

Abstract
UDP-galactopyranose mutase (UGM) plays an essential role in galactofuranose biosynthesis in microorganisms by catalyzing the conversion of UDP-galactopyranose to UDP-galactofuranose. The enzyme has gained attention recently as a promising target for the design of new antifungal, antitrypanosomal, and antileishmanial agents. Here we report the first crystal structure of UGM complexed with its redox partner NAD(P)H. Kinetic protein crystallography was used to obtain structures of oxidized Aspergillus fumigatus UGM (AfUGM) complexed with NADPH and NADH, as well as reduced AfUGM after dissociation of NADP+. NAD(P)H binds with the nicotinamide near the FAD isoalloxazine and the ADP moiety extending toward the mobile 200s active site flap. The nicotinamide riboside binding site overlaps that of the substrate galactopyranose moiety, thus NADPH and substrate binding are mutually exclusive. On the other hand, the pockets for the adenine of NADPH and uracil of the substrate are distinct and separated by only 6 Å, which raises the possibility of designing novel inhibitors that bind both sites. All twelve residues that contact NADP(H) are conserved among eukaryotic UGMs. Residues that form the AMP pocket are absent in bacterial UGMs, which suggests that eukaryotic and bacterial UGMs have different NADP(H) binding sites. The structures address the longstanding question of how UGM binds NAD(P)H and provide new opportunities for drug discovery.
INTRODUCTION
UDP-galactopyranose mutase (UGM) catalyzes the conversion of UDP-galactopyranose (UDP-Galp) to UDP-galactofuranose (UDP-Galf), which is a central step in Galf biosynthesis (Figure 1A).1 The enzyme has attracted interest as a drug target because Galf is an essential building block of the cell wall and extracellular matrix of many bacteria, fungi, and protozoa, but Galf and UGM are absent in humans. In particular, eukaryotic UGMs have emerged recently as targets for the design of antifungal, antitrypanosomal, and antileishmanial agents.2–4 For example, gene deletion studies have shown that UGM is essential for the virulence of the pathogenic fungus Aspergillus fumigatus and the protozoan parasite Leishmania major.5,6 Thus, there is interest in characterizing the ligand binding sites of UGMs to facilitate drug discovery.
Figure 1.
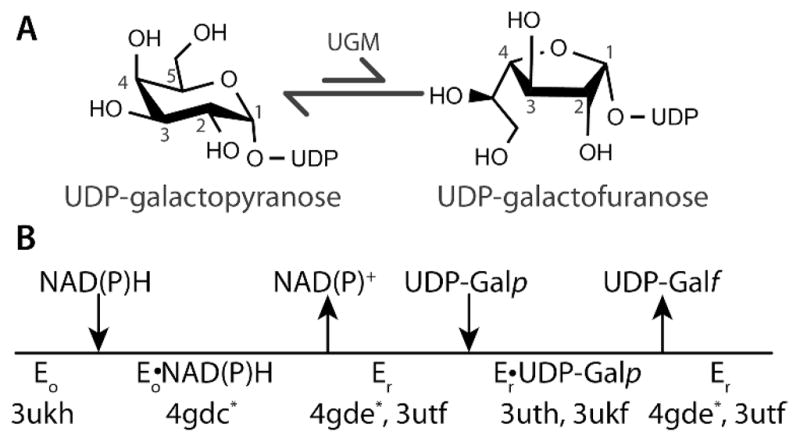
(A) Reaction catalyzed by UGM. (B) Catalytic scheme indicating the PDB codes of AfUGM crystal structures. Asterisks denote the structures reported here.
UGM is an atypical flavoenzyme in that it does not catalyze an oxidation-reduction reaction, and thus the flavin redox state is unchanged by the transformation of substrate to product.7 Rather than serving as a redox center, the flavin cofactor in UGM is a nucleophile that attacks the anomeric carbon atom of the substrate (C1 in Figure 1A).8–10 The role as nucleophile requires that the flavin be reduced (FADH−) in the resting state of the enzyme.
The requirement of FADH− in UGM raises the question of what is the physiological reductant of the enzyme. Reduced nicotinamide adenine dinucleotides (NAD(P)H) are logical candidates because of their ubiquity in biology and known reactivity with a multitude of flavoenzymes, including flavin reductases and flavin monooxygenases.11 Indeed, early studies TABLE 1. showed that NADPH and NADH reduce UGM from Klebsiella pneumoniae reacts, albeit relatively slowly.12,13 More recently, we showed that NADPH and NADH are effective redox partners of Trypanosoma cruzi UGM (TcUGM).10 These studies suggest that reduced nicotinamide adenine dinucleotides may be the physiological reductant of some UGMs. However, an NAD(P)H binding motif is not evident in UGM sequences, and the location of the NAD(P)H binding site has remained elusive despite the availability of dozens of crystal structures of bacterial14–19 and eukaryotic20–22 UGMs. To address this outstanding issue of UGM biochemistry, we have determined the crystal structures of UGM from the pathogenic fungus Aspergillus fumigatus (AfUGM) complexed with NADPH and NADH.
Table 1.
X-ray Diffraction Data Collection and Refinementa
| AfUGMo-NADPH | AfUGMo-NADH | AfUGMr | |
|---|---|---|---|
| Data collection
| |||
| Soaking ligand | NADPH | NADH | NADPH |
| Soaking time (min.) | 1 | 1 | 30 |
| Flavin redox state | oxidized | oxidized | reduced |
| Space group | P6522 | P6522 | P6522 |
| Unit cell parameters (Å) | a =218.3, c = 319.2 | a =218.3, c = 318.1 | a = 217.4, c = 319.9 |
| Wavelength (Å) | 0.979 | 0.979 | 0.979 |
| Resolution (Å) | 163-2.75 (2.90-2.75) | 162-2.75 (2.90-2.75) | 50-2.20 (2.32-2.20) |
| Observations | 851567 | 982507 | 984169 |
| Unique reflections | 116001 | 115207 | 222394 |
| Rmerge(I)b | 0.137 (0.885) | 0.158 (1.216) | 0.093 (0.673) |
| Rmeas(I)b | 0.147 (0.967) | 0.168 (1.297) | 0.106 (0.778) |
| Rpim(I)b | 0.054 (0.382) | 0.054 (0.429) | 0.049 (0.383) |
| Mean I/σ | 11.6 (2.2) | 10.9 (2.1) | 9.8 (2.0) |
| Completeness (%) | 99.9 (99.9) | 99.6 (99.9) | 99.7 (99.8) |
| Multiplicity | 7.3 (6.1) | 8.5 (7.9) | 4.4 (4.0) |
|
| |||
| Refinement
| |||
| Resolution (Å) | 163-2.75 (2.78-2.75) | 162-2.75 (2.78-2.75) | 48-2.20 (2.28-2.20) |
| No. of protein residues | 1987 | 1989 | 2017 |
| No. of protein atoms | 15008 | 15164 | 15526 |
| No. of FAD atoms | 212 | 212 | 212 |
| No. of NAD(P)H molecules/atoms | 4/158 | 2/88 | 0/0 |
| NAD(P)H occupancyc | 0.7–0.9 | 0.9 | - |
| No. of water molecules | 16 | 26 | 435 |
| Rcryst | 0.210 (0.299) | 0.212 (0.404) | 0.201 (0.293) |
| Rfreed | 0.246 (0.318) | 0.245 (0.391) | 0.227 (0.343) |
| rmsd bond lengths (Å)e | 0.008 | 0.008 | 0.007 |
| rmsd bond angles (°)e | 1.17 | 1.19 | 1.05 |
| Ramachandran plotf | |||
| Favored (no. residues) | 1908 | 1919 | 1978 |
| Allowed (no. residues) | 59 | 50 | 31 |
| Outliers (no. residues) | 0 | 0 | 0 |
| Average B-factor (Å2) | |||
| Protein | 53 | 51 | 41 |
| FAD | 52 | 53 | 37 |
| NAD(P)Hc | 48–72 | 50–52 | - |
| Water | 36 | 41 | 37 |
| Coordinate error (Å)g | 0.41 | 0.51 | 0.36 |
| PDB code | 4gdc | 4gdd | 4gde |
Values for the outer resolution shell of data are given in parenthesis.
Definitions of Rmerge, Rmeas, and Rpim can be found in Weiss.23
Range reflects the ligands bound to different chains of the tetramer.
A common set of test reflections (5 %) was used for refinement of all structures.
Compared to the parameters of Engh and Huber.24
The Ramachandran plot was generated with RAMPAGE.25
Maximum likelihood-based coordinate error estimate from PHENIX.
EXPERIMENTAL SECTION
Crystal Soaking Experiments
Methods for the crystallization of oxidized AfUGM have been described;20 see Supporting Information (SI) for details. Kinetic protein crystallography26 was used to determine structures of oxidized AfUGM (AfUGMo) complexed with NADPH or NADH, and reduced AfUGM (AfUGMr) after dissociation of NADP+. The soaking time and reductant concentration were varied to find appropriate soaking conditions. The AfUGMo–NAD(P)H complexes were obtained by soaking the crystals for ~1 minute in cryobuffer (1.4 M ammonium sulfate, 0.1 M sodium acetate, and 25% ethylene glycol at pH 4.5) containing 120 – 140 mM NADPH or NADH prior to flash-cooling in liquid nitrogen. The crystals remained yellow during this relatively short soaking time, which is consistent with the oxidized state of the FAD. The structure of AfUGMr without NADP+ bound was determined from a crystal soaked for 30 minutes in the cryobuffer containing 100 mM NADPH. Over this timescale, the yellow color was completely bleached, which is indicative of full reduction of the crystalline enzyme.
X-ray Diffraction Data Collection and Refinement
Diffraction data were collected on beamlines 24-ID-C and 24-IDE of the Advanced Photon Source. The data were integrated using XDS27 and scaled with SCALA.28 Data processing statistics are listed in Table 1.
Crystallographic refinement calculations were initiated from coordinates derived from the 2.25 Å resolution structure of AfUGM (Protein Data Bank (PDB) code 3utf). These calculations were performed with PHENIX,29 and the B-factor model consisted of an isotropic B-factor for each atom and TLS refinement using one TLS group per protein chain. NCS restraints were used in the refinement of the 2.75 Å resolution structures. COOT was used for model building.30 The ligand occupancy (q) was estimated by inspecting the ligand B-factors after refinements performed with the ligand occupancy fixed at various values. The optimal occupancy was chosen as that which produced ligand B-factors that were commensurate with the average B-factor of the structure. Refinement statistics are listed in Table 1. Coordinates and structure factor amplitudes have been deposited in the PDB under the accession numbers listed in Table 1.
Mutagenesis and Kinetics
Site-directed mutants of AfUGM were created using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) following the protocol supplied by the manufacturer. All the mutant enzymes were expressed and purified following the procedures for AfUGM.31
AfUGM and AfUGM mutant enzymes were analyzed using stopped-flow kinetics experiments as described previously for TcUGM.10 In these experiments, the enzyme and NAD(P)H were mixed under anaerobic conditions at 15 °C and pH 7.0, and the reaction was monitored using the flavin absorbance at 452 nm. The time course of the absorbance was fit to a single exponential to obtain an observed rate constant, kobs. Kinetic parameters for the reduction of the enzyme by NAD(P)H were obtained by fitting the kobs values to the equation, kobs = kred[NAD(P)H]/(Kd + [NAD(P)H]), where kred is the rate constant for flavin reduction, and Kd is the dissociation constant for NAD(P)H. The synthesis of deuterated coenzymes for kinetic isotope effect (KIE) measurements is described in SI.
RESULTS
Kinetics of Enzyme Activation by NAD(P)H
We first established that NAD(P)H reduces AfUGM (Table 2). The rate constant for reduction (kred) by NADPH is 3 s−1, and the dissociation constant for NADPH (Kd) is 25 μM. The catalytic efficiency for NADPH is thus 120000 M−1s−1, which is 20 times higher than that of TcUGM10 (kred = 0.6 s−1, Kd = 98 μM, kred/Kd = 6000 M−1s−1). The kinetic constants for the reaction of AfUGM with NADH are kred of 0.2 s−1 and Kd of 260 μM (kred/Kd=670 M−1s−1). These results show that NAD(P)H activates AfUGM in vitro and that AfUGM, like TcUGM, exhibits a preference for NADPH.
TABLE 2.
Kinetic Parameters for the Reduction of AfUGM Mutant Enzymes by NAD(P)H
| kred (s−1) | Kd (μM) | kred/Kd (M−1 s−1) | Variational Effect (kred/Kd of variant)/(kred/Kd of AfUGM) | |
|---|---|---|---|---|
| NADPH | ||||
| AfUGM | 2.98 ± 0.078 | 25 ± 2 | 120000 ± 10000 | |
| R447A | 0.00240 ± 0.00007 | 39 ± 6 | 60 ± 10 | 0.0005 ± 0.0001 |
| R91A | 0.313 ± 0.008 | 330 ± 20 | 980 ± 70 | 0.008 ± 0.001 |
| S93A | 0.17 ± 0.01 | 21 ± 8 | 8000 ± 3000 | 0.07 ± 0.03 |
| Y317A | 3.36 ± 0.09 | 100 ± 10 | 34000 ± 3000 | 0.28 ± 0.03 |
| Y104A | 2.20 ± 0.04 | 54 ± 4 | 41000 ± 3000 | 0.34 ± 0.04 |
| NADH | ||||
| AfUGM | 0.172 ± 0.003 | 260 ± 20 | 670 ± 50 | |
The stereochemistry of hydride transfer for AfUGM was determined using KIE measurements (Table S1). When the pro-R H atom was substituted with D, KIE values on flavin reduction of 3.0 ± 0.1 and 6.0 ± 0.7 were observed for NADPH and NADH, respectively. In contrast, a KIE value of 0.93 ± 0.03 was measured when pro-S NADPD was used to reduce AfUGM. These results show that the pro-R hydride of NADPH is transferred to AfUGM. KIE measurements also indicate pro-R stereochemistry for TcUGM (Table S1).
The NAD(P)H Binding Site
Kinetic protein crystallography26 was used to obtain structures of AfUGM relevant to enzyme activation by NADP(H) (Figure 1B). Short soaks (< 1 min.) of crystals in ~ 100 mM NAD(P)H followed by freeze-trapping in liquid nitrogen yielded 2.75 Å resolution structures of the oxidized enzyme (AfUGMo) complexed with NADPH and NADH. The two structures are very similar and have a root mean square deviation (RMSD) of 0.31 Å for the Cα atoms of the tetramer. The pairwise RMSDs for the individual chains of the two structures span the range 0.17 – 0.51 Å.
Electron density maps clearly indicated the presence of NADPH bound in the active site (Figure 2B). The occupancy of NADPH varied among the protomers of the tetramer, with the strongest density appearing in chains A and B. We note that this pattern of differential ligand occupancy (high in protomers A and B, low in protomers C and D) was observed in our studies of substrate and inhibitor binding to AfUGM, which also involved soaking the P6522 crystal form used here.20 The maps allowed the building of complete models of NADPH at nearly full occupancy (q = 0.9) in chains A and B (Figure 2B). In chains C and D of the tetramer, the density was strong for the ADP group but weaker for the nicotinamide riboside group. Therefore, only the ADP part of NADPH (q = 0.7) was modeled in chains C and D.
Figure 2.

Crystal structure of AfUGMo complexed with NADPH. (A) Protomer structure. FAD and NADPH are colored yellow and pink, respectively. Domains 1, 2, and 3 are colored blue, yellow, and green, respectively. (B) Stereographic view of the NADPH binding site. The cage represents a simulated annealing σA-weighted Fo − Fc omit map (3.0 σ). Prior to calculating the map, NADPH was omitted and simulated annealing refinement was performed.
Strong density was also observed for NADH (Figure S1A). As in the AfUGMo-NADPH structure, differential ligand occupancy was evident. Complete models of NADH were built at q = 0.9 in chains A and B, while NADH was omitted in chains C and D.
NAD(P)H binds to AfUGMo at the intersection of the three protein domains with the nicotinamide near the re face of the isoalloxazine and the rest of the dinucleotide directed toward the 200s substrate-binding flap (Figure 2A). The nicotinamide riboside binding site overlaps that of the Galp moiety of the substrate (Figure 3A), thus NAD(P)H and substrate binding are mutually exclusive. This result is expected since both NAD(P)H and UDP-Galp require access to the flavin N5 atom at the re face of the isoalloxazine. On the other hand, the AMP group of NAD(P)H and the UMP group of the substrate bind in different pockets on either side of Tyr104 (Figure 3A).
Figure 3.

Spatial proximity of the NADPH and substrate binding sites (stereographic views). In both panels, the view is from the outside of the enzyme looking into the active site. (A) The active site surface of AfUGMo-NADPH. FAD and NADPH are colored yellow and pink, respectively. For reference, the AfUGMo-NADPH structure has been overlaid with the AfUGMr-UDP-Galp complex (PDB code 3uth20) and the UDP-Galp (green) included in this image. The dashed yellow line denotes the 6.3 Å distance between the adenine and uracil sites. (B) The active site surface of AfUGMr. For reference, the AfUGMo-NADPH structure has been overlaid with AfUGMr and the NADPH (pink) included in this image. Note that the adenosine pocket is also present in the reduced enzyme.
NAD(P)H forms several interactions with AfUGMo (Figures 2B and S1). The adenosine moiety occupies a hydrophobic pocket formed by Ile65, Phe66, Tyr104, and the non-polar chain of Arg91. The latter residue serves as the floor of the adenosine binding pocket along with the main chain of Ile92 and the side chain of Ser93. The adenine base forms hydrogen bonds with Ser93 and Tyr317. Tyr104 of the pocket makes a hydrogen bond with the 2′-phosphoryl of NADPH. This side chain is rotated slightly in the NADH complex because of the absence of the 2′-phosphoryl in NADH (Figure S1B). Otherwise, the binding sites of the NADPH and NADH complexes are identical within experimental error (Figure S1B). The pyrophosphate is stabilized by electrostatic interactions with His68 and Asn457. Finally, the nicotinamide ring is wedged between Tyr453 and Asn457, and its carboxamide forms hydrogen bonds with Arg447 and Tyr419.
The nicotinamide is not optimally aligned for hydride transfer in the AfUGMo-NADPH and AfUGMo-NADH complexes. For reference, glutathione reductase, which also binds FAD using a Rossmann fold domain, demonstrates the expected orientation of the nicotinamide relative to the isoalloxazine (PDB code 1grb32). In glutathione reductase, the nicotinamide stacks in parallel with the middle ring of the isoalloxazine such that the hydride transfer partners - C4 of NADPH and N5 of FAD - are separated by 3.2 Å. In AfUGM, the nicotinamide and isoalloxazine rings are not parallel, and the C4-N5 distance is 5.8 Å (Figures 2B and S1). The observed arrangement of the hydride transfer partners may result from the adventitious binding of a sulfate ion in the active site (Figures 2B and S1). Alternatively, the observed complex could represent a transient species that precedes the active hydride transfer complex. This possibility is supported by the observation that the 30-min. soak in NADPH generated the reduced enzyme with the dinucleotide dissociated (vide infra), implying that NADPH moves from the observed conformation to the active one. Baeyer-Villiger monooxygenases provide precedent for this phenomenon, which is known as the “sliding cofactor” model.33,34
Guided by the constraint of pro-R stereochemistry determined from KIE measurements, we generated a model of the hydride transfer complex. Using only dihedral angle rotations and keeping the ADP fixed, it was possible to bring the re face of the nicotinamide in contact with the isoalloxazine such that the hydride transfer partners are separated by 3.1 Å (Figure S2).
Assignment of the Flavin Redox State
As described previously, the conformations of oxidized and dithionite-reduced AfUGM are dramatically different, and these structural differences can be used to deduce the flavin redox state in lieu of microspectrophotometry.20–22 Briefly, the oxidation state of the FAD can be deduced from the conformation of the conserved histidine loop (Gly61-Gly62-His63), the identity of the hydrogen-bonding partner of the flavin N5 (Arg327 for FAD, Gly62 for FADH−), the orientation of Trp315, and the curvature of the isoalloxazine (planar for FAD, bent by 7° for FADH−).
To establish the conformation of the NADPH-reduced enzyme, the 2.2 Å resolution structure of AfUGM was determined from a crystal of the oxidized enzyme that had been soaked for 30 minutes in 100 mM NADPH. The yellow color of the crystal was completely bleached, which is indicative of full reduction of the crystalline enzyme. The NADPH-reduced and dithionite-reduced (PDB code 3utf) structures are very similar (RMSD = 0.24 Å). Furthermore, the structural indicators of the flavin redox state in all four chains of the NADPH-reduced enzyme are consistent with the reduced flavin state. In particular, His63 is located on the si face of the isoalloxazine, where it forms hydrogen bonds with Gly61 and the ribityl chain (Figure 4A). Furthermore, Gly62 accepts a hydrogen bond from the flavin N5 atom. This interaction is diagnostic of the reduced flavin because the main chain carbonyl is an obligate hydrogen bond acceptor, and N5 is a donor only in the reduced state. Lastly, at 2.2 Å resolution it is possible to discern the 7° bend of the isoalloxazine (Figure 4A). All of these structural features are also observed in the structure of dithionite-reduced AfUGM, as shown by an overlay of the NADPH-reduced and dithionite-reduced active sites (Figure 4A).
Figure 4.

Structural features used to assign the flavin redox state (stereographic views). In panels A and B, the cage represents a simulated annealing σA-weighted Fo − Fc omit map (3.0 σ). Prior to calculating the maps, the flavin, His loop, Trp315, Arg327, and Tyr419 were omitted and simulated annealing refinement was performed. (A) Superposition of NADPH-reduced AfUGM (gray) and dithionite-reduced AfUGM (orange, PDB code 3utf). The hydrogen bond between Gly62 and the flavin N5 atom is diagnostic of the reduced state.20,21 (B) AfUGMo-NADPH. The Arg327-N5 hydrogen bond and the location of His63 near Trp315 are indicative of the oxidized enzyme.20,21 (C) Superposition of AfUGMr (gray) and AfUGMo-NADPH (yellow). Black and yellow dashes indicate hydrogen bonds in AfUGMr and AfUGMo-NADPH, respectively.
The structural indicators of the redox state clearly indicate that the flavin is oxidized the NAD(P)H complexes. At 2.75 Å resolution, it is difficult to discern the curvature of the iso-alloxazine, so the protein conformation is a better indicator of the flavin redox state. In particular, His63 is near the pyrimidine portion of the isoalloxazine and oriented parallel to Trp315 (Figure 4B). As noted by van Straaten et al.22, this particular arrangement of these two side chain occurs in AfUGMo. We note that the position of His63 in AfUGMo and AfUGMr differs by 7 Å (Figure 4C), which is clearly resolvable at 2.75 Å resolution. Furthermore, Arg327 donates a hydrogen bond to the flavin N5 in AfUGMo-NADPH (Figure 4B). This hydrogen bond is diagnostic of the oxidized enzyme because Arg is an obligate hydrogen bond donor, and N5 is an obligate acceptor only in the oxidized state. In summary, the flavin is oxidized in the structures determined from the short soaks in NAD(P)H, implying that the trapped species represents the oxidized enzyme complexed with the reduced coenzyme.
Mutagenesis Studies
Site-directed mutagenesis (to Ala) was used to assess the importance of selected residues in the NAD(P)H binding site (Table 2). Three mutations have pronounced effects on the kinetics of reduction (R447A, R91A, and S93A). Mutation of Arg447, which interacts with the nicotinamide carboxamide, lowers the catalytic efficiency by a factor of 2000. This result is consistent with Arg447 helping to guide the nicotinamide into position for hydride transfer. Arg91 and Ser93 are on the 90s loop, which forms the base of the adenine binding pocket. The R91A and S93A mutations decrease efficiency 125-fold and 14-fold, respectively. These results are consistent with the 90s loop helping to anchor the adenosine moiety. Mutation of Tyr104 or Tyr317, which interact with the adenine base, decreases the catalytic efficiency 3-fold. This result is consistent with these residues playing ancillary roles in binding the adenine. In summary, Arg447 and the 90s loop, which are located at opposite ends of the NADPH binding site, appear to be essential for efficient activation of AfUGM by NADPH.
DISCUSSION
The structures reported here provide the first images of a UGM complexed with NAD(P)H. All of the residues within 3.9 Å of NADPH are identically conserved in other eukaryotic UGMs, including TcUGM and Leishmania major UGM, two enzymes that are of interest for the design of drugs to treat Chagas disease and leishmaniasis, respectively (Figure S3). This level of sequence conservation suggests that the identified binding site is physiologically relevant and present in other eukaryotic UGMs. Site-directed mutagenesis provides additional validation of the site; mutation of residues contacting NADPH decreases the catalytic efficiency of FAD reduction by factors of 3 – 2000 (Table 2).
The NAD(P)H site of AfUGM is probably different from that of bacterial UGMs. Notably, none of the residues that contact the AMP half of the dinucleotide are present in the sequences of bacterial UGMs. Furthermore, overlaying NADPH from our structure onto K. pneumoniae UGM reveals severe steric clashes with an active site loop (Figure S4). The absence of the high affinity NADPH site described here for AfUGM may explain the low activity of bacterial UGMs13 with NADPH. For example, we have estimated kred for Mycobacterium tuberculosis UGM to be only 0.00002 s−1 (Figure S5), which is several orders of magnitude slower than AfUGM and TcUGM.
Finally, the AfUGMo-NADPH structure provides new opportunities for inhibitor design. One possible strategy is to target the NADPH site of AfUGMo in order to lock the enzyme in the inactive state. Alternatively, the active, reduced enzyme could be targeted because the ADP site is also present in AfUGMr (Figure 3B). Retention of the ADP site in the reduced enzyme reflects the fact that several residues that contact the ADP moiety have similar conformations in AfUGMo and AfUGMr, including Ph66, His68, Arg91, Ser93, Tyr104, Tyr317, Asn457, and His460. Both strategies are attractive because one could repurpose existing adenosine analogs. Lastly, although the binding of NADPH and UDP-Galp is mutually exclusive, the adenine and uracil pockets are distinct and separated by 6 Å (Figure 3). Thus, it may be possible to design novel bidentate compounds that simultaneously access both pockets.
Supplementary Material
Acknowledgments
We thank Dr. Jonathan Schuermann for help with X-ray data collection. This work is based upon research conducted at the Advanced Photon Source on the NE-CAT beamlines, which are supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Funding Sources
Research reported in this publication was supported by the National Institute of General Medical Science of the National Institutes of Health under award number R01GM094469 (to P.S. and J.J.T).
ABBREVIATIONS
- UGM
UDP-galactopyranose mutase
- UDP-Galp
UDP-galactopyranose
- UDP-Galf
UDP-galactofuranose
- TcUGM
UDP-galactopyranose mutase from Trypanosoma cruzi
- AfUGM
UDP-galactopyranose mutase from Aspergillus fumigatus
- AfUGMo
oxidized UDP-galactopyranose mutase from Aspergillus fumigatus
- AfUGMr
reduced UDP-galactopyranose mutase from Aspergillus fumigatus
- KIE
kinetic isotope effect
- PDB
Protein Data Bank
- RMSD
root mean square deviation
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information. Additional experimental methods; table of KIE data; supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Richards MR, Lowary TL. Chembiochem. 2009;10:1920. doi: 10.1002/cbic.200900208. [DOI] [PubMed] [Google Scholar]
- 2.Kizjakina K, Tanner JJ, Sobrado P. Curr Pharm Des. 2012 doi: 10.2174/1381612811319140007. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oppenheimer M, Valenciano AL, Sobrado P. Enzyme Res. 2011;2011:1. doi: 10.4061/2011/415976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tefsen B, Ram AF, van Die I, Routier FH. Glycobiology. 2012;22:456. doi: 10.1093/glycob/cwr144. [DOI] [PubMed] [Google Scholar]
- 5.Schmalhorst PS, Krappmann S, Vervecken W, Rohde M, Muller M, Braus GH, Contreras R, Braun A, Bakker H, Routier FH. Eukaryot Cell. 2008;7:1268. doi: 10.1128/EC.00109-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleczka B, Lamerz AC, van Zandbergen G, Wenzel A, Gerardy-Schahn R, Wiese M, Routier FH. J Biol Chem. 2007;282:10498. doi: 10.1074/jbc.M700023200. [DOI] [PubMed] [Google Scholar]
- 7.Bornemann S. Nat Prod Rep. 2002;19:761. doi: 10.1039/b108916c. [DOI] [PubMed] [Google Scholar]
- 8.Soltero-Higgin M, Carlson EE, Gruber TD, Kiessling LL. Nat Struct Mol Biol. 2004;11:539. doi: 10.1038/nsmb772. [DOI] [PubMed] [Google Scholar]
- 9.Sun HG, Ruszczycky MW, Chang WC, Thibodeaux CJ, Liu HW. J Biol Chem. 2012;287:4602. doi: 10.1074/jbc.M111.312538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oppenheimer M, Valenciano AL, Kizjakina K, Qi J, Sobrado P. PLoS One. 2012;7:e32918. doi: 10.1371/journal.pone.0032918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Massey V. Biochem Soc Trans. 2000;28:283. [PubMed] [Google Scholar]
- 12.Koplin R, Brisson JR, Whitfield C. J Biol Chem. 1997;272:4121. doi: 10.1074/jbc.272.7.4121. [DOI] [PubMed] [Google Scholar]
- 13.Barlow JN, Marcinkeviciene J, Blanchard JS. In: Enzymatic Mechanisms. Frey PA, Northrop DB, editors. IOS Press; Amsterdam: 1999. p. 98. [Google Scholar]
- 14.Sanders DA, Staines AG, McMahon SA, McNeil MR, Whitfield C, Naismith JH. Nat Struct Biol. 2001;8:858. doi: 10.1038/nsb1001-858. [DOI] [PubMed] [Google Scholar]
- 15.Beis K, Srikannathasan V, Liu H, Fullerton SW, Bamford VA, Sanders DA, Whitfield C, McNeil MR, Naismith JH. J Mol Biol. 2005;348:971. doi: 10.1016/j.jmb.2005.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gruber TD, Borrok MJ, Westler WM, Forest KT, Kiessling LL. J Mol Biol. 2009;391:327. doi: 10.1016/j.jmb.2009.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gruber TD, Westler WM, Kiessling LL, Forest KT. Biochemistry. 2009;48:9171. doi: 10.1021/bi901437v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Partha SK, van Straaten KE, Sanders DA. J Mol Biol. 2009;394:864. doi: 10.1016/j.jmb.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 19.Partha SK, Sadeghi-Khomami A, Slowski K, Kotake T, Thomas NR, Jakeman DL, Sanders DA. J Mol Biol. 2010;403:578. doi: 10.1016/j.jmb.2010.08.053. [DOI] [PubMed] [Google Scholar]
- 20.Dhatwalia R, Singh H, Oppenheimer M, Karr DB, Nix JC, Sobrado P, Tanner JJ. J Biol Chem. 2012;287:9041. doi: 10.1074/jbc.M111.327536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhatwalia R, Singh H, Oppenheimer M, Sobrado P, Tanner JJ. Biochemistry. 2012;51:4968. doi: 10.1021/bi300498c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Straaten KE, Routier FH, Sanders DA. J Biol Chem. 2012;287:10780. doi: 10.1074/jbc.M111.322974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weiss M. J Appl Cryst. 2001;34:130. [Google Scholar]
- 24.Engh RA, Huber R. Acta Cryst. 1991;A47:392. [Google Scholar]
- 25.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Proteins. 2003;50:437. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 26.Bourgeois D, Royant A. Curr Opin Struct Biol. 2005;15:538. doi: 10.1016/j.sbi.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 27.Kabsch W. Acta Crystallogr D Biol Crystallogr. 2010;66:125. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans P. Acta Cryst. 2006;D62:72. [Google Scholar]
- 29.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr, Sect D. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Acta Cryst. 2004;D60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Oppenheimer M, Poulin MB, Lowary TL, Helm RF, Sobrado P. Arch Biochem Biophys. 2010;502:31. doi: 10.1016/j.abb.2010.06.035. [DOI] [PubMed] [Google Scholar]
- 32.Karplus PA, Schulz GE. J Mol Biol. 1989;210:163. doi: 10.1016/0022-2836(89)90298-2. [DOI] [PubMed] [Google Scholar]
- 33.Mirza IA, Yachnin BJ, Wang S, Grosse S, Bergeron H, Imura A, Iwaki H, Hasegawa Y, Lau PC, Berghuis AM. J Am Chem Soc. 2009;131:8848. doi: 10.1021/ja9010578. [DOI] [PubMed] [Google Scholar]
- 34.Franceschini S, van Beek HL, Pennetta A, Martinoli C, Fraaije MW, Mattevi A. J Biol Chem. 2012;287:22626. doi: 10.1074/jbc.M112.372177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.