledgments
The authors would like to thank Florian Puls and Jan Becker for initial help with retrieving and reviewing samples included in this study.
Funding Statement
This work was supported by a grant from the German Research Council within the Hannover-Heidelberg Transregio-SFB TRR 77 “Liver cancer” (Project B1). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Knudson AG (2002) Cancer genetics. Am J Med Genet 111: 96–102. [DOI] [PubMed] [Google Scholar]
- 2. Iacobuzio-Donahue CA (2008) Epigenetic Changes in Cancer. Annu Rev Pathol [DOI] [PubMed] [Google Scholar]
- 3. Morison IM, Ramsay JP, Spencer HG (2005) A census of mammalian imprinting. Trends in genetics : TIG 21: 457–465. [DOI] [PubMed] [Google Scholar]
- 4. Varmuza S, Mann M (1994) Genomic imprinting–defusing the ovarian time bomb. Trends Genet 10: 118–123. [DOI] [PubMed] [Google Scholar]
- 5. Youngson NA, Whitelaw E (2008) Transgenerational epigenetic effects. Annu Rev Genomics Hum Genet 9: 233–257. [DOI] [PubMed] [Google Scholar]
- 6. Morison IM, Becroft DM, Taniguchi T, Woods CG, Reeve AE (1996) Somatic overgrowth associated with overexpression of insulin-like growth factor II. Nat Med 2: 311–316. [DOI] [PubMed] [Google Scholar]
- 7. Weksberg R, Shuman C, Beckwith JB (2010) Beckwith-Wiedemann syndrome. Eur J Hum Genet 18: 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Engel JR, Smallwood A, Harper A, Higgins MJ, Oshimura M, et al. (2000) Epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. Journal of medical genetics 37: 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schulz R, McCole RB, Woodfine K, Wood AJ, Chahal M, et al. (2009) Transcript- and tissue-specific imprinting of a tumour suppressor gene. Hum Mol Genet 18: 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DeVeale B, van der Kooy D, Babak T (2012) Critical evaluation of imprinted gene expression by RNA-Seq: a new perspective. PLoS Genet 8: e1002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Greger V, Passarge E, Hopping W, Messmer E, Horsthemke B (1989) Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet 83: 155–158. [DOI] [PubMed] [Google Scholar]
- 12. Schroeder WT, Chao LY, Dao DD, Strong LC, Pathak S, et al. (1987) Nonrandom loss of maternal chromosome 11 alleles in Wilms tumors. Am J Hum Genet 40: 413–420. [PMC free article] [PubMed] [Google Scholar]
- 13. Yang JD, Roberts LR (2010) Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol 7: 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Braconi C, Kogure T, Valeri N, Huang N, Nuovo G, et al. (2011) microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene 30: 4750–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang J, Zhang X, Zhang M, Zhu JD, Zhang YL, et al. (2007) Up-regulation of DLK1 as an imprinted gene could contribute to human hepatocellular carcinoma. Carcinogenesis 28: 1094–1103. [DOI] [PubMed] [Google Scholar]
- 16. Kim KS, Lee YI (1997) Biallelic expression of the H19 and IGF2 genes in hepatocellular carcinoma. Cancer Lett 119: 143–148. [DOI] [PubMed] [Google Scholar]
- 17. Li X, Nong Z, Ekstrom C, Larsson E, Nordlinder H, et al. (1997) Disrupted IGF2 promoter control by silencing of promoter P1 in human hepatocellular carcinoma. Cancer Res 57: 2048–2054. [PubMed] [Google Scholar]
- 18. Midorikawa Y, Yamamoto S, Ishikawa S, Kamimura N, Igarashi H, et al. (2006) Molecular karyotyping of human hepatocellular carcinoma using single-nucleotide polymorphism arrays. Oncogene 25: 5581–5590. [DOI] [PubMed] [Google Scholar]
- 19. Poirier K, Chalas C, Tissier F, Couvert P, Mallet V, et al. (2003) Loss of parental-specific methylation at the IGF2 locus in human hepatocellular carcinoma. J Pathol 201: 473–479. [DOI] [PubMed] [Google Scholar]
- 20. Scelfo RA, Schwienbacher C, Veronese A, Gramantieri L, Bolondi L, et al. (2002) Loss of methylation at chromosome 11p15.5 is common in human adult tumors. Oncogene 21: 2564–2572. [DOI] [PubMed] [Google Scholar]
- 21. Schwienbacher C, Gramantieri L, Scelfo R, Veronese A, Calin GA, et al. (2000) Gain of imprinting at chromosome 11p15: A pathogenetic mechanism identified in human hepatocarcinomas. Proc Natl Acad Sci U S A 97: 5445–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takeda S, Kondo M, Kumada T, Koshikawa T, Ueda R, et al. (1996) Allelic-expression imbalance of the insulin-like growth factor 2 gene in hepatocellular carcinoma and underlying disease. Oncogene 12: 1589–1592. [PubMed] [Google Scholar]
- 23. Uchida K, Kondo M, Takeda S, Osada H, Takahashi T, et al. (1997) Altered transcriptional regulation of the insulin-like growth factor 2 gene in human hepatocellular carcinoma. Mol Carcinog 18: 193–198. [PubMed] [Google Scholar]
- 24. Wu J, Qin Y, Li B, He WZ, Sun ZL (2008) Hypomethylated and hypermethylated profiles of H19DMR are associated with the aberrant imprinting of IGF2 and H19 in human hepatocellular carcinoma. Genomics 91: 443–450. [DOI] [PubMed] [Google Scholar]
- 25. Astuti D, Latif F, Wagner K, Gentle D, Cooper WN, et al. (2005) Epigenetic alteration at the DLK1-GTL2 imprinted domain in human neoplasia: analysis of neuroblastoma, phaeochromocytoma and Wilms' tumour. Br J Cancer 92: 1574–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao J, Dahle D, Zhou Y, Zhang X, Klibanski A (2005) Hypermethylation of the promoter region is associated with the loss of MEG3 gene expression in human pituitary tumors. J Clin Endocrinol Metab 90: 2179–2186. [DOI] [PubMed] [Google Scholar]
- 27. Zhou Y, Zhang X, Klibanski A (2012) MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol 48: R45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, et al. (2007) Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 9: 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kagami M, O'Sullivan MJ, Green AJ, Watabe Y, Arisaka O, et al. (2010) The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet 6: e1000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kagami M, Sekita Y, Nishimura G, Irie M, Kato F, et al. (2008) Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat Genet 40: 237–242. [DOI] [PubMed] [Google Scholar]
- 31. Miyoshi N, Wagatsuma H, Wakana S, Shiroishi T, Nomura M, et al. (2000) Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells 5: 211–220. [DOI] [PubMed] [Google Scholar]
- 32. Irahara N, Nosho K, Baba Y, Shima K, Lindeman NI, et al. (2010) Precision of pyrosequencing assay to measure LINE-1 methylation in colon cancer, normal colonic mucosa, and peripheral blood cells. J Mol Diagn 12: 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stoot JH, Coelen RJ, De Jong MC, Dejong CH (2010) Malignant transformation of hepatocellular adenomas into hepatocellular carcinomas: a systematic review including more than 1600 adenoma cases. HPB (Oxford) 12: 509–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nahm CB, Ng K, Lockie P, Samra JS, Hugh TJ (2011) Focal nodular hyperplasia–a review of myths and truths. J Gastrointest Surg 15: 2275–2283. [DOI] [PubMed] [Google Scholar]
- 35. Wilkins RJ (1988) Genomic imprinting and carcinogenesis. Lancet 1: 329–331. [DOI] [PubMed] [Google Scholar]
- 36. Mai M, Qian C, Yokomizo A, Tindall DJ, Bostwick D, et al. (1998) Loss of imprinting and allele switching of p73 in renal cell carcinoma. Oncogene 17: 1739–1741. [DOI] [PubMed] [Google Scholar]
- 37. Tessema M, Langer F, Bock O, Seltsam A, Metzig K, et al. (2005) Down-regulation of the IGF-2/H19 locus during normal and malignant hematopoiesis is independent of the imprinting pattern. Int J Oncol 26: 499–507. [PubMed] [Google Scholar]
- 38. Mai M, Yokomizo A, Qian C, Yang P, Tindall DJ, et al. (1998) Activation of p73 silent allele in lung cancer. Cancer Res 58: 2347–2349. [PubMed] [Google Scholar]
- 39. Park YN (2011) Update on precursor and early lesions of hepatocellular carcinomas. Archives of pathology & laboratory medicine 135: 704–715. [DOI] [PubMed] [Google Scholar]
- 40. Trankenschuh W, Puls F, Christgen M, Albat C, Heim A, et al. (2010) Frequent and distinct aberrations of DNA methylation patterns in fibrolamellar carcinoma of the liver. PLoS One 5: e13688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, et al. (2008) Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res 68: 6779–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roessler S, Jia HL, Budhu A, Forgues M, Ye QH, et al. (2010) A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res 70: 10202–10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Benetatos L, Hatzimichael E, Dasoula A, Dranitsaris G, Tsiara S, et al. (2009) CpG methylation analysis of the MEG3 and SNRPN imprinted genes in acute myeloid leukemia and myelodysplastic syndromes. Leuk Res 34: 148–153. [DOI] [PubMed] [Google Scholar]
- 44. Luk JM, Burchard J, Zhang C, Liu AM, Wong KF, et al. (2011) DLK1-DIO3 genomic imprinted microRNA cluster at 14q32.2 defines a stemlike subtype of hepatocellular carcinoma associated with poor survival. J Biol Chem 286: 30706–30713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khoury H, Suarez-Saiz F, Wu S, Minden MD (2010) An upstream insulator regulates DLK1 imprinting in AML. Blood 115: 2260–2263. [DOI] [PubMed] [Google Scholar]
- 46. Lehmann U, Hasemeier B, Lilischkis R, Kreipe H (2001) Quantitative analysis of promoter hypermethylation in laser-microdissected archival specimens. Lab Invest 81: 635–638. [DOI] [PubMed] [Google Scholar]
- 47. Potapova A, Albat C, Hasemeier B, Haeussler K, Lamprecht S, et al. (2011) Systematic cross-validation of 454 sequencing and pyrosequencing for the exact quantification of DNA methylation patterns with single CpG resolution. BMC biotechnology 11: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Royo JL, Galan JJ (2009) Pyrosequencing for SNP genotyping. Methods Mol Biol 578: 123–133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of microarray datasets retrieved from Oncomine.
(DOC)
List of significantly up- or down-regulated imprinted loci in human HCC.
(DOC)
Patient data.
(DOC)
List of all primers used in this study.
(DOC)
DNA methylation levels at IG-DMR (1 and 2) and MEG3-DMR (1, 2 and 3) in a panel of human HCC cell lines (HLE, HLF, Huh7, HepG2, Hep3B, SNU182, SNU387) and immortalized hepatocytes (THLE2, THLE3).
(TIF)
DNA methylation of the IG-DMR 1 and 2 and MEG3-DMR 1, 2, and 3 as well as DLK1 promoter in primary human HCC. Display of all individual quantitative measurements.
(TIF)
Reduction of DNMT1 protein (A) and mRNA (B) after siRNA-mediated knock down.
(TIF)
Correlation between DLK1/MEG3 DMRs and global methylation (measured as LINE-1 methylation values).
(TIF)
DNA methylation analysis of DLK1/MEG3 imprinting locus in benign liver tumours. IG-DMR 1 (A), IG-DMR 2 (B), MEG3-DMR 1(C), MEG3-DMR 2 (D), MEG3-DMR 3 (E), and DLK1 promoter (F).
(TIF)
LINE-1 methylation in HCA (n = 10) and FNH (n = 5) and in corresponding adjacent liver tissue as well as in unrelated healthy liver tissues (n = 5).
(TIF)
Sanger sequencing of cDNA confirming gain of bi-allelic expression (A) and allelic switching (B) in tumour samples.
(TIF)
Quantitative SNP analysis of the genomic and cDNAs from the tumour specimens displaying allelic switching for MEG3 (A) and DLK1 (B–F).
(TIF)
Bisulfite sequencing of MEG3 DMR1 – 3 for the 8 tumours displaying allelic switching.
(TIF)
Methylation analysis using MSP with primers described by Benetatos et al. [43] , and also used by Braconi et al. [14] .
(DOC)