

Abstract
Activation of adrenergic receptors (ARs) represents the primary mechanism to increase cardiac performance under stress. Activated βARs couple to Gs proteins, leading to adenylyl cyclase (AC)-dependent increases in secondary-messenger cyclic adenosine monophosphate (cAMP) to activate activation of protein kinase A (PKA). The increased PKA activities promote phosphorylation of diversified substrates ranging from the receptor and its associated partners, to proteins involved in increases in contractility and heart rate. Recent progress with live-cell imaging has drastically advanced our understanding of the βAR-induced cAMP and PKA activities that are precisely regulated in a spatiotemporal fashion in highly differentiated myocytes. Several features stand out: membrane location of βAR and its associated complexes dictates the cellular compartmentalization of signaling; βAR agonist dose-dependent equilibrium between cAMP production and cAMP degradation shapes persistent increases in cAMP signals for sustained cardiac contraction response; and arrestin acts as an agonist dose-dependent master switch to promote cAMP diffusion and propagation into intracellular compartments by sequestrating phosphodiesterase (PDE) isoforms associated with the βAR signaling cascades. These features and the underlying molecular mechanisms of the dynamic regulation of βAR complexes with AC and PDE enzymes and the implication in heart failure will be discussed.
Keywords: adrenergic receptor, phosphodiesterase, cAMP, protein kinase A
Introduction
cAMP/PKA activation represents a key signaling mechanism upon stimulation of G protein-coupled receptors for cardiac contraction and energy metabolism under stress conditions. Activation of βARs, a group of prototypical G protein-coupled receptors, is one of the major neurohormonal mechanisms controlling cAMP/PKA activities for physiological responses in animal hearts. 1–6 β1 and β2AR are highly homologous receptors expressed in animal hearts and are responsible for enhancing cardiac performance. β1AR plays a dominant role in increasing chronotropy and ionotropy in cardiac myocytes, whereas β2AR produces only modest chronotropic effects. 1, 2 In addition, a minor β3AR subtype is also expressed in myocardium and modulates myocyte function. 7 Upon activation of βARs via ligand binding, the receptors undergo conformational changes that lead to coupling and activation of Gs protein, which in turn stimulates ACs for cAMP production. The small molecule cAMP functions as a second messenger that diffuses into distinct subcellular locations/compartments 8, 9 and activates the locally anchored/tethered PKA. 10–12 Thus, the specificity of substrate phosphorylation is achieved for cellular functions such as contractile responses. One of the emerging mechanisms that safeguard the specificity of G protein-coupled receptor/cAMP signaling is the control of cAMP transients in space and time via degradation by cyclic nucleotide PDEs. 8, 9, 13, 14
The concept of spatiotemporal regulation of cellular cAMP and PKA activities provides new insights into understanding how cAMP/PKA signaling is translated into physiological contraction response in highly organized muscle cells. 8, 9, 14, 15 In this paradigm, PKA is anchored to distinct subcellular structures through a family of proteins named A-kinase anchoring proteins (AKAPs). In contrast, correlating to the distribution of most ACs, cellular cAMP is primarily confined along the plasma membrane under neurohormonal stimulation. 15 Despite being a diffusible small molecule, the distribution and diffusion of cAMP is rather limited because of cAMP degradation mediated by PDEs. 8, 9, 14, 16, 17 Under a specific hormonal stimulation, individual PKAs anchored at different subcellular compartments will be selectively activated to phosphorylate a local pool of proteins for specific cellular processes. 10, 18 A spatial distribution of cAMP/PKA signaling regulated by ACs and PDEs is therefore essential for selective phosphorylation of substrates important in myocyte contraction. This is critical considering that a wide range of different neurohormonal chemicals can stimulate cardiac myocytes, and many of these agonists lead to increases in intracellular cAMP, raising the point that cardiac myocytes must be able to segregate all these signals and prevent unnecessary phosphorylation under a specific stimulus. Consequently, precisely fine-tuning the βAR signaling for cardiac contractile performance is a vital mechanism to allow the body to adjust to stress. Clinically, dysfunction of the adrenergic signaling pathway contributes to cardiac arrhythmia, 19, 20 and cardiac remodeling including myocyte apoptosis 21–24 and myocyte hypertrophic growth 25, 26 in diseases such as myocardium infarction and heart failure.
Highly differentiated cardiac myocytes have several unique membrane structure properties. Recent studies have significantly advanced our understanding of these structures in the spatiotemporal regulation of βAR signaling in cardiac myocytes. First, myocytes contain abundant lipid rafts, specialized regions of the plasma membrane enriched in cholesterol and other lipids, and caveolae, a subset of lipid rafts that form flask-shaped invaginations of the plasma membrane enriched in particular proteins such as caveolins 27. Second, myocytes have an extensive t-tubular structure network that results from invagination and extension of the plasma membrane into the internal space of the cell bodies. Third, myocytes are innervated by sympathetic ganglia neurons to form adrenergic synapse, which induces highly specialized post-synaptic regions on the plasma membrane. 28 The relative distribution and enrichment of βAR subtypes in these specialized membrane structures facilitate recruitment and association of other signaling components in a location dependent manner, and leverage a strong impact on the production of cAMP and signaling efficiency and specificity in cardiac myocytes.
Biochemical characterization has also advanced our understanding of the organization of ACs and PDEs associated with βARs in cardiac myocytes. AC5/6 has been shown to be tethered by scaffold protein AKAP79 in rat brain tissues. 29 AKAP79 is known to bind to β1 and β2ARs, as well as PKA and downstream effectors such as ion channels in various tissues. 30–32 In cardiac myocytes, AC6 can be coimmunoprecipitated with β1AR 33, suggesting that the receptor and AC6 could be assembled into a complex via AKAP scaffold proteins. Conversely, a group of phosphodiesterase 4Ds (PDE4Ds) selectively binding to βARs 33–35 play significant roles in regulating the βAR subtype-induced neonatal myocyte contraction rate response. 36 These receptor-associated PDE4Ds play critical roles in controlling cAMP and PKA activities in the vicinity of the receptors as well as the diffusion of cAMP for differential cardiac responses under βAR stimulation. 33–37 Overall, a balance between AC-dependent cAMP production and PDE-dependent cAMP degradation in an agonist dose-dependent manner differentially regulates cAMP/PKA signaling in cardiac myocytes (Figure 1). Moreover, at increasing concentrations of isoproterenol, arrestin plays a master role in switching the cAMP signals from a transient response to a sustained response under βAR stimulation. The temporal profile of cAMP signaling dictates signaling distribution, PKA substrate-specificity, and myocyte contraction responses. 33, 38 This review focuses on recent evidence unraveling the mechanisms that modulate adrenergic receptor-induced cAMP and PKA activities in space and time to promote specific cardiac contractile responses.
Figure 1.
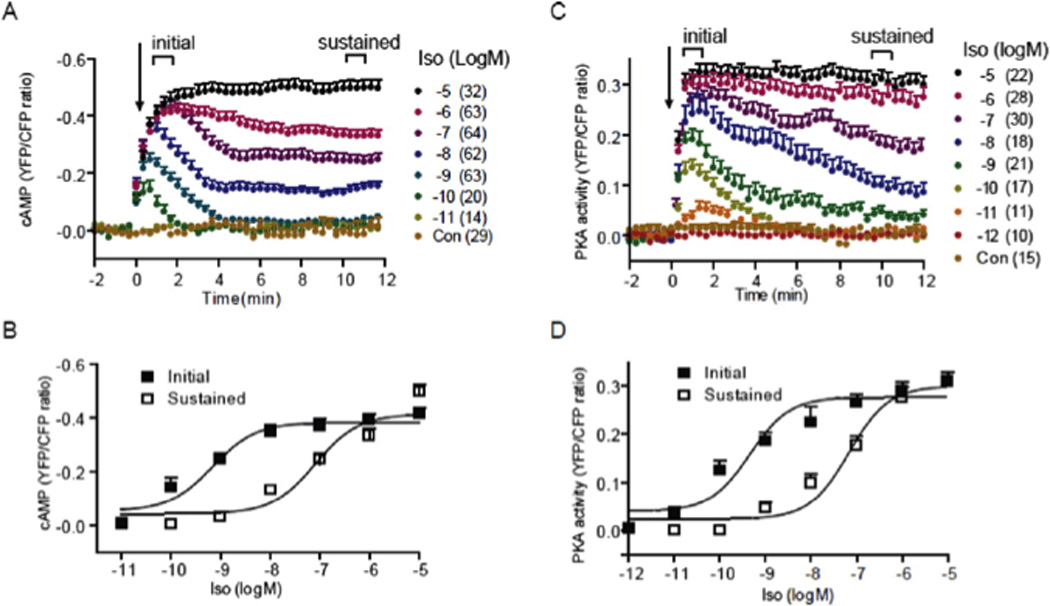
Activation of βARs induces a dose-dependent increase in cAMP ICUE3 and PKA AKAR3 FRET ratio in cardiac myocyte (Reprint with permission from Molecular Pharmacology). A and B, the cAMP biosensor ICUE3 was expressed in wild-type myocytes. Cells were treated with isoproterenol at different concentrations. Changes in cAMP ICUE3 FRET ratio (an indication of cAMP activity) were measured. A, time courses of changes in cAMP FRET ratio were calculated and normalized against the baseline levels. B, the initial peak increases (EC50 6.86 × 10−10 M) and the sustained increases (EC50 7.99 × 10−8 M) in cAMP FRET ratio were plotted. C and D, the PKA biosensor AKAR3 was expressed in wild-type myocytes. Cells were treated with isoproterenol at different concentrations. Changes in PKA AKAR3 FRET ratio (an indication of PKA activity) were calculated and normalized against the baseline levels. C, time courses of changes in PKA FRET ratio were plotted. D, the initial peak increases (EC50 4.53 × 10−10 M) and the sustained increases (EC50 6.77 × 10−8 M) in PKA FRET ratio were plotted. Only the sustained cAMP/PKA activities promote cardiac contractile responses in neonatal and adult myocytes.
Membrane Localization in Cellular Compartmentalization of Signaling
Cardiac myocytes are highly differentiated cells with several unique membrane structure properties implicated in mediating adrenergic signaling transduction. 27, 39 First, cardiac myocytes contain abundant lipid rafts, specialized regions of the plasma membrane enriched in cholesterol and other lipids, and caveolae, a subset of lipid rafts that form flask-shaped invaginations of the plasma membrane enriched with particular proteins such as caveolins. 27 Early studies indicate that β2ARs are highly enriched in the caveolae/lipid rafts and induce local cAMP and PKA signals in cardiac myocytes. 40, 41 In contrast, β1ARs are distributed throughout both caveolae/lipid rafts and non-lipid raft membrane domains, and induce a global cAMP and PKA signals with a much broader reach to intracellular compartments. 40 Meanwhile, other GPCR signaling proteins including G proteins, ACs, G-protein receptor kinases (GRKs), AKAP79, PKA, protein phosphatase 2A, as well as L-type calcium channel are also enriched in lipid rafts and caveolae in cardiac myocytes. 27, 39, 42, 43 Accordingly, the β2AR-induced signaling for downstream calcium channel activation is sensitive to disruption of caveolae via extraction of cholesterol by detergent, whereas the β1AR-induced effect is not altered by extraction of cholesterol in lipid rafts/caveolae. 39 Accordingly, stimulation of β2AR leads to activation of L-type calcium channels in a local vicinity, 44–46 whereas stimulation of β1AR leads to activation of L-type calcium channels in the distance 45, 46.
Second, myocytes have an extensive t-tubular structure network that results from invagination and extension of the plasma membrane into the internal space of the cell body. Using real time imaging in live myocytes, Nikolaev et. al. observed that the cAMP induced by β2AR is confined in the t-tubular structure in adult myocytes. 48 In contrast, the cAMP induced by β1AR is distributed in both plasma membrane and t-tubular structure. 48 Moreover, a local stimulation of β1AR at one end of elongated adult myocytes leads to a far-reaching cAMP diffusion inside of cells, whereas stimulation of β2AR leads to very confined cAMP signal at the stimulation site. 47 Together, these studies indicate that activation of β1AR promotes a broad distribution of intracellular cAMP signal, whereas the β2AR actions are local. Given that the β2AR signaling is confined in t-tubular structure, and sensitive to disruption of caveolae, it would be interesting to examine whether the caveolae membrane is enriched in the t-tubular structure in cardiac myocytes.
Third, upon formation of adrenergic synapses between myocytes and sympathetic ganglia neurons, a highly specialized adrenergic synaptic region is formed on the plasma membrane of myocytes. 28 Shcherbakova, et. al. has analyzed the distribution of βAR subtypes on myocytes relative to innervation of sympathetic ganglia neurons. Both β1 and β2ARs are highly enriched at postsynaptic regions on cardiac myocytes, which are also enriched with the scaffold proteins AKAP79 and SAP97. 28 The distribution of βAR subtypes within the innervated cardiac myocytes is further confirmed with an elegant in vitro co-culture model of sympathetic ganglia neurons and cardiac myocytes. Upon stimulation of sympathetic ganglia neurons, the released catecholamines stimulate both β1 and β2ARs in the co-cultured cardiac myocytes and induce distinct trafficking patterns. While β1ARs remains enriched at the post-synaptic region, β2ARs are redistributed from the postsynaptic membrane, 28 presumably via receptor internalization. Together, the observed segregation of βARs plays critical roles in organizing the receptor signaling complexes in cardiac myocytes.
Organization of βAR/Gs/AC complexes for cAMP production
Upon activation, βARs couple to Gs protein, which leads to activation of AC for cAMP production. The enrichment of βARs and immediate downstream components in local plasma membrane domains such as caveolae suggests that they may be preassembled into macromolecular complexes to facilitate signaling transduction specificity and efficiency. ACs is a family of diversified genes that display different regulatory mechanisms and interaction with other signaling pathways. 15, 49, 50 In cardiac myocytes AC5 and AC6 represent the dominant isoforms, 50, 51 and can be activated by βAR stimulation.
Accumulating evidence supports the idea that the βAR, Gs protein, and AC form preassembled complexes to facilitate signaling transduction. First, it is well known that βAR can be preassembled with Gs protein, which is also referred to as a G-protein precoupled receptor. 52–54 This form of receptor displays higher binding affinity to ligands than that of receptor in a G protein-free form 55. Stripping G proteins from membrane preparations containing βARs abolishes the high affinity binding sites. 56, 57 The preassembled βARs in complex with Gs proteins represent a receptor population that is ready for agonist stimulation, and is also sensitive to low concentrations of agonist due to the high affinity binding sites. Second, the βARs are known to bind a variety of scaffold proteins. β2AR binds to AKAP79 and AKAP250 in various tissues. 58, 59 β2AR also binds to Na-H exchanger regulatory factor 60 and N-ethylmaleimide-sensitive factor. 61, 62 In contrast, β1AR binds to AKAP79 32 and PDZ domain containing proteins such as synaptic associated proteins (SAPs) 63, GAIP-interacting protein C-terminus (GIPC) 64, and membrane-associated guanylate kinase inverted (MAGI) proteins 65. Both AKAPs and SAPs are well-known scaffold proteins that can tether additional signaling proteins such as kinases, phosphatases, and other regulatory proteins to the adrenergic receptors. 11, 66 In the case of β1AR, AKAP79 and SAP97 form a tertiary complex that facilitates PKA phosphorylation of the activated receptor and promotes receptor recycling after internalization in HEK293 cells. 67 In brain tissues, AKAP79 directly binds to AC5 and AC6, 29 which serve as a coordinator to mediate PKA phosphorylation of the ACs. The PKA phosphorylation inhibits AC activities, which functions as a negative feedback mechanism to attenuate cAMP production under βAR stimulation. 29 In cardiac myocytes, AC6 can be coimmunoprecipitated with overexpressed β1AR, 33 indicating that β1AR could be connected to ACs via AKAP79 to regulate AC activities for cAMP production. 68 Finally, in a co-culture of sympathetic ganglia neurons and neonatal cardiac myocytes, both β1AR and β2ARs are enriched in the postsynaptic regions on the plasma membrane of cardiac myocytes. 28 These receptors are also co-localized with AKAP79 and SAP97, 28 supporting again the concept that the βARs could exist in a preassembled complex containing G proteins and scaffold proteins that connect to AC and PKA, and are ready to respond to catecholamine released from the nerve terminus.
βAR-associated PDEs mediate cAMP degradation
Over the past decades, a series of studies have shown the dynamics of cAMP and PKA signaling in different subcellular compartments upon βAR stimulation. 16, 17, 33, 37, 38, 69–71 The duration and distribution of cAMP signals are disrupted/altered upon inhibition of PDEs with 3-isobutyl-1-methylxanthine, IBMX. 37, 69 These studies point out the critical roles of PDEs in confining the cAMP in space and time in cardiac myocytes under adrenergic stimulation. PDEs include 11 families based on their amino acid sequence homology, substrate specificities, and pharmacological properties. 72 Each of the 11 PDE families has one to four distinct genes. In addition, most PDE genes encode multiple splicing variants through the usage of different promoters and alternative splicing. At least six different PDE families are expressed in animal hearts, including PDE1, 2, 3, 4, 5, and 8 (see review 73). PDE1, 2, and 3 can hydrolyze both cAMP and cyclic guanosine monophosphate (cGMP). PDE5 specifically hydrolyzes cGMP whereas PDE4 and PDE8 are specific for cAMP degradation. The relative expression of each PDE family varies between human and rodents, and during developmental and disease stages. In rodent hearts, PDE4 and PDE3 are the two major families, which account for more than 90% of PDE activities. 74 In particular, PDE4D genes have been shown associated with βAR subtypes, and regulate the receptor-induced cAMP signaling in cardiac myocytes. 34–36 Although PDE3 and PDE4 families account much less portion of overall PDE activities in human myocardium, the expression and function of PDE4 genes are well conserved in human hearts, 75 underscoring the critical roles of these genes in regulating βAR signaling properties in cardiac myocytes across different mammalian species. Besides PDE3 and PDE4, other PDE family such as PDE2 and PDE8 are also implicated in cAMP metabolism. PDE2 is activated by cGMP to enhance cAMP degradation, which negatively regulates the βAR/Gs-induced cAMP signaling. 71 In contrast, deletion of PDE8A displays a greater increase in calcium signaling including L-type calcium channel and calcium spark activities as well as calcium transients. 76 The mechanism of how PDE8 is involved in the cAMP-mediated calcium handling for cardiac contraction is not clear yet. Together, under adrenergic stimulation, these PDEs play distinct roles in maintaining subcellular specificity of cAMP signaling by preventing diffusion of cAMP from one microdomain to another in cardiac myocytes. 36, 37
The PDE3 family consists of PDE3A and PDE3B genes whereas the PDE4 family contains PDE4A, PDE4B, PDE4C, and PDE4D genes. 72 At resting states, both PDE3 and PDE4 activities modulate a basal intracellular concentration of cAMP by continuously hydrolyzing the cAMP synthesized by constitutively active adenylyl cyclases, 70 thus maintaining a tonic PKA activity in cardiac myocytes. 38 Upon adrenergic stimulation, PDEs play a role in controlling the duration and amplitude of cAMP signals. 38, 70, 77 Using a cardiac contraction rate assay, the PDE4 family was found to attenuate the adrenergic stimulation of cAMP/PKA signal for enhancing contraction response. 36 Further analysis with gene deficiency reveals that the PDE4D isoforms are critical for regulating the βAR-induced cAMP signals for contractile response in cardiac myocytes. 36 Deletion of PDE4D, but not PDE4A and PDE4B genes, enhances the βAR-induced cAMP signals in mouse embryonic fibroblasts, 78 and contraction rate response in neonatal cardiac myocytes. 36 Probing cAMP activities in living myocytes confirms the critical role of PDE4 family genes in the control of cAMP generated by βAR stimulation in both neonatal and adult cardiac myocytes. 16, 17, 33, 37, 38, 69–71 In comparison, PDE3 isoforms appear to be involved in regulating cAMP content in a functionally distinct pool, 38, 70, 77 which may control the cAMP activities in the SR for calcium cycling, 79 as well as adrenergic stimulation-induced cardiac myocyte apoptosis via inhibiting the expression of inducible cAMP early repressor. 80, 81 Thus, PDE3s and PDE4s play distinct roles in modulating the cAMP signals in myocytes. 17, 33, 38 In this review, I will focus on the PDE4D genes due to their direct association with βAR subtypes in cardiac myocytes and prominent role in regulating cAMP signaling under adrenergic stimulation.
Agonist dose-dependent association and sequestration between PDE4D isoforms and βARs
The function of individual PDEs in adrenergic signal transduction is also dependent on their distribution within the three-dimensional matrix of the cell. Indeed, membrane fractionation studies show that both PDE3 and PDE4 are highly enriched in the membrane fraction, and are resistant to detergent extraction. 72, 82 Thus, PDEs are usually tightly anchored/tethered to the membrane or protein complexes in respect to other regulatory and effector elements. Subsequently, two groups have independently characterized the selective association of PDE4D8 and β1AR. 33, 34 Richter et. al. have shown that PDE4D8 directly binds to β1AR via the C-terminal of receptor, though the receptor binding sites on PDE4D8 are yet to be identified. 34 Conversely, β1AR is shown to coimmunoprecipitate together with the N-terminal region of PD4D8 in neonatal cardiac myocytes, 33 indicating that the variable N-terminal region contains the information for selective binding to the receptor. However, activation of β1AR preferentially enhances the activity of PDE4D8 and PDE4D9 in HEK293 cells, 34 indicating that β1AR can potentially associate with other PDE4D isoforms besides PDE4D8. Moreover, inhibition of PDE4D8 activities with overexpression of the catalytically inactive form of PDE4D8 or the unique N-terminal domain is sufficient to enhance both cAMP production and myocyte contraction rate response upon β1AR stimulation. 35
In comparison, the association between β2AR and PDE4D isoforms are much more complex. 35 In neonatal cardiac myocytes, β2AR displays a broad binding to different isoforms in PDE4D family including PDE4D3, 5, 7, 8, and 9 with a preferential binding to PDE4D9, and to lesser extent, PDE4D8. 35, 83, 84 Interestingly, the basal levels of binding between β2AR and PDE4D are reduced in cell lacking both arrestin 2 and arrestin 3 genes, 35 indicating a possible role of arrestin in organizing the β2AR/PDE4D complexes. Supporting this notion, arrestin 3 is shown to organize a stable complex between PDE4D3 and relaxin family peptide receptor 1. 85 Selective inhibition of individual PDE4D isoforms shows that PDE4D9, as well as PDE4D5 and PDE4D8, can affect either the basal contraction rate or the β2AR signaling-induced contraction rate response in neonatal cardiac myocytes. 35 The complex effect of different PDE4D isoforms on β2AR signaling is dependent on dynamic association between individual isoforms with the activated receptors.
Stimulation with subnanomolar of agonist: A transient cAMP is dominated by PDE4D activities within the preassembled βAR complexes
With the advancement of live-cell imaging approaches, recent characterizations of cardiac adrenergic signaling have generated evidence to not only solidify the concept of spatial distribution and regulation of cAMP signals in subcellular organelles, but also provide evidence for mechanisms to address the sustained and agonist-dose dependent contractile responses in cardiac myocytes. 16, 17, 33, 37, 38, 69–71 These studies suggest that well-orchestrated receptor association with ACs and PDEs, as well as activation of these enzymes plays a regulatory role in fine-tuning cardiac contractile responses. In this new paradigm, the AC-dependent cAMP production and the PDE-dependent cAMP degradation dictate an agonist dose dependent equilibrium of cAMP activities, which produces a transient cAMP signal at minimal concentration of agonist, or a sustained increase at the different levels over the baseline at higher concentration of agonist stimulation (Figure 1). 33, 38
At subnanomolar concentrations of isoproterenol stimulation, the cAMP production is rapidly accumulated. 33, 38 This high potency of isoproterenol suggests that the cAMP response is induced by preassembled βAR/Gs/AC complexes with high affinity binding for ligands. 55 As a result, the cAMP production is rapidly saturated at 10−8 M of isoproterenol, a concentration that is well above the binding constant at the high affinity sites. 55 Accordingly, the activation of receptor and G protein occurs almost simultaneously within 50 milliseconds, whereas the cAMP production is produced within 2 seconds due to rapid activation of AC in the complex. 86 In addition, the expression of AC is the rate-limiting factor for the βAR/Gs/AC-induced cAMP production in cardiac myocytes. 33, 87 In agreement, overexpression of AC6 but not the β1AR significantly enhances the maximal cAMP synthesis in cardiac myocytes. 33 These observations challenge the traditional view that cAMP production can be further enhanced at higher concentrations of βAR agonist, due to recruitment of Gs proteins to available βARs on the cell surface.
However, at this concentration range, the βAR-induced cAMP signals are very transient in cardiac myocytes. One of the major reasons is that the cAMP signals lead to activation of PKA in the same complexes, which phosphorylates and activates at least two distinct negative feedback mechanisms to attenuate signaling. One mechanism is to phosphorylate and inhibit AC activity for cAMP synthesis, 29 and another is to phosphorylate and activate PDE to enhance cAMP degradation. 69 Consequently, inhibition of PKA via either a PKA inhibitor, knocking down the expression of AKAP79, or displacing PKA holoenzymes from AKAP79 significantly prolongs cAMP signals under βAR stimulation. 29, 69 Between these two negative feedback mechanisms, the PDE4-mediated cAMP degradation seems to play a dominant role in modulating the cAMP signals. Although overexpression of AC6 significantly enhances the peak level induced by adrenergic stimulation, 33 the overall cAMP signals still display a transient response with a rapid attenuation. 33 In contrast, over-expression of PDE4D8 is sufficient to completely inhibit cAMP signaling induced by saturated doses of agonist in neonatal cardiac myocytes. 33 Moreover, inhibition of PDE4 is sufficient to convert a transient cAMP response to a sustained signal. 33 All this evidence argues that the PDE4D-mediated cAMP degradation serves as the major mechanism to attenuate the receptor stimulated cAMP signals. 33, 38 These observations also argue the limited impact of PKA phosphorylation of AC on the cAMP equilibrium when compared to the dominant role of cAMP degradation by PDE. Together, the balance between syntheses vs. degradation of cAMP dictates both peak levels and duration. The PDE4-mediated cAMP degradation is so overwhelmingly dominant that, when overexpressed, it completely degrades any cAMP produced from βAR stimulation in cardiac myocytes.
More importantly, the transient cAMP signals are confined in the local domain surrounding the βARs on the plasma membrane (Figure 2). Using PKA-based cAMP biosensors Zaccolo et. al. have shown that cAMP accumulation is, indeed, confined along t-tubular structures in rat neonatal cardiac myocytes. 37 Inhibition of PDE with IBMX produces a much broader distribution of cAMP signals. 37 This is also consistent with early biochemical evidence showing that activation of βARs selectively stimulates type II PKA in cardiac tissues, and the majority of type II PKA is membrane bound in cardiac myocytes. 88 Therefore, the cAMP produced by βAR activation is highly localized and targeted to type II PKA for substrate phosphorylation.
Figure 2.
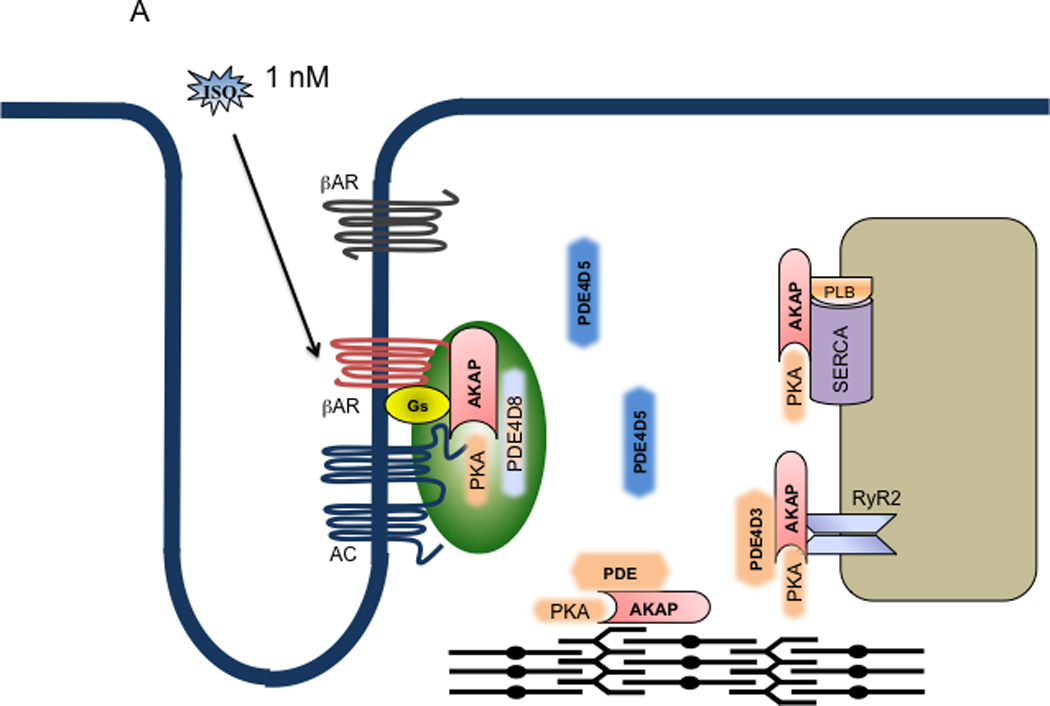
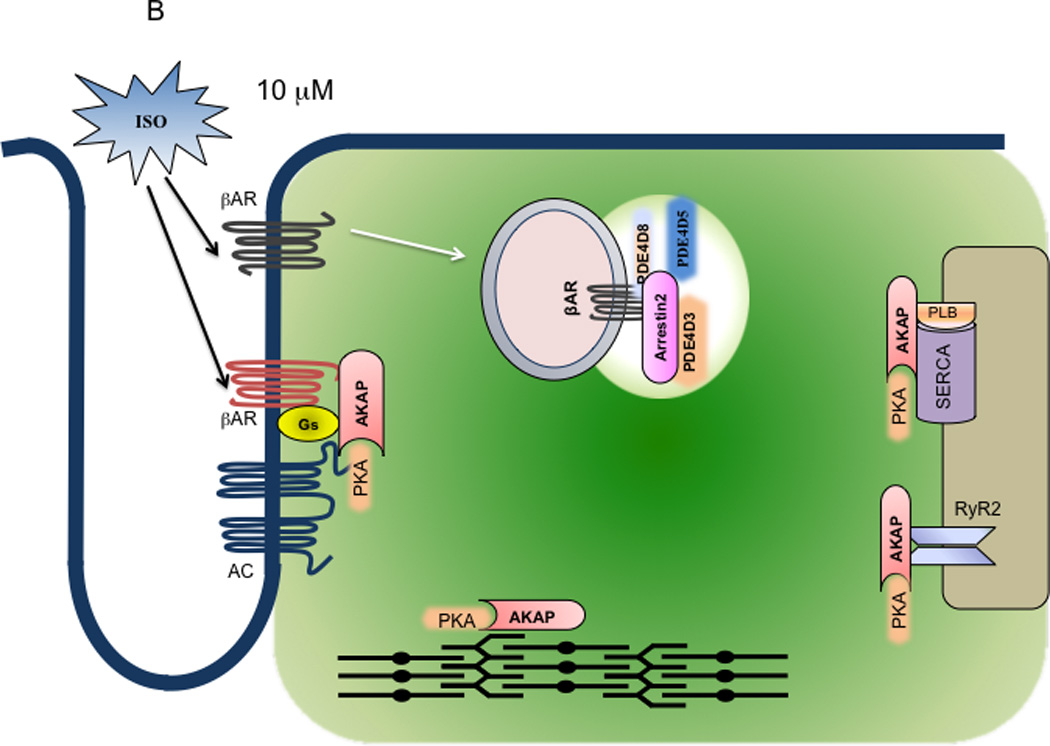
Model of dual mechanistic regulation of cAMP/PKA activities by AC and PDE4D under different doses of adrenergic stimulation. A. At 10−9 M isoproterenol, only the G-protein precoupled βAR is activated (red), which leads to activation of AC in the same complex to produce cAMP (the gas pedal is on). The cAMP signal leads to PKA activation for phosphorylation of the receptor and receptor-associated PDE that negatively feeds back to attenuate cAMP signal (the brake is still on). As a result, the cAMP signal is transient and restricted at the vicinity of the receptor for local PKA phosphorylation. B. At 10−5 M isoproterenol, both G-protein precoupled (red) and G-protein free (dark brown) βARs are activated. Activation of the G-protein precoupled receptors leads to AC-mediated cAMP production (the gas pedal is on) whereas activation of G-protein free receptors leads to arrestin-mediated internalization. Meanwhile, the PDE4D isoforms are dissociated from the precoupled receptor complexes (the brake is off), which are facilitated by sequestration of PDE4Ds to the internalized receptor/arrestin complexes on endosomes. Therefore, the cAMP can diffuse and propagate to access PKA in different subcellular compartments. Moreover, the PDEs such as PDE4D3 from the SR may be also sequestrated on endosome via binding the receptor/arrestin complexes; this allows the accumulation of cAMP on the SR membrane for persistent PKA activation and phosphorylation of substrates such as PLB. As a result, the cAMP and PKA can phosphorylate both local (near the receptor) and distant substrates such as PLB and TnI for myocyte contraction responses.
The transient and local cAMP signals lead to phosphorylation of the receptors and their associated proteins, but have limited access to distant substrates, such as PKA targets on the sarcoplasmic reticulum membrane as well as targets associated with myofibrils. The opposing actions on cAMP signals by both AC and PDE4 in local compartments argue that both enzymes are localized in the proximity, or potentially in direct association with the same βAR/G protein complex. Evidence for a direct βAR/AC/PDE complex is missing in cardiac myocytes. However, a recent study has already provided the first example of coexistence of AC and PDE in the same relaxin family peptide receptor 1 complex for sustained tonic cAMP/PKA activities under subpicomolar concentration of agonist stimulation. 85
Due to its limited access to distant substrates, such as PKA targets on the sarcoplasmic reticulum membrane or associated with myofibrils, the transient cAMP signals have minimal impact on the contractility of ventricular myocytes. However, these local cAMP/PKA signals should be accessible to other local effectors such as ion channels on the plasma membrane in cardiac myocytes. Both the PDZ proteins (SAPs and Na-H exchanger regulatory factors) and the AKAP proteins are shown to organize the βAR complexes with downstream targets including L-type calcium channels, cystic fibrosis transmembrane conductance regulator, and Na-H exchanger in different tissues and cells. 60, 89, 90 Physical associations between the βARs and these ion channels enable the local cAMP signals to be effective in modulating channel activities in the local vicinity. For example, in sinoatrial node cells, these cAMP activities may be able to stimulate contractile responses via activating cyclic nucleotide-gated ion channels in the vicinity. 91 In neonatal ventricular cardiac myocytes, the same signaling machinery probably regulates both cyclic nucleotide-gated ion channels and L-type calcium channels for enhancing contraction rate. However, in adult cardiac myocytes, the stimulation of local cAMP signaling has minimal effect on contractile shortening. 38
Stimulation with micromolar of agonist: A dose-dependent sustained increase of cAMP signaling is shaped by sequestration of PDE4 from the βAR/Gs/AC complexes
Upon further increases in agonist concentration (from 10−9 M to 10−5 M), the cAMP signaling is gradually shifted from transient responses to saturated and sustained responses (Figure 1). This shifting is accompanied with an agonist dose-dependent dissociation of PDE4D8 from the β1ARs. 33–35 This dissociation of PDE4s from the activated βARs leads to a shift in balance between cAMP production and cAMP degradation. As a result, after transient initial rapid decreases, the cAMP signals are maintained at incremental levels above the baselines in an agonist-dose dependent manner 33. Using another biosensor to directly measure PKA activities, De Arcangelis et. al. have showed that the activation of downstream PKA also displays similar sustained responses at high concentrations of agonist (Figure 1). 33 This is the first evidence to show that stimulation of βARs induces sustained cAMP and PKA signals, a feature correlated to the physiological contractile responses that are persistent under the very same stimulation. 33
The prominent role of PDE4 in shaping the duration of cAMP response is also supported by a series studies with pulse stimulation of βAR agonists. 16, 17, 37, 38, 69–71 In these studies, a pulse stimulation of βARs is introduced to cardiac myocytes with a short perfusion of agonists following an extended washing. Such a short stimulation induces a transient response of cAMP signaling; and the decreases are significantly attenuated by inhibition of PDE4, or by inhibition of PKA. 16, 17, 37, 38, 69–71 Meanwhile, as discussed previously, overexpression of PDE4D8, a β1AR-associated isoform, is sufficient to abolish cAMP response even under saturated concentrations of isoproterenol. 33
This elegant cAMP equilibrium under adrenergic stimulation also displays several interesting features. First, the system is self-adjustable to establish a balance between cAMP production and cAMP degradation maintained at different levels. While the dissociation of PDEs appears to be the most critical factor in determining the equilibrium levels, other factors can also weigh in. These include receptor phosphorylation for desensitization and internalization, 92, 93 G-protein inactivation through GTP hydrolysis 94, 95, and PKA phosphorylation and inactivation of AC, 29 as well as PKA phosphorylation and activation of PDEs. 96, 97 Second, a continuous cAMP production is obligatory to maintain an equilibrium; which is due to continuous presence of agonist within the extracellular space. Removal of agonist or addition of a βAR antagonist leads to rapid attenuation of cAMP signals to the baseline levels. 33, 38 Therefore, a sustained cAMP response is dependent on continuous activation of the βAR/Gs/AC system to maintain the cAMP production; this cAMP production is balanced by the PDE-mediated degradation in an agonist concentration-dependent fashion. Third, the sustained cAMP activities are able to diffuse out of the confinement of the βAR microdomains, which permits access to the PKA enzymes anchored on different subcellular structures including the sarcoplasmic reticulum and myofibrils in cardiac myocytes. 33, 38 These PKA activities lead to persistent phosphorylation of substrates such as phospholamban (PLB) on the sarcoplasmic reticulum membrane and troponin I (TnI) associated with myofibrils for sustained contractile responses in both neonatal and adult cardiac myocytes. 33, 38 Last, the increases in sustained cAMP levels display a close correlation with the increases in persistent contraction rate responses in neonatal myocytes in an agonist dose-dependent fashion. 33, 77 Together, these data have, for the first time, demonstrated specific pools of cAMP activities capable of modulating the βAR agonist dose-dependent contractile response in cardiac myocytes.
Biochemically, to maintain a sustained cAMP production, a pool of βAR/G-protein complexes needs to be occupied by ligand constantly to stimulate the associated ACs. Since the β1AR does not undergo agonist-induced internalization in cardiac myocytes, 98 the observations suggest a rapid uncoupling/recoupling cycle to maintain a pool of the receptor in a complex form associated with Gs proteins and ACs. Alternatively, it could simply be due to a stable β1AR/Gs/AC complex under stimulation. In agreement with these notions, the association of AC6 and the β1AR is not altered upon stimulation of either 10−9 M or 10−5 M of isoproterenol. 33 In comparison, the PDE4D8 selectively dissociates from the β1AR at much higher doses of agonist stimulation. 33–35
Arrestin mediates sequestration of PDE4D for cAMP diffusion and propagation
As of today, the mechanisms for transportation of PDE4D isoforms in cardiac myocytes are not clear. In one possible scenario, the desensitized and internalized β2ARs serve as the initiator for complex formation between the receptor, arrestin, and different PDE4D isoforms. The β2ARs could keep shuttling between the G-protein complexes and the arrestin-complexes via rapid internalization and recycling 99, 100 to maintain the equilibrium. Alternatively, since GRK phosphorylation happens on the receptor at high concentration of agonist stimulation 101, 102, it indicates that these receptors do not form complexes with G proteins, and have low affinity-binding sites for agonist (Figure 2B). In this scenario, two different pools of βARs are presented, a G protein-precoupled pool and a G protein-free pool. At low concentrations, only the precoupled receptor/G protein complexes are activated due to their high affinity binding to ligand; and the PDE4D isoforms associated with the same complexes confined the cAMP signals within the vicinities of the complexes (Figure 2A). However, at high concentrations, the G protein-free receptors are also activated via low affinity binding to ligand, and selectively phosphorylated by GRKs for internalization, which serve as the sites to sequestrate PDE4Ds via arrestin binding (Figure 2B). As a result, the cAMP production machinery (G protein-precoupled receptors) is segregated from the cAMP degradation machinery (PDE4Ds). In this scenario, arrestins function as a master regulator to switch off cAMP degradation, which permits accumulation and diffusion of cAMP in agonist dose-dependent fashion. Only the accumulated and diffusible cAMP is sufficient in promoting PKA phosphorylation of proteins such as TnI and PLB for cardiac contraction.
In agreement with this hypothesis, upon stimulation with catecholamines, PDE4D8 dissociates from the activated β1AR in both HEK293 cells and neonatal cardiac myocytes in an agonist concentration-dependent manner. 33, 34 At minimal 10−7 M of isoproterenol, PDE4D8 displays dissociation from the receptor in HEK293 cells, which reaches the peak level at 10 minutes of stimulation 33. Conversely, β2AR displays a much more complex association with different PDE4D isoforms. While β2AR binds to primarily PDE4D9 and PDE4D8 at resting state, PDE4D9 dissociates from the receptor and PDE4D8 is recruited to the receptor after a transient dissociation upon agonist stimulation. 35 In addition, PDE4D3 and PDE4D5 are also recruited to the activated β2AR. 35, 83, 84 The recruitment/sequestration of different PDE4D isoforms is dependent on the formation of β2AR and arrestin complexes. In a series of recent studies, the arrestin binding sites have been mapped on the C-terminal region, which is conserved throughout the PDE4D family, as well as the unique N-terminal domain of PDE4D5. 103 Under adrenergic stimulation, PDE4D5 can also be ubiquitinated by the E3-ubiquitin ligase, Mdm2 that is scaffolded by arrestin. Ubiquitination of PDE4D5 elicits an increase in the fraction of PDE4D5 sequestration by arrestin in cells, thus potentially decreasing the fraction of PDE4D5 associated with β1AR as well as receptor for activated protein kinase C 1. 104 Meanwhile, PDE4D5 can also be modified by sumoylation, which enhances PKA-mediated activation, but attenuates the ERK-mediated inhibition of the enzyme activities. 105 Together, these data argue a critical role of arrestin in switching off the PDE4D isoform-dependent cAMP degradation under adrenergic stimulation in agonist dose-dependent fashion. It is thus critical to further explore molecular and cellular mechanisms of how arrestin affects the selective association of PDE4D isoforms with different receptor complexes, and shuttling these complexes in distinct cellular organelles in cardiac myocytes.
PDE4 controls cAMP access to AKAP-localized PKA in distinct cellular organelles
The spatiotemporal regulation of βAR signaling in cardiac myocytes is also dependent on the spatial distribution of PKA in distinct subcellular compartments. PKA is consisted of two regulatory subunits and two catalytic subunits. Upon cAMP binding, the catalytic subunits are released from the regulatory subunits, and activated to phosphorylate downstream targets. PKA is tethered to subcellular organelles as well as the cytoskeletal system via binding AKAPs. AKAPs are a large family of sequentially and structurally divergent genes that share a common region in binding to the regulatory subunits of PKA. Due to the ability of AKAPs to bind different cellular proteins/structures in distinct subcellular compartments, PKA is therefore anchored to these locations. 10 This is critical to facilitate the proximity between PKA and its targets, leading to preferential phosphorylation of a local pool of substrates for specific cellular function such as myocyte contraction.
In addition to the aforementioned βAR-associated AKAPs, a growing list of AKAPs is expressed in cardiac tissues, and these AKAPs regulate both βAR signaling and myocyte contraction. 10 For example, AKAP18 isoforms are differentially associated with L-type calcium channels and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) in cardiac myocytes. 106–108 Disruption of PKA anchoring to the L-type calcium channel by AKAP18α significantly inhibits the βAR-induced regulation of the channel activities. 106 In comparison, AKAP18δ scaffolds PKA together with PLB and SERCA to conduct PKA phosphorylation of PLB under adrenergic stimulation, 109 thus regulating SERCA-mediated calcium uptake into the sarcoplasmic reticulum in cardiac myocytes.
Another cardiac AKAP, mAKAP is expressed in cardiac myocytes and interact directly with ryanodine receptor 2 (RyR2) on the sarcoplasmic reticulum. mAKAPs also scaffolds PKA as well as phosphatase 2A, PDE4D3, which forms a signaling module to tightly regulate RyR2 phosphorylation and activity for calcium release from the sarcoplasmic reticulum. 19, 110 mAKAP is also implicated in cross-regulating protein kinase G signaling in cardiac myocytes for phosphorylation of TnI and myocyte contraction. 111, 112 In addition, mAKAP is shown to localize at the nuclear envelope of cardiac myocytes 113 to coordinate transmission of cAMP/PKA signals as well as other signaling into nucleus for cardiac remodeling via phosphorylation of histone deacetylase 5. 114, 115
Upon dissociation and sequestration of PDEs, the βAR-induced cAMP is capable of diffusing into cardiac myocytes from the plasma membrane to reach the PKA anchored onto the different cellular organelles. Because most PDE4s and PDE3s are membrane bound 72, 116, it is plausible that the diffusion of cAMP through the intracellular space is relatively easy with limited restriction. Supporting this notion, a local release of caged cAMP leads to PKA activation at a distance. 117 In another study, using a local perfusion to activate βARs, the cAMP is detected almost throughout the body of myocytes. 47 However, this scenario can be complicated by a couple of other factors. First, myocytes have an extensive t-tubular structure throughout the cell body, and both βARs are presented on the t-tubular membrane. Therefore, agonist can theoretically diffuse through the t-tubular structure to activate the signaling machinery for cAMP production in different locations, rendering the necessity for long-distance intracellular cAMP diffusion. Supporting this notion, AC5 is anchored by mAKAP on the sarcoplasmic reticulum membrane. 118 While the architecture of the complex formation remains to be characterized, these physical associations raise a possibility that cAMP can be produced at the vicinity of the sarcoplasmic reticulum. Meanwhile, membrane t-tubular structures and myofibrils form extensive physical “barriers’ throughout the cell body of cardiac myocytes. These “barriers” limit the cAMP diffusion induced by a local stimulation of βARs. 47 The development of t-tubular structures also underscores the difference between neonatal myocytes and adult myocytes, indicating a much more tightly controlled cAMP diffusion in adult cardiac myocytes. 38 In the future, direct measurement of cAMP and PKA activities with locally tethered biosensors in a subcellular compartment like the sarcoplasmic reticulum 119 will be crucial to analyze the spatiotemporal regulation of cAMP for cardiac contractile responses under adrenergic stimulation.
Since PDEs are also present at different cellular organelles, and in the light of the observation that mAKAP directly tethers PDE4D3 in the same complex with PKA, 120 arrival of cAMP signals to these locations/organelles would be short-lived for PKA activation unless the local PDEs are inhibited or sequestrated. Interestingly, PDE4D3 displays increased binding to βAR/arrestin complexes under saturated βAR stimulation in cardiac myocytes. 84 These observations suggest that the βAR stimulation could also sequester the PDEs away from the mAKAP-anchored PKA on the sarcoplasmic reticulum membrane (Figure 2B) as well as other different organelles such as myofibrils. The sequestration of the sarcoplasmic reticulum membrane-bound PDEs then allows accumulation of cAMP on the membrane, which promotes sustained PKA activities and phosphorylation of RyR2 and SERCA-associated PLB to maintain elevated SR calcium cycling for contractile response. In agreement with the notion, both forskolin and isoproterenol induce similar cAMP and PKA activities in cytosol, 16, 17, 38, 70, 71 but the βAR-induced PKA activities in the sarcoplasmic reticulum are much higher than those induced by forskolin. 119 This suggests that activation of βAR machinery preferentially promotes cAMP accumulation on the sarcoplasmic reticulum for PKA activation. Moreover, high concentration of agonist stimulation also induces sustained PKA activities on the sarcoplasmic reticulum, which are dependent on continuous occupation of the receptor with ligands. 119 Therefore, these data support the notion that, despite a close proximity between the βAR/Gs/AC machinery on the t-tubular membrane and the PKA substrates on the sarcoplasmic reticulum, the βAR-induced cAMP appears to undergo at least two independent steps to activate PKA on the sarcoplasmic reticulum: sequestration of PDE from βAR/Gs/AC complexes to open the gate for cAMP diffusion, and removal or inhibition of PDEs from the sarcoplasmic reticulum to allow accumulation of cAMP for sustained PKA activation and phosphorylation of targeted proteins (Figure 2B).
In addition, AKAPs can also scaffold phosphatases in the complex with PKA, which serves as another negative feedback loop to counterbalance the PKA activities. Recent studies show that phosphatases 2A are transported away from myofibrils under βAR stimulation. 121 This observation supports the idea that the negative feedback phosphatases, like PDEs, are sequestrated from the targeted organelles to maintain sustained PKA phosphorylation for cardiac contractile response. However, phosphatases can also be recruited to the PKA/AKAP complexes 77 and activated by PKA 122 to counter the PKA activities. This negative feedback may be critical when the cAMP activities are extremely high in cardiac myocytes, such as after PDEs are artificially inhibited. 77 Under these conditions, phosphatases serve as a downstream protective mechanism to prevent hyperphosphorylation of substrates such as PLB and TnI by PKA, and prevent myocyte overstimulation. 77 Clinically, both hyperphosphorylation 123 and decreased phosphorylation 124 have been observed in many proteins in failing hearts, which underscores the important role of PDEs and phosphatases in maintaining normal cardiac function. 125
Divergent β1 and β2AR signaling in cardiac myocytes
While both β1 and β2ARs are significantly expressed in cardiac myocytes, activation of individual subtypes in myocardium exerts divergent and sometimes even opposing effects on cardiac function. Activation of the β1AR leads to increase both contractile response and rate in vitro 38, 126 and deletion of the β1AR in myocardium completely abolished the contractile responses upon perfusion of isoproterenol. 127, 128 In contrast, activation of the β2AR has minimal effects on both contraction rate and contractility in vitro, 38, 126 and deletion of the β2AR does not affect the contractile responses to perfusion of isoproterenol. 128, 129 These observations suggest that β1 and β2ARs induce distinct cellular signaling for specific function in cardiac myocytes.
The β1 and β2AR-induced signals are probed in the myocytes lacking individual β1 and β2AR genes. In myocytes lacking the β2AR, stimulation of β1AR induces sustained cAMP and PKA signals that are able to promote PKA phosphorylation of PLB and TnI, and contraction responses in both neonatal and adult cardiac myocytes. 38 In myocytes lacking the β1AR, stimulation of β2AR induces transient cAMP and PKA signals, which can promote PKA phosphorylation of the receptor, but has limited access to PLB and TnI in neonatal cardiac myocytes for small increase in contraction rate response. 38 The duration of cAMP and PKA activities in adult myocytes under β2AR stimulation are even shorter, 38 indicating a tight segregation of cAMP signals. As a consequence, the β2AR stimulation fails to promote cardiac myocyte contractile shortening response. 38
In addition, the cAMP signal induced by β2AR can be further shaped by the receptor coupling to Gi. 38, 101 Interestingly, the β2AR/Gi coupling is dependent on both PKA and GRK phosphorylation, and requires transportation of activated receptor via internalization and recycling. 41, 101, 130–132 Therefore, inhibition of Gi does not alter the cAMP response at 10−9 M of isoproterenol stimulation, a concentration not sufficient to promote GRK phosphorylation. 101 At saturated concentrations of isoproterenol (10−5 M), inhibition of Gi promotes duration of cAMP signal, and PKA phosphorylation on the sarcoplasmic reticulum and on the myofibrils. 101 Consequently, inhibition of Gi also enables the β2AR stimulation to promote calcium signaling, 133–135 and myocyte contractile responses. 101
The observations of dissociation of PDE4D8 from the β1AR and association of PDE4D8 to the β2AR/arrestin complex under high concentrations of agonist stimulation raise a possibility for synergistic effects on cAMP accumulation and diffusion when both receptors are co-activated in cardiac myocytes. In this model (Figure 2B), the β2AR/arrestin serves as the sequestration mechanism not only for PDE4D8 dissociated from the β1AR, but also PDE4D5 from the cytosol and PDE4D3 from the sarcoplasmic reticulum membrane. Thus, the cAMP produced from β1AR activation has a clear path to travel from the plasma membrane to the sarcoplasmic reticulum. Several lines of evidence support this hypothesis. First, β1AR is the major subtype to promote cardiac contractile responses whereas β2AR has minimal role in promoting contractile responses. 38, 126, 127, 129 Second, β1AR displays limited internalization whereas β2AR undergoes robust internalization upon agonist stimulation. 98 The sequestration of PDE4 by β2AR/arrestin complex on the endosome 35, 83 facilitates segregation of the cAMP production by β1AR/Gs/AC machinery from the cAMP degradation enzymes. Therefore, co-stimulation of β1 and β2ARs at the postsynapse could lead to transportation of PDE4D8 from the β1AR/Gs/AC complex at the postsynapse to the β2AR/arrestin complex on the endosome, opening the “gate” for the receptor-induced cAMP diffusion and propagation into the organelles such as the sarcoplasmic reticulum in cardiac myocytes.
Crosstalk among βARs and other GPCRs for cAMP/PKA signaling transduction
Although the expression of β3AR is relative low in myocardium, the expression is detected in different mammalian species from human to rodents and regulates cardiac function. 7 Activation of β3AR induces different signaling pathway in cell lines or primary tissues, including Gs, AMP-activated protein kinase, and endothelial nitric oxide synthase. 7 Early studies with functional characterization have shown that activation of β3AR leads to contradictory observations ranging from significantly enhanced contractile response to minimal or even reduced contractile responses. 136 Using myocytes lacking both β1 and β2ARs, Devic et. al. have showed that stimulation with βAR-specific agonist isoproterenol or β3AR-specific agonist CL-316243 induced a small decrease in rate in spontaneously contracting mouse neonatal myocytes. 126 This observation indicates that β3AR directly exerts a negative effect on contraction responses in murine cardiac myocytes. Recent studies have revealed that cross-talk between β3AR-induced cGMP and other βAR-induced cAMP in mouse cardiac myocytes. 9, 71 The β3AR-induced cGMP activates PDE2 to enhance its catalytic activity for cAMP, which negatively modulates the cAMP induced by Gs signaling. 9, 71 In this case, inhibition of β3AR significantly enhances the maximal cAMP accumulation after stimulation of wild type myocyte with norepinephrine. 71 Meanwhile, inhibition of PDE2 also promotes cAMP accumulation induced by norepinephrine. 71 Interestingly, a third generation of cardiac-specific and β1-selective blocker nebivolol also stimulates endothelial nitric oxide synthase, 137 which can enhance nitric oxide/cGMP activities to attenuate the cAMP signaling for myocytes contractile function. Further examination of this signaling crosstalk may help us to better understand how drugs like nebivolol work in heart failure patients. Therefore, under stimulation of isoproterenol and norepinephrine, β3AR-induced cGMP has a negative impact on cAMP accumulation, thus reduces the contraction response.
The fact that many GPCRs expressed in myocytes display relative segregation indicates that they have distinct functionality. However, many receptors are also localized/enriched in lipid rafts/caveolae, 27 raising the opportunities for signaling cross-talk between other GPCRs and adrenergic signaling. 138, 139 For example, muscarinic stimulation can attenuate cAMP signaling induced by βARs. This is dependent on the ligand occupation of muscarinic receptor; the inhibition was rapidly reversed when the agonist is removed. 139 The mechanism underlying the cross-talk for the observed response in cAMP signals is not entirely clear. The coupling of muscarinic receptor to Gi is involved in the cross-talk. 140 However, the rapid reverse of the inhibitory effect suggests that additional modulation of other regulators such as PDEs could play a role in the process. 140 Further analysis with live-cell imaging will help to understand how other neurohormonal stimulation affects the βAR signaling cascades under various physiological and clinic cardiac conditions.
Clinical implication of spatiotemporal regulation of adrenergic signaling in cardiac myocytes
Downregulation of the β1AR and adrenergic response are hallmarks of human heart failure, as a result of chronic stimulation under elevated circulating catecholamines in plasma and increased sympathetic tone. Recently, evidence has emerged that the adrenergic receptor-induced cAMP/PKA signaling is also altered in cardiac myocytes under chronic conditions. These alterations probably occur before downregulation of the β1AR, an indicator of the ending stage in heart failure. Using a sophisticated imaging technique, Nikolaev observed that β2AR is redistributed from t-tubular structure to the plasma membrane in failing cardiac myocytes, thus enhancing the cAMP distribution under agonist stimulation. 48 In hypertrophic or failing hearts, the expression of PDE3A, PDE4A, 4B, 4D, and PDE5A are down regulated, 19, 80, 81, 141, 142 although the increase in expression of PDE5A is also reported in different studies. 143, 144 In comparison, the PDE1A and PD2A expression are increased in hypertrophic heart. 145, 146 The contradictory reports on expression levels of PDE genes in these studies are likely due to the detection of the proteins at different stage of diseases as well as using different animal models. For example, a recent report shows that the expression of PDE4 as well as other PDEs such as PDE1, PDE2, PDE5 is broadly increased in the early stage of cardiac hypertrophy induced by chronic Angiotensin II perfusion. 147 Of these observed changes, the decrease expression of PDE4D3 and its association with RyR2 is particularly interesting since it causes elevated cAMP/PKA activities in local domain for hyperphorphorylation of RyR2. 19 Such a hyperphosphorylation leads to increase of channel activities, contributing leaking of calcium from the sarcoplasmic reticulum, as well as arrhythmia and sudden death in heart failure patients. 19 Together, these data underscore the evolving expression of PDE isoforms in compensating the alteration of adrenergic signaling during the development of heart failure, thus offering potential targets for clinical therapy.
Conclusion and Remarks
Recent advances in biochemical characterization of βAR signaling complexes and the development of live-cell imaging of spatiotemporal regulation of βAR signaling in cardiac myocytes offer new paradigms to understand how signaling transduction is translated into physiological contractile response. The above discussion on separation of cAMP production vs. cAMP accumulation and diffusion, as well as the equilibrium between cAMP production and degradation offers new concepts to dissect how the βAR/cAMP signaling is transduced in the highly differentiated and structurally rigid cardiac myocytes in an agonist dose-dependent manner. This information also offers new directions to analyze subtle alterations during development of pathological conditions. Several key questions remain to be addressed. The compositions of the βAR complexes need to be further characterized, in particular, whether PDEs and ACs are associated with the same receptor complexes, and whether different pools of βARs exist in a single cell. The mechanism of PDE dissociation from the activated receptors and the master role of arrestin as a switch for cAMP accumulation and diffusion at high concentration of agonist stimulation remain to be further characterized. These are critical steps for accumulation and propagation of cAMP signals for physiological cardiac contraction. Moreover, the role of PDE3 in adrenergic signaling and cardiac contractile regulation remains to be investigated. Last and most importantly, an understanding of how the precisely controlled spatiotemporal cAMP signals are altered is essential during early adaption in myocardium in chronic conditions, such as diabetes and chronic inflammation, and during development of heart failure. Any insights in understanding these processes will potentially have tremendous impact on the research direction and clinical practice.
Acknowledgements
I thank members of Xiang lab for critical reading and comments and Sungjin Kim for preparation the figures.
Source of Funding
This study is supported by NIH grant HL082846.
Abbreviations
- AR
adrenergic receptor
- AC
adenylyl cyclase
- PDE
phosphodiesterase
- PKA
protein kinase A
- AKAP
A-kinase anchoring protein
- SAP
synaptic associated protein
- IBMX
3-isobutyl-1-methylxanthine
- cAMP
cyclic adenosine monophosphate
- cGMP
cyclic guanosine monophosphate
- PLB
phospholamban
- TnI
troponin I
- GRK
G-protein receptor kinase
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
- RyR2
ryanodine receptor 2
- mAKAP
muscle A kinase–anchoring protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None
References
- 1.Xiang Y, Kobilka BK. Myocyte adrenoceptor signaling pathways. Science. 2003;300:1530–1532. doi: 10.1126/science.1079206. [DOI] [PubMed] [Google Scholar]
- 2.Xiao RP, Zhu W, Zheng M, Cao C, Zhang Y, Lakatta EG, Han Q. Subtype-specific alpha1- and beta-adrenoceptor signaling in the heart. Trends Pharmacol Sci. 2006;27:330–337. doi: 10.1016/j.tips.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Lefkowitz RJ. Seven transmembrane receptors: Something old, something new. Acta Physiol (Oxf) 2007;190:9–19. doi: 10.1111/j.1365-201X.2007.01693.x. [DOI] [PubMed] [Google Scholar]
- 4.Salazar NC, Chen J, Rockman HA. Cardiac gpcrs: Gpcr signaling in healthy and failing hearts. Biochim Biophys Acta. 2007;1768:1006–1018. doi: 10.1016/j.bbamem.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dorn GW, 2nd, Liggett SB. Pharmacogenomics of beta-adrenergic receptors and their accessory signaling proteins in heart failure. Clin Transl Sci. 2008;1:255–262. doi: 10.1111/j.1752-8062.2008.00059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pleger ST, Boucher M, Most P, Koch WJ. Targeting myocardial beta-adrenergic receptor signaling and calcium cycling for heart failure gene therapy. J Card Fail. 2007;13:401–414. doi: 10.1016/j.cardfail.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Rasmussen HH, Figtree GA, Krum H, Bundgaard H. The use of beta3-adrenergic receptor agonists in the treatment of heart failure. Curr Opin Investig Drugs. 2009;10:955–962. [PubMed] [Google Scholar]
- 8.Zaccolo M. Camp signal transduction in the heart: Understanding spatial control for the development of novel therapeutic strategies. Br J Pharmacol. 2009;158:50–60. doi: 10.1111/j.1476-5381.2009.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaccolo M, Movsesian MA. Camp and cgmp signaling cross-talk: Role of phosphodiesterases and implications for cardiac pathophysiology. Circ Res. 2007;100:1569–1578. doi: 10.1161/CIRCRESAHA.106.144501. [DOI] [PubMed] [Google Scholar]
- 10.McConnachie G, Langeberg LK, Scott JD. Akap signaling complexes: Getting to the heart of the matter. Trends Mol Med. 2006;12:317–323. doi: 10.1016/j.molmed.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 11.Carnegie GK, Means CK, Scott JD. A-kinase anchoring proteins: From protein complexes to physiology and disease. IUBMB Life. 2009;61:394–406. doi: 10.1002/iub.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mauban JR, O'Donnell M, Warrier S, Manni S, Bond M. Akap-scaffolding proteins and regulation of cardiac physiology. Physiology (Bethesda) 2009;24:78–87. doi: 10.1152/physiol.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinberg SF, Brunton LL. Compartmentation of g protein-coupled signaling pathways in cardiac myocytes. Annu Rev Pharmacol Toxicol. 2001;41:751–773. doi: 10.1146/annurev.pharmtox.41.1.751. [DOI] [PubMed] [Google Scholar]
- 14.Houslay MD, Baillie GS, Maurice DH. Camp-specific phosphodiesterase-4 enzymes in the cardiovascular system: A molecular toolbox for generating compartmentalized camp signaling. Circ Res. 2007;100:950–966. doi: 10.1161/01.RES.0000261934.56938.38. [DOI] [PubMed] [Google Scholar]
- 15.Cooper DM. Compartmentalization of adenylate cyclase and camp signalling. Biochem Soc Trans. 2005;33:1319–1322. doi: 10.1042/BST0331319. [DOI] [PubMed] [Google Scholar]
- 16.Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechene P, Mazet JL, Conti M, Fischmeister R, Vandecasteele G. Spatiotemporal dynamics of beta-adrenergic camp signals and l-type ca2+ channel regulation in adult rat ventricular myocytes: Role of phosphodiesterases. Circ Res. 2008;102:1091–1100. doi: 10.1161/CIRCRESAHA.107.167817. [DOI] [PubMed] [Google Scholar]
- 17.Rochais F, Vilardaga JP, Nikolaev VO, Bunemann M, Lohse MJ, Engelhardt S. Real-time optical recording of beta1-adrenergic receptor activation reveals supersensitivity of the arg389 variant to carvedilol. J Clin Invest. 2007;117:229–235. doi: 10.1172/JCI30012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jarnaess E, Tasken K. Spatiotemporal control of camp signalling processes by anchored signalling complexes. Biochem Soc Trans. 2007;35:931–937. doi: 10.1042/BST0350931. [DOI] [PubMed] [Google Scholar]
- 19.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4d deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the kcnq1-kcne1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 21.Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. 1998;98:1329–1334. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- 22.Singh K, Xiao L, Remondino A, Sawyer DB, Colucci WS. Adrenergic regulation of cardiac myocyte apoptosis. J Cell Physiol. 2001;189:257–265. doi: 10.1002/jcp.10024. [DOI] [PubMed] [Google Scholar]
- 23.Singh K, Communal C, Sawyer DB, Colucci WS. Adrenergic regulation of myocardial apoptosis. Cardiovasc Res. 2000;45:713–719. doi: 10.1016/s0008-6363(99)00370-3. [DOI] [PubMed] [Google Scholar]
- 24.Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, Devic E, Kobilka BK, Cheng H, Xiao RP. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase a-independent activation of ca2+/calmodulin kinase ii. J Clin Invest. 2003;111:617–625. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi DJ, Rockman HA. Beta-adrenergic receptor desensitization in cardiac hypertrophy and heart failure. Cell Biochem Biophys. 1999;31:321–329. doi: 10.1007/BF02738246. [DOI] [PubMed] [Google Scholar]
- 26.Sugden PH. Signalling pathways in cardiac myocyte hypertrophy. Ann Med. 2001;33:611–622. [PubMed] [Google Scholar]
- 27.Patel HH, Murray F, Insel PA. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu Rev Pharmacol Toxicol. 2008;48:359–391. doi: 10.1146/annurev.pharmtox.48.121506.124841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shcherbakova OG, Hurt CM, Xiang Y, Dell'Acqua ML, Zhang Q, Tsien RW, Kobilka BK. Organization of beta-adrenoceptor signaling compartments by sympathetic innervation of cardiac myocytes. J Cell Biol. 2007;176:521–533. doi: 10.1083/jcb.200604167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW, Scott JD. Dynamic regulation of camp synthesis through anchored pka-adenylyl cyclase v/vi complexes. Mol Cell. 2006;23:925–931. doi: 10.1016/j.molcel.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraser ID, Cong M, Kim J, Rollins EN, Daaka Y, Lefkowitz RJ, Scott JD. Assembly of an a kinase-anchoring protein-beta(2)-adrenergic receptor complex facilitates receptor phosphorylation and signaling. Curr Biol. 2000;10:409–412. doi: 10.1016/s0960-9822(00)00419-x. [DOI] [PubMed] [Google Scholar]
- 31.Malbon CC. A-kinase anchoring proteins: Trafficking in g-protein-coupled receptors and the proteins that regulate receptor biology. Curr Opin Drug Discov Devel. 2007;10:573–579. [PubMed] [Google Scholar]
- 32.Gardner LA, Tavalin SJ, Goehring AS, Scott JD, Bahouth SW. Akap79-mediated targeting of the cyclic amp-dependent protein kinase to the beta1-adrenergic receptor promotes recycling and functional resensitization of the receptor. J Biol Chem. 2006;281:33537–33553. doi: 10.1074/jbc.M601809200. [DOI] [PubMed] [Google Scholar]
- 33.De Arcangelis V, Liu S, Zhang D, Soto D, Xiang YK. Equilibrium between adenylyl cyclase and phosphodiesterase patterns adrenergic agonist dose-dependent spatiotemporal camp/protein kinase a activities in cardiomyocytes. Mol Pharmacol. 2010;78:340–349. doi: 10.1124/mol.110.064444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richter W, Day P, Agrawal R, Bruss MD, Granier S, Wang YL, Rasmussen SG, Horner K, Wang P, Lei T, Patterson AJ, Kobilka B, Conti M. Signaling from beta1- and beta2-adrenergic receptors is defined by differential interactions with pde4. Embo J. 2008;27:384–393. doi: 10.1038/sj.emboj.7601968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Arcangelis V, Liu R, Soto D, Xiang Y. Differential association of phosphodiesterase 4d isoforms with beta2-adrenoceptor in cardiac myocytes. J Biol Chem. 2009;284:33824–33832. doi: 10.1074/jbc.M109.020388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiang Y, Naro F, Zoudilova M, Jin SL, Conti M, Kobilka B. Phosphodiesterase 4d is required for {beta}2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc Natl Acad Sci U S A. 2005 doi: 10.1073/pnas.0405263102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaccolo M, Pozzan T. Discrete microdomains with high concentration of camp in stimulated rat neonatal cardiac myocytes. Science. 2002;295:1711–1715. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- 38.Soto D, De Arcangelis V, Zhang J, Xiang Y. Dynamic protein kinase a activities induced by beta-adrenoceptors dictate signaling propagation for substrate phosphorylation and myocyte contraction. Circ Res. 2009;104:770–779. doi: 10.1161/CIRCRESAHA.108.187880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac l-type ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc Natl Acad Sci U S A. 2006;103:7500–7505. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rybin VO, Xu X, Lisanti MP, Steinberg SF. Differential targeting of beta -adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the camp signaling pathway. J Biol Chem. 2000;275:41447–41457. doi: 10.1074/jbc.M006951200. [DOI] [PubMed] [Google Scholar]
- 41.Xiang Y, Rybin VO, Steinberg SF, Kobilka B. Caveolar localization dictates physiologic signaling of beta 2- adrenoceptors in neonatal cardiac myocytes. J Biol Chem. 2002;277:34280–34286. doi: 10.1074/jbc.M201644200. [DOI] [PubMed] [Google Scholar]
- 42.Ostrom RS, Insel PA. The evolving role of lipid rafts and caveolae in g protein-coupled receptor signaling: Implications for molecular pharmacology. Br J Pharmacol. 2004;143:235–245. doi: 10.1038/sj.bjp.0705930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Penela P, Ribas C, Mayor F., Jr Mechanisms of regulation of the expression and function of g protein-coupled receptor kinases. Cell Signal. 2003;15:973–981. doi: 10.1016/s0898-6568(03)00099-8. [DOI] [PubMed] [Google Scholar]
- 44.Jurevicius J, Skeberdis VA, Fischmeister R. Role of cyclic nucleotide phosphodiesterase isoforms in camp compartmentation following beta2-adrenergic stimulation of ica,l in frog ventricular myocytes. J Physiol. 2003;551:239–252. doi: 10.1113/jphysiol.2003.045211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao R-P, Lakatta E. S1-adrenoceptor stimulation and β2-adrenoceptor stimulation differ in their effects on contraction, cytosolic ca++, and ca++ current in single rat ventricular cells. Circ Res. 1993;73:286–300. doi: 10.1161/01.res.73.2.286. [DOI] [PubMed] [Google Scholar]
- 46.Chen-Izu Y, Xiao RP, Izu LT, Cheng H, Kuschel M, Spurgeon H, Lakatta EG. G(i)-dependent localization of beta(2)-adrenergic receptor signaling to l-type ca(2+) channels. Biophys J. 2000;79:2547–2556. doi: 10.1016/S0006-3495(00)76495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nikolaev VO, Bunemann M, Schmitteckert E, Lohse MJ, Engelhardt S. Cyclic amp imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ Res. 2006;99:1084–1091. doi: 10.1161/01.RES.0000250046.69918.d5. [DOI] [PubMed] [Google Scholar]
- 48.Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE, Gorelik J. Beta2-adrenergic receptor redistribution in heart failure changes camp compartmentation. Science. 2010;327:1653–1657. doi: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 49.Willoughby D, Cooper DM. Organization and ca2+ regulation of adenylyl cyclases in camp microdomains. Physiol Rev. 2007;87:965–1010. doi: 10.1152/physrev.00049.2006. [DOI] [PubMed] [Google Scholar]
- 50.Dessauer CW. Adenylyl cyclase--a-kinase anchoring protein complexes: The next dimension in camp signaling. Mol Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Defer N, Best-Belpomme M, Hanoune J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am J Physiol Renal Physiol. 2000;279:F400–F416. doi: 10.1152/ajprenal.2000.279.3.F400. [DOI] [PubMed] [Google Scholar]
- 52.Qin K, Sethi PR, Lambert NA. Abundance and stability of complexes containing inactive g protein-coupled receptors and g proteins. Faseb J. 2008;22:2920–2927. doi: 10.1096/fj.08-105775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hein P, Bunemann M. Coupling mode of receptors and g proteins. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:435–443. doi: 10.1007/s00210-008-0383-7. [DOI] [PubMed] [Google Scholar]
- 54.Tian WN, Duzic E, Lanier SM, Deth RC. Determinants of alpha 2-adrenergic receptor activation of g proteins: Evidence for a precoupled receptor/g protein state. Mol Pharmacol. 1994;45:524–531. [PubMed] [Google Scholar]
- 55.Weitl N, Seifert R. Distinct interactions of human beta1- and beta2-adrenoceptors with isoproterenol, epinephrine, norepinephrine, and dopamine. J Pharmacol Exp Ther. 2008;327:760–769. doi: 10.1124/jpet.108.143412. [DOI] [PubMed] [Google Scholar]
- 56.Seifert R, Wenzel-Seifert K, Lee TW, Gether U, Sanders-Bush E, Kobilka BK. Different effects of gsα splice variants on β2-adrenoreceptor- mediated signaling. The β2-adrenoreceptor coupled to the long splice variant of gsα has properties of a constitutively active receptor. J Biol Chem. 1998;273:5109–5116. [PubMed] [Google Scholar]
- 57.Seifert R, Wenzel-Seifert K, Kobilka BK. Gpcr-galpha fusion proteins: Molecular analysis of receptor-g-protein coupling. Trends Pharmacol Sci. 1999;20:383–389. doi: 10.1016/s0165-6147(99)01368-1. [Record as supplied by publisher] [DOI] [PubMed] [Google Scholar]
- 58.Lin F, Wang H, Malbon CC. Gravin-mediated formation of signaling complexes in beta 2-adrenergic receptor desensitization and resensitization. J Biol Chem. 2000;275:19025–19034. doi: 10.1074/jbc.275.25.19025. [DOI] [PubMed] [Google Scholar]
- 59.Cong M, Perry SJ, Lin FT, Fraser ID, Hu LA, Chen W, Pitcher JA, Scott JD, Lefkowitz RJ. Regulation of membrane targeting of the g protein-coupled receptor kinase 2 by protein kinase a and its anchoring protein akap79. J Biol Chem. 2001;276:15192–15199. doi: 10.1074/jbc.M009130200. [DOI] [PubMed] [Google Scholar]
- 60.Hall RA, Premont RT, Chow CW, Blitzer JT, Pitcher JA, Claing A, Stoffel RH, Barak LS, Shenolikar S, Weinman EJ, Grinstein S, Lefkowitz RJ. The beta2-adrenergic receptor interacts with the na+/h+-exchanger regulatory factor to control na+/h+ exchange. Nature. 1998;392:626–630. doi: 10.1038/33458. [DOI] [PubMed] [Google Scholar]
- 61.Cong M, Perry SJ, Hu LA, Hanson PI, Claing A, Lefkowitz RJ. Binding of the beta2 adrenergic receptor to n-ethylmaleimide-sensitive factor regulates receptor recycling. J Biol Chem. 2001;276:45145–45152. doi: 10.1074/jbc.M106087200. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y, Lauffer B, Von Zastrow M, Kobilka BK, Xiang Y. N-ethylmaleimide-sensitive factor regulates beta2 adrenoceptor trafficking and signaling in cardiomyocytes. Mol Pharmacol. 2007;72:429–439. doi: 10.1124/mol.107.037747. [DOI] [PubMed] [Google Scholar]
- 63.Hu LA, Tang Y, Miller WE, Cong M, Lau AG, Lefkowitz RJ, Hall RA. Beta 1-adrenergic receptor association with psd-95. Inhibition of receptor internalization and facilitation of beta 1-adrenergic receptor interaction with n-methyl-d-aspartate receptors. J Biol Chem. 2000;275:38659–38666. doi: 10.1074/jbc.M005938200. [DOI] [PubMed] [Google Scholar]
- 64.Hu LA, Chen W, Martin NP, Whalen EJ, Premont RT, Lefkowitz RJ. Gipc interacts with the beta1-adrenergic receptor and regulates beta1-adrenergic receptor-mediated erk activation. J Biol Chem. 2003;278:26295–26301. doi: 10.1074/jbc.M212352200. [DOI] [PubMed] [Google Scholar]
- 65.Xu J, Paquet M, Lau AG, Wood JD, Ross CA, Hall RA. Beta 1-adrenergic receptor association with the synaptic scaffolding protein membrane-associated guanylate kinase inverted-2 (magi-2). Differential regulation of receptor internalization by magi-2 and psd-95. J Biol Chem. 2001;276:41310–41317. doi: 10.1074/jbc.M107480200. [DOI] [PubMed] [Google Scholar]
- 66.Fujita A, Kurachi Y. Sap family proteins. Biochem Biophys Res Commun. 2000;269:1–6. doi: 10.1006/bbrc.1999.1893. [DOI] [PubMed] [Google Scholar]
- 67.Gardner LA, Naren AP, Bahouth SW. Assembly of an sap97-akap79-camp-dependent protein kinase scaffold at the type 1 psd-95/dlg/zo1 motif of the human beta(1)-adrenergic receptor generates a receptosome involved in receptor recycling and networking. J Biol Chem. 2007;282:5085–5099. doi: 10.1074/jbc.M608871200. [DOI] [PubMed] [Google Scholar]
- 68.Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of pka to glutamate receptors through a maguk-akap complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 69.Willoughby D, Wong W, Schaack J, Scott JD, Cooper DM. An anchored pka and pde4 complex regulates subplasmalemmal camp dynamics. Embo J. 2006;25:2051–2061. doi: 10.1038/sj.emboj.7601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD, Zaccolo M. Fluorescence resonance energy transfer-based analysis of camp dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004;95:67–75. doi: 10.1161/01.RES.0000134629.84732.11. [DOI] [PubMed] [Google Scholar]
- 71.Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, Pozzan T, Kass DA, Paolocci N, Houslay MD, Zaccolo M. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an no/cgmp-dependent pathway. Circ Res. 2006;98:226–234. doi: 10.1161/01.RES.0000200178.34179.93. [DOI] [PubMed] [Google Scholar]
- 72.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- 73.Miller CL, Yan C. Targeting cyclic nucleotide phosphodiesterase in the heart: Therapeutic implications. J Cardiovasc Transl Res. 2010;3:507–515. doi: 10.1007/s12265-010-9203-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Richter W, Jin SL, Conti M. Splice variants of the cyclic nucleotide phosphodiesterase pde4d are differentially expressed and regulated in rat tissue. Biochem J. 2005;388:803–811. doi: 10.1042/BJ20050030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Richter W, Xie M, Scheitrum C, Krall J, Movsesian MA, Conti M. Conserved expression and functions of pde4 in rodent and human heart. Basic research in cardiology. 2011;106:249–262. doi: 10.1007/s00395-010-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Patrucco E, Albergine MS, Santana LF, Beavo JA. Phosphodiesterase 8a (pde8a) regulates excitation-contraction coupling in ventricular myocytes. Journal of molecular and cellular cardiology. 2010;49:330–333. doi: 10.1016/j.yjmcc.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.De Arcangelis V, Soto D, Xiang Y. Phosphodiesterase 4 and phosphatase 2a differentially regulate camp/protein kinase a signaling for cardiac myocyte contraction under stimulation of beta1 adrenergic receptor. Mol Pharmacol. 2008;74:1453–1462. doi: 10.1124/mol.108.049718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bruss MD, Richter W, Horner K, Jin SL, Conti M. Critical role of pde4d in beta2-adrenoceptor-dependent camp signaling in mouse embryonic fibroblasts. J Biol Chem. 2008;283:22430–22442. doi: 10.1074/jbc.M803306200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kerfant BG, Zhao D, Lorenzen-Schmidt I, Wilson LS, Cai S, Chen SR, Maurice DH, Backx PH. Pi3kgamma is required for pde4, not pde3, activity in subcellular microdomains containing the sarcoplasmic reticular calcium atpase in cardiomyocytes. Circ Res. 2007;101:400–408. doi: 10.1161/CIRCRESAHA.107.156422. [DOI] [PubMed] [Google Scholar]
- 80.Ding B, Abe J, Wei H, Xu H, Che W, Aizawa T, Liu W, Molina CA, Sadoshima J, Blaxall BC, Berk BC, Yan C. A positive feedback loop of phosphodiesterase 3 (pde3) and inducible camp early repressor (icer) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci U S A. 2005;102:14771–14776. doi: 10.1073/pnas.0506489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ding B, Abe J, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, Yan C. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: Implication in heart failure. Circulation. 2005;111:2469–2476. doi: 10.1161/01.CIR.0000165128.39715.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Conti M, Richter W, Mehats C, Livera G, Park JY, Jin C. Cyclic amp-specific pde4 phosphodiesterases as critical components of cyclic amp signaling. J Biol Chem. 2003;278:5493–5496. doi: 10.1074/jbc.R200029200. [DOI] [PubMed] [Google Scholar]
- 83.Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ. Targeting of cyclic amp degradation to beta 2-adrenergic receptors by beta-arrestins. Science. 2002;298:834–836. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- 84.Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD. Beta-arrestin-mediated pde4 camp phosphodiesterase recruitment regulates beta-adrenoceptor switching from gs to gi. Proc Natl Acad Sci U S A. 2003;100:940–945. doi: 10.1073/pnas.262787199. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85.Halls ML, Cooper DM. Sub-picomolar relaxin signalling by a pre-assembled rxfp1, akap79, ac2, beta-arrestin 2, pde4d3 complex. Embo J. 2010;29:2772–2787. doi: 10.1038/emboj.2010.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hein P, Rochais F, Hoffmann C, Dorsch S, Nikolaev VO, Engelhardt S, Berlot CH, Lohse MJ, Bunemann M. Gs activation is time-limiting in initiating receptor-mediated signaling. J Biol Chem. 2006;281:33345–33351. doi: 10.1074/jbc.M606713200. [DOI] [PubMed] [Google Scholar]
- 87.Ostrom RS, Gregorian C, Drenan RM, Xiang Y, Regan JW, Insel PA. Receptor number and caveolar co-localization determine receptor coupling efficiencyto adenylyl cyclase. J Biol Chem. 2001;276:42063–42069. doi: 10.1074/jbc.M105348200. [DOI] [PubMed] [Google Scholar]
- 88.Corbin JD, Keely SL. Characterization and regulation of heart adenosine 3':5'-monophosphate-dependent protein kinase isozymes. J Biol Chem. 1977;252:910–918. [PubMed] [Google Scholar]
- 89.Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. A beta2 adrenergic receptor signaling complex assembled with the ca2+ channel cav1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- 90.Naren AP, Cobb B, Li C, Roy K, Nelson D, Heda GD, Liao J, Kirk KL, Sorscher EJ, Hanrahan J, Clancy JP. A macromolecular complex of beta 2 adrenergic receptor, cftr, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by pka. Proc Natl Acad Sci U S A. 2003;100:342–346. doi: 10.1073/pnas.0135434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barbuti A, DiFrancesco D. Control of cardiac rate by "funny" channels in health and disease. Ann N Y Acad Sci. 2008;1123:213–223. doi: 10.1196/annals.1420.024. [DOI] [PubMed] [Google Scholar]
- 92.Shenoy SK, Lefkowitz RJ. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem J. 2003;375:503–515. doi: 10.1042/BJ20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ferguson SS, Barak LS, Zhang J, Caron MG. G-protein-coupled receptor regulation: Role of g-protein-coupled receptor kinases and arrestins. Can J Physiol Pharmacol. 1996;74:1095–1110. doi: 10.1139/cjpp-74-10-1095. [DOI] [PubMed] [Google Scholar]
- 94.Cassel D, Selinger Z. Catecholamine-stimulated gtpase activity in turkey erythrocyte membranes. Biochim Biophys Acta. 1976;452:538–551. doi: 10.1016/0005-2744(76)90206-0. [DOI] [PubMed] [Google Scholar]
- 95.Brandt DR, Ross EM. Catecholamine-stimulated gtpase cycle. Multiple sites of regulation by beta-adrenergic receptor and mg2+ studied in reconstituted receptor-gs vesicles. J Biol Chem. 1986;261:1656–1664. [PubMed] [Google Scholar]
- 96.Shakur Y, Holst LS, Landstrom TR, Movsesian M, Degerman E, Manganiello V. Regulation and function of the cyclic nucleotide phosphodiesterase (pde3) gene family. Prog Nucleic Acid Res Mol Biol. 2001;66:241–277. doi: 10.1016/s0079-6603(00)66031-2. [DOI] [PubMed] [Google Scholar]
- 97.MacKenzie SJ, Baillie GS, McPhee I, MacKenzie C, Seamons R, McSorley T, Millen J, Beard MB, van Heeke G, Houslay MD. Long pde4 camp specific phosphodiesterases are activated by protein kinase a-mediated phosphorylation of a single serine residue in upstream conserved region 1 (ucr1) Br J Pharmacol. 2002;136:421–433. doi: 10.1038/sj.bjp.0704743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xiang Y, Devic E, Kobilka B. The pdz binding motif of the beta 1 adrenergic receptor modulates receptor trafficking and signaling in cardiac myocytes. J Biol Chem. 2002;277:33783–33790. doi: 10.1074/jbc.M204136200. [DOI] [PubMed] [Google Scholar]
- 99.Gage RM, Kim KA, Cao TT, von Zastrow M. A transplantable sorting signal that is sufficient to mediate rapid recycling of g protein-coupled receptors. J Biol Chem. 2001;276:44712–44720. doi: 10.1074/jbc.M107417200. [DOI] [PubMed] [Google Scholar]
- 100.Yudowski GA, Puthenveedu MA, Henry AG, von Zastrow M. Cargo-mediated regulation of a rapid rab4-dependent recycling pathway. Mol Biol Cell. 2009;20:2774–2784. doi: 10.1091/mbc.E08-08-0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu R, Ramani B, Soto D, De Arcangelis V, Xiang Y. Agonist dose-dependent phosphorylation by protein kinase a and g protein-coupled receptor kinase regulates beta2 adrenoceptor coupling to g(i) proteins in cardiomyocytes. J Biol Chem. 2009;284:32279–32287. doi: 10.1074/jbc.M109.021428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tran TM, Friedman J, Qunaibi E, Baameur F, Moore RH, Clark RB. Characterization of agonist stimulation of camp-dependent protein kinase and g protein-coupled receptor kinase phosphorylation of the beta2-adrenergic receptor using phosphoserine-specific antibodies. Mol Pharmacol. 2004;65:196–206. doi: 10.1124/mol.65.1.196. [DOI] [PubMed] [Google Scholar]
- 103.Bolger GB, Baillie GS, Li X, Lynch MJ, Herzyk P, Mohamed A, Mitchell LH, McCahill A, Hundsrucker C, Klussmann E, Adams DR, Houslay MD. Scanning peptide array analyses identify overlapping binding sites for the signalling scaffold proteins, beta-arrestin and rack1, in camp-specific phosphodiesterase pde4d5. The Biochemical journal. 2006;398:23–36. doi: 10.1042/BJ20060423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li X, Baillie GS, Houslay MD. Mdm2 directs the ubiquitination of beta-arrestin-sequestered camp phosphodiesterase-4d5. J. Biol. Chem. 2009;284:16170–16182. doi: 10.1074/jbc.M109.008078. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 105.Li X, Vadrevu S, Dunlop A, Day J, Advant N, Troeger J, Klussmann E, Jaffrey E, Hay RT, Adams DR, Houslay MD, Baillie GS. Selective sumo modification of camp-specific phosphodiesterase-4d5 (pde4d5) regulates the functional consequences of phosphorylation by pka and erk. The Biochemical journal. 2010;428:55–65. doi: 10.1042/BJ20091672. [DOI] [PubMed] [Google Scholar]
- 106.Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. Beta-adrenergic regulation requires direct anchoring of pka to cardiac cav1.2 channels via a leucine zipper interaction with a kinase-anchoring protein 15. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2003;100:13093–13098. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hulme JT, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation of serine 1928 in the distal c-terminal domain of cardiac cav1.2 channels during beta1-adrenergic regulation. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2006;103:16574–16579. doi: 10.1073/pnas.0607294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gray PC, Johnson BD, Westenbroek RE, Hays LG, Yates JR, 3rd, Scheuer T, Catterall WA, Murphy BJ. Primary structure and function of an a kinase anchoring protein associated with calcium channels. Neuron. 1998;20:1017–1026. doi: 10.1016/s0896-6273(00)80482-1. [DOI] [PubMed] [Google Scholar]
- 109.Lygren B, Carlson CR, Santamaria K, Lissandron V, McSorley T, Litzenberg J, Lorenz D, Wiesner B, Rosenthal W, Zaccolo M, Tasken K, Klussmann E. Akap complex regulates ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007;8:1061–1067. doi: 10.1038/sj.embor.7401081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. Pka phosphorylation dissociates fkbp12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 111.Cuello F, Bardswell SC, Haworth RS, Yin X, Lutz S, Wieland T, Mayr M, Kentish JC, Avkiran M. Protein kinase d selectively targets cardiac troponin i and regulates myofilament ca2+ sensitivity in ventricular myocytes. Circulation research. 2007;100:864–873. doi: 10.1161/01.RES.0000260809.15393.fa. [DOI] [PubMed] [Google Scholar]
- 112.Haworth RS, Roberts NA, Cuello F, Avkiran M. Regulation of protein kinase d activity in adult myocardium: Novel counter-regulatory roles for protein kinase cepsilon and protein kinase a. Journal of molecular and cellular cardiology. 2007;43:686–695. doi: 10.1016/j.yjmcc.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 113.Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS. The makap complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. Journal of cell science. 2005;118:5637–5646. doi: 10.1242/jcs.02675. [DOI] [PubMed] [Google Scholar]
- 114.Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase a anchoring protein makap coordinates two integrated camp effector pathways. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases c and d mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Molecular and cellular biology. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.De Arcangelis V, Coletti D, Conti M, Lagarde M, Molinaro M, Adamo S, Nemoz G, Naro F. Igf-i-induced differentiation of l6 myogenic cells requires the activity of camp-phosphodiesterase. Mol Biol Cell. 2003;14:1392–1404. doi: 10.1091/mbc.E02-03-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Saucerman JJ, Zhang J, Martin JC, Peng LX, Stenbit AE, Tsien RY, McCulloch AD. Systems analysis of pka-mediated phosphorylation gradients in live cardiac myocytes. Proc Natl Acad Sci U S A. 2006;103:12923–12928. doi: 10.1073/pnas.0600137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kapiloff MS, Piggott LA, Sadana R, Li J, Heredia LA, Henson E, Efendiev R, Dessauer CW. An adenylyl cyclase-makapbeta signaling complex regulates camp levels in cardiac myocytes. J Biol Chem. 2009;284:23540–23546. doi: 10.1074/jbc.M109.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu S, Zhang J, Xiang YK. Fret-based direct detection of dynamic protein kinase a activity on the sarcoplasmic reticulum in cardiomyocytes. Biochem Biophys Res Commun. 2011;404:581–586. doi: 10.1016/j.bbrc.2010.11.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, Hill EV, Houslay MD, Langeberg LK, Scott JD. Makap assembles a protein kinase a/pde4 phosphodiesterase camp signaling module. Embo J. 2001;20:1921–1930. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yin X, Cuello F, Mayr U, Hao Z, Hornshaw M, Ehler E, Avkiran M, Mayr M. Proteomics analysis of the cardiac myofilament subproteome reveals dynamic alterations in phosphatase subunit distribution. Mol Cell Proteomics. 2010;9:497–509. doi: 10.1074/mcp.M900275-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dodge-Kafka KL, Bauman A, Mayer N, Henson E, Heredia L, Ahn J, McAvoy T, Nairn AC, Kapiloff MS. Camp-stimulated protein phosphatase 2a activity associated with muscle a kinase-anchoring protein (makap) signaling complexes inhibits the phosphorylation and activity of the camp-specific phosphodiesterase pde4d3. J Biol Chem. 2010;285:11078–11086. doi: 10.1074/jbc.M109.034868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marks AR, Reiken S, Marx SO. Progression of heart failure: Is protein kinase a hyperphosphorylation of the ryanodine receptor a contributing factor? Circulation. 2002;105:272–275. [PubMed] [Google Scholar]
- 124.Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: Dephosphorylation of ser23/24 on troponin i could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol. 2007;42:247–259. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 125.Rapundalo ST. Cardiac protein phosphorylation: Functional and pathophysiological correlates. Cardiovasc Res. 1998;38:559–588. doi: 10.1016/s0008-6363(98)00063-7. [DOI] [PubMed] [Google Scholar]
- 126.Devic E, Xiang Y, Gould D, Kobilka B. Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from beta(1) and beta(2) adrenoceptor knockout mice. Mol Pharmacol. 2001;60:577–583. [PubMed] [Google Scholar]
- 127.Rohrer DK, Desai KH, Jasper JR, Stevens ME, Regula DP, Jr, Barsh GS, Bernstein D, Kobilka BK. Targeted disruption of the mouse beta1-adrenergic receptor gene: Developmental and cardiovascular effects. Proc Natl Acad Sci U S A. 1996;93:7375–7380. doi: 10.1073/pnas.93.14.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rohrer DK, Chruscinski A, Schauble EH, Bernstein D, Kobilka BK. Cardiovascular and metabolic alterations in mice lacking both beta1- and beta2-adrenergic receptors. J Biol Chem. 1999;274:16701–16708. doi: 10.1074/jbc.274.24.16701. [DOI] [PubMed] [Google Scholar]
- 129.Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, Bernstein D, Kobilka BK. Targeted disruption of the beta2 adrenergic receptor gene. J Biol Chem. 1999;274:16694–16700. doi: 10.1074/jbc.274.24.16694. [DOI] [PubMed] [Google Scholar]
- 130.Xiang Y, Kobilka B. The pdz-binding motif of the beta2-adrenoceptor is essential for physiologic signaling and trafficking in cardiac myocytes. Proc Natl Acad Sci U S A. 2003;100:10776–10781. doi: 10.1073/pnas.1831718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang Y, Lauffer B, Von Zastrow M, Kobilka B, Xiang Y. Nsf regulates {beta}2 adrenoceptors trafficking and signaling in cardiomyocytes. Mol Pharmacol. 2007 doi: 10.1124/mol.107.037747. [DOI] [PubMed] [Google Scholar]
- 132.Wang Y, De Arcangelis V, Gao X, Ramani B, Jung YS, Xiang Y. Norepinephrine- and epinephrine-induced distinct beta2-adrenoceptor signaling is dictated by grk2 phosphorylation in cardiomyocytes. J Biol Chem. 2008;283:1799–1807. doi: 10.1074/jbc.M705747200. [DOI] [PubMed] [Google Scholar]
- 133.Skeberdis VA, Jurevicius J, Fischmeister aR. Beta-2 adrenergic activation of l-type ca++ current in cardiac myocytes. J Pharmacol Exp Ther. 1997;283:452–461. [PubMed] [Google Scholar]
- 134.Zhou YY, Cheng H, Bogdanov KY, Hohl C, Altschuld R, Lakatta EG, Xiao RP. Localized camp-dependent signaling mediates beta 2-adrenergic modulation of cardiac excitation-contraction coupling. Am J Physiol. 1997;273:H1611–H1618. doi: 10.1152/ajpheart.1997.273.3.H1611. [DOI] [PubMed] [Google Scholar]
- 135.Kuschel M, Zhou YY, Cheng H, Zhang SJ, Chen Y, Lakatta EG, Xiao RP. G(i) protein-mediated functional compartmentalization of cardiac beta(2)-adrenergic signaling. J Biol Chem. 1999;274:22048–22052. doi: 10.1074/jbc.274.31.22048. [DOI] [PubMed] [Google Scholar]
- 136.Gauthier C, Langin D, Balligand JL. Beta3-adrenoceptors in the cardiovascular system. Trends Pharmacol Sci. 2000;21:426–431. doi: 10.1016/s0165-6147(00)01562-5. [DOI] [PubMed] [Google Scholar]
- 137.Munzel T, Gori T. Nebivolol: The somewhat-different beta-adrenergic receptor blocker. Journal of the American College of Cardiology. 2009;54:1491–1499. doi: 10.1016/j.jacc.2009.05.066. [DOI] [PubMed] [Google Scholar]
- 138.Ma H, Green RD. Modulation of cardiac cyclic amp metabolism by adenosine receptor agonists and antagonists. Mol Pharmacol. 1992;42:831–837. [PubMed] [Google Scholar]
- 139.Warrier S, Belevych AE, Ruse M, Eckert RL, Zaccolo M, Pozzan T, Harvey RD. Beta-adrenergic- and muscarinic receptor-induced changes in camp activity in adult cardiac myocytes detected with fret-based biosensor. Am J Physiol Cell Physiol. 2005;289:C455–C461. doi: 10.1152/ajpcell.00058.2005. [DOI] [PubMed] [Google Scholar]
- 140.Iancu RV, Ramamurthy G, Harvey RD. Spatial and temporal aspects of camp signalling in cardiac myocytes. Clin Exp Pharmacol Physiol. 2008;35:1343–1348. doi: 10.1111/j.1440-1681.2008.05020.x. [DOI] [PubMed] [Google Scholar]
- 141.Abi-Gerges A, Richter W, Lefebvre F, Mateo P, Varin A, Heymes C, Samuel JL, Lugnier C, Conti M, Fischmeister R, Vandecasteele G. Decreased expression and activity of camp phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic camp signals. Circulation research. 2009;105:784–792. doi: 10.1161/CIRCRESAHA.109.197947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Salloum FN, Abbate A, Das A, Houser JE, Mudrick CA, Qureshi IZ, Hoke NN, Roy SK, Brown WR, Prabhakar S, Kukreja RC. Sildenafil (viagra) attenuates ischemic cardiomyopathy and improves left ventricular function in mice. American journal of physiology. Heart and circulatory physiology. 2008;294:H1398–H1406. doi: 10.1152/ajpheart.91438.2007. [DOI] [PubMed] [Google Scholar]
- 143.Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, St Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Dyck JR, Michelakis ED. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007;116:238–248. doi: 10.1161/CIRCULATIONAHA.106.655266. [DOI] [PubMed] [Google Scholar]
- 144.Pokreisz P, Vandenwijngaert S, Bito V, Van den Bergh A, Lenaerts I, Busch C, Marsboom G, Gheysens O, Vermeersch P, Biesmans L, Liu X, Gillijns H, Pellens M, Van Lommel A, Buys E, Schoonjans L, Vanhaecke J, Verbeken E, Sipido K, Herijgers P, Bloch KD, Janssens SP. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation. 2009;119:408–416. doi: 10.1161/CIRCULATIONAHA.108.822072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Miller CL, Oikawa M, Cai Y, Wojtovich AP, Nagel DJ, Xu X, Xu H, Florio V, Rybalkin SD, Beavo JA, Chen YF, Li JD, Blaxall BC, Abe J, Yan C. Role of ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circulation research. 2009;105:956–964. doi: 10.1161/CIRCRESAHA.109.198515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yanaka N, Kurosawa Y, Minami K, Kawai E, Omori K. Cgmp-phosphodiesterase activity is up-regulated in response to pressure overload of rat ventricles. Biosci Biotechnol Biochem. 2003;67:973–979. doi: 10.1271/bbb.67.973. [DOI] [PubMed] [Google Scholar]
- 147.Mokni W, Keravis T, Etienne-Selloum N, Walter A, Kane MO, Schini-Kerth VB, Lugnier C. Concerted regulation of cgmp and camp phosphodiesterases in early cardiac hypertrophy induced by angiotensin ii. PLoS One. 2010;5:e14227. doi: 10.1371/journal.pone.0014227. [DOI] [PMC free article] [PubMed] [Google Scholar]