

Abstract
Structure-based design, synthesis and X-ray structure of protein-ligand complexes of exceptionally potent and selective β-secretase inhibitors are described. The inhibitors are designed specifically to interact with S1′ active site residues to provide selectivity over memapsin 1 and cathepsin D. Inhibitor 5 has exhibited exceedingly potent inhibitory activity (Ki = 17 pM) and high selectivity over BACE 2 (>7,000-fold) and cathepsin D (>250,000-fold). A protein-ligand crystal structure revealed important molecular insight into these selectivities. These interactions may serve as an important guide to design selectivity over the physiologically important aspartic acid proteases.
Introduction
Alzheimer’s disease is a progressive, degenerative brain disorder with no effective treatment to date and the development of new drugs is an urgent priority in medicine.1 The hallmark of Alzheimer’s disease (AD) is the formation of neutritic plaques containing 40/42 residue amyloid-β (Aβ) peptides and neurofibrillary tangles in the brain.2 β-Secretase (memapsin 2, BACE 1) is one of two proteases which cleaves β-amyloid precursor protein (APP) and generates Aβ and its aggregation product.3 There is considerable evidence that excess Aβ leads to brain inflammation, neuronal death, and AD.4 Consequently, β-secretase has become a major therapeutic target for drug development.5,6 Since our design of initial transition-state inhibitor (1, Figure 1) and subsequent determination of inhibitor-bound memapsin 2 X-ray structure, nearly a decade ago, steady progress has been made towards the evolution of small molecule potent and brain-penetrable inhibitor drugs.7,8 Recently, we have shown that administration of β-secretase inhibitor 2 rescued cognitive decline in transgenic AD mice, validating β-secretase as an important drug design target.9,10 However, the development of clinical β-secretase inhibitor drug is faced with numerous formidable challenges, including lack of selectivity against other physiologically important aspartic acid proteases and issues of poor pharmacological profiles including blood-brain penetration.7,8 In our continuing work towards the design of small molecule potent and selective inhibitors, we have been particularly interested in developing tools for selectivity against relevant physiologically important aspartic acid proteases especially cathepsin D and BACE 2. BACE 2 has specificity similarity to BACE 1 and this is known to have important physiological functions.11 Cathepsin D plays a key role in important biological functions like protein catabolism.12 The abundance of cathepsin D in various cells, especially in CNS tissue cells, is very high. Furthermore, cathepsin D gene knock-out studies in mice showed marked phenotypic response including high mortality rate.13 Therefore, the selective inhibition of β-secretase over cathepsin D and BACE 2 is very critical to reduce toxicity and other side effects of β-secretase inhibitor drugs.
Figure 1.

Structures of β-secretase inhibitors 1-3.
As described by us previously, the X-ray crystal structure of inhibitor 1-bound β-secretase showed an interesting hydrogen bonding between P2′-carbonyl and the hydroxyl of Tyr-198, forming a rare kink at the P2′ site.8 We have exploited this interaction in the design and synthesis of very potent and highly selective β-secretase inhibitors such as 3 by incorporating hydroxyethylene isosteres.14 However, cellular β-secretase inhibitory activity of this class of inhibitors was only in micromolar range. In an attempt to design small molecule inhibitors with improved selectivity and cellular activity exploiting this unique interaction, we have further explored β-secretase inhibitors with a reduced amide isostere and incorporated functionality to improve potency and selectivity. The basic amine functionality in the reduced amide isostere may also improve cell permeability.15 Herein, we report our structure-based design and synthesis of very potent and exceptionally selective inhibitors with excellent cellular inhibitory properties. A protein-ligand X-ray structure provided important molecular insight into the specific cooperative ligand-binding site interactions for selectivity.
The inhibitors containing reduced amide isostere have been reported however, they exhibited only marginal selectivity against memapsin 1 (BACE 2).6,16 A reduced amide β–secretase inhibitor 4 was synthesized by us and this compound has exhibited a BACE 1 Ki of 27 nM and marginal selectivity against BACE 2 and cathepsin D in our in-house enzyme inhibitory assays. An energy-minimized model of 4 was created based upon the protein-ligand X-ray structure of 2-bound β-secretase.9 Our preliminary model suggested that an introduction of a hydroxyl group with S-configuration on the ethyl group of homo-alanine moiety would make enhanced interactions in the active site and possibly enhance selectivity. Based upon this molecular insight, we have designed, synthesized and evaluated inhibitors 5 and 6 (Figure 2) to investigate the influence of the hydroxyl group and also the role of stereochemistry on the potency and selectivity. We have also synthesized and evaluated inhibitors 7–9 to examine the importance of various functional groups at the P1′ and P2′ sites on the potency.
Figure 2.

Structure of isophthalamide-derived β-secretase inhibitors 4–9
Chemistry
The synthesis of inhibitors 4–6 is shown in scheme 1. Amino acids 10a–c were coupled with isobutyl amine in the presence of EDC, HOBt and iPr2NEt to afford the corresponding amides in 71–91% yield. Removal of the Boc group with trifluoroacetic acid followed by reductive amination of the resulting amine with aldehyde 1117 provided compounds 12a-c in 49–76% yields. Treatment of 12a–c with trifluoroacetic acid followed by coupling with acid 1318 provided the inhibitors 4–6 in 68–75% yields.
Scheme 1.

Synthesis of inhibitors 4–6.
Synthesis of inhibitors 7–9 are shown in Schemes 2 and 3. Coupling of Boc protected allothreonine derivative 10b with N-isobutyl-N-methyl amine using EDC, HOBt, and iPr2NEt followed by Boc removal using trifluoroacetic acid afforded (2S, 3S)-2-amino-3-hydroxy-N-isobutyl-N-methyl-butanamide in 49 % yield over two steps. Reductive amination of this amine with known aldehyde 11, provided 12d in 30% yields. Inhibitor 7 was synthesized in 61% yields from 12d by Boc removal using trifluoroacetic acid followed by coupling of the resulting amine with the known acid 13 (Scheme 2). As shown in scheme 2, inhibitor 8 was prepared from 15 following the similar reaction sequence utilized for the synthesis of inhibitors 4–7 (schemes 1 and 2). Compound 15, in turn, has been synthesized by O-methylation of 14 using MeI and Ag2O19 followed by hydrolysis using aqueous LiOH.
Scheme 2.

Synthesis of inhibitors 7 and 8
Scheme 3.

Synthesis of inhibitor 9
Synthesis of inhibitor 9 has been carried-out as shown in scheme 3. Treatment of 14 with TBSOTf in the presence of Et3N followed by reaction with DiBALH provided the corresponding aldehyde. Reductive amination of this crude aldehyde with isobutyl amine afforded 16 in 66% yields over three steps. Removal of the Boc group using trifluoroacetic acid followed by reductive amination of the resulting amine with the aldehyde 11 afforded compound 17.
Reaction of compound 17 with trifluoroacetic acid followed by coupling with acid 13 using EDC, HOBt, and iPr2NEt provided TBS protected inhibitor which was upon treating with TBAF gave the inhibitor 9 in 46 % yields over three steps.
Based on the results obtained from these inhibitors, we have directed our attention to reduce the peptidic nature of 5 by keeping all the key hydrogen bonding interactions of the prime region intact. In this direction, we have designed and synthesized the inhibitors 18–22 (Fig. 3) containing 7,6,5-tricyclic indole moieties as P2 ligands.
Figure 3.

Structure of indole-derived β-secretase inhibitors 18–22
For synthesis of inhibitor 18, known indolecarboxylic acid 23 was prepared as described in the literature.20 The synthesis of α,α-dimethyl tricyclic indole derivative 25, present in inhibitor 20, was synthesized as shown in scheme 4. Reaction of the known tricyclic indole derivative 2421 with MeI in the presence of NaH provided the corresponding α,α-dimethyl derivative. Saponification of the resulting ester with aqueous NaOH afforded α,α-dimethylindole carboxylic acid 25 (37% yields over 2 steps). Inhibitor 18 was synthesized by treatment of Boc-derivative 12b with trifluoroacetic acid followed by coupling of the resulting amine with acid 2320 (68% yield). Similarly, coupling of the above amine with cyclopropyl indole derivative 30 provided inhibitor 21.
Scheme 4.

Synthesis of carboxylic acid 25 and inhibitors 18 and 21.
Synthesis of tricyclic indole derivative in inhibitors 21 and 22 has been carried out from butyl 3-chloropropanesulfonate 26 as shown in Scheme 5. In a one-pot two step reaction, 26 was treated first with BuLi followed by treatment again with BuLi and BOMCl provided the corresponding butyl 1-(benzyloxymethyl)cyclopropanesulfonate in 80% yield. Hydrolysis of this sulfonate with KSCN followed by refluxing of the resulting potassium sulfonate with SOCl2 afforded sulfonyl chloride 27 in 91% yield over two steps. Reaction of indole derivative 2822 with 27 in the presence of pyridine and catalytic amount of DMAP furnished sulfonamide 29 in 74% yields. Hydrogenolysis of 29, followed by mesylation of the resulting alcohol using mesyl chloride and Et3N afforded the corresponding mesylate. This mesylate was subjected to a one pot cyclization and N-methylation using NaH and MeI followed by hydrolysis in the presence of aqueous NaOH to obtain the desired carboxylic acid 30 in 65% yields over two steps.
Scheme 5.

Synthesis of carboxylic acid 30.
Synthesis of (2S, 3S)-2-amino-3-hydroxy-N-isobutyl (or isopropyl)hexanamide moiety present in the prime region of the inhibitors 19, 20 and 22 has been carried out as shown in Scheme 6. Asymmetric dihydroxylation of 31 using AD-mix α followed by reaction of the resulting dihydroxy compound with p-nitrobenzenesulfonyl chloride in the presence of Et3N afforded nosylate 32 in 32% yields over two steps.23 Treatment of 32 with NaN3 followed by hydrogenation of the corresponding azide using 10% Pd-C in the presence of (Boc)2O afforded 33 in 80% yields. Hydrolysis using aqueous LiOH followed by coupling with isobutyl amine and isopropyl amine using EDC, HOBt and iPr2NEt afforded 34a and 34b respectively. Boc removal using trifluoroacetic acid followed by reductive amination of the resulting amine with aldehyde 11 gave 35a and 35b in 49% and 74% yields respectively. Inhibitors 19, 20 and 22 were synthesized by coupling of the amine, obtained by Boc removal of 35a and 35b using trifluoroacetic acid, with tricyclic indole derivatives 23 or 25 or 30.
Scheme 6.

Synthesis of inhibitors 19, 20 and 22
Results and Discussions
The BACE 1 inhibitory activity of synthetic inhibitors 4–9 was determined against recombinant β-secretase using our previously reported assay protocols.24 The results are shown in Table 1. As can be seen, inhibitor 4 with a homoalanine P1′-side chain has shown a Ki of 27 nM (entry 1). Inhibitor 5 with an allothreonine P1′-side chain has exhibited remarkable BACE 1 inhibitory activity with a Ki of 17 pM (entry 2). Inhibitor 6 with a threonine P1′-side chain has shown significant reduction of BACE 1 activity over the deoxyderivative 4 or the inhibitor 5 with an allothreonine P1′-side chain (entries 1–3). We have also evaluated the cellular inhibition of β-secretase in neuroblastoma cells.25 Consistent with potent BACE 1 inhibitory activity, inhibitor 5 exhibited an average cellular IC50 value of 1 nM. The corresponding inhibitor 6 with a threonine P1′-side chain has shown a cellular IC50 value of >1 µM in the same assay. Inhibitor 7 with a N-methyl amide abolished all BACE 1 inhibitory activity. Inhibitor 8 with an ‘OMe’ group in the P1′-region also showed substantial reduction in inhibitory potency (entry 5) compared to inhibitor 5 with hydroxyl group. Inhibitor 9 with a reduced amide in the P2′-region resulted in total loss in potency. These results clearly demonstrate the significance of hydrogen bonding interactions of inhibitor 5 with the prime region of the BACE 1 active site.
Table 1.
BACE 1 inhibitory and cellular activity of inhibitors 4–9.
| Entry | Inhibitor | Ki (nM) | IC50 (nM)a |
|---|---|---|---|
| 1 | 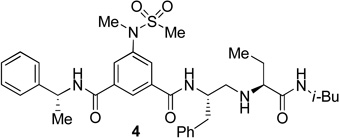 |
27.12 | 9.5 |
| 2 | 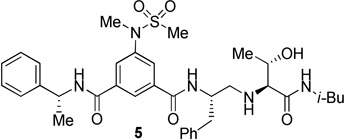 |
0.017 | 1 |
| 3. | 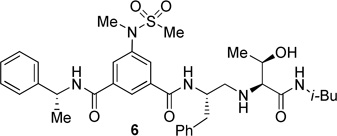 |
98.8 | >1000 |
| 4. | 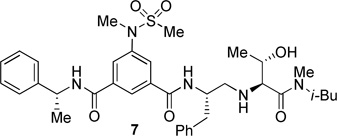 |
>1000 | --- |
| 5. | 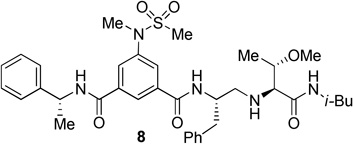 |
25 | --- |
| 6. | 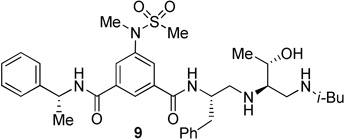 |
>1000 | --- |
IC50 was determined in neuroblastoma cells. GRL-8234 exhibited Ki = 1.8 nM, IC50 = 2.5 nM in this assay.
To gain further molecular insight, we have determined the X-ray structure of 5-bond to β-secretase at a 2.2 Å resolution. As shown in Figure 4, the amine functionality of the reduced amide isostere forms two tight hydrogen bonds (2.4 and 2.7 Å bond distances) with active site aspartic acid Asp228.7 Interestingly, the other active site Asp32 is not directly interacting with the reduced amide isostere. Asp32 is extensively hydrogen bonded to three groups: the amide nitrogen of Gly34 (2.8 Å), the hydroxyl group of Ser35 (2.5 Å to 3 Å with rotations of the involved groups), and the amide nitrogen of Gly230 (3.3 Å). These interactions appear to lock Asp32 in a rigid conformation thus its hydrogen bonding to Asp228 produced a network of hydrogen bond interactions to include the interaction of the inhibitor and the protease. A similar inhibitor-enzyme interacting pattern has been reported for the crystal structure of reduced amide isosteres.16b The fact that inhibitors 2 and 5 are highly potent suggests that the interaction of both active site carboxyls with the transition-state isostere is not a necessary feature for the design of potent inhibitors. The P3-phenyl ring occupies a unique position which spans S3 and S4 subsites and causes a significant positional shift of a protein loop containing residues from 8 to 13 (the 10’s loop)26 located in the S3/S4 pocket similar to inhibitor 2. This flexible part of the active site cleft can be further exploited for ligand design. The P2′-carbonyl as well as P2′-NH are within proximity to form hydrogen bonds with Thr72 and Gly34 respectively. Most significantly, the allothreonine hydroxyl group is oriented toward Tyr-198 hydroxyl group. This interaction is presumably absent in inhibitor 4. Also, the P1′-hydroxyl group stereochemistry is optimal for critical hydrogen bonding with Tyr-198. The combinations of active site interactions are responsible for potency and selectivity of inhibitor 5.
Figure 4.

The X-ray structure of 5 (green)-bound-β-secretase complex. Hydrogen bonds are shown in dotted line (pdb id: 4GID).
The X-ray crystal structure of 5 bound memapsin 2 demonstrates the importance of allothreonine moiety as it forms key hydrogen bonding interactions with the prime region of memapsin 2. Therefore, inhibitors 18–22, with 7,6,5 tricyclicindole moiety as P2 ligand, were designed with a view to reduce labile amide bonds in isophthalic acid amide-derived ligand. Synthetic inhibitors 18–22 were evaluated against recombinant BACE 1 and the results are summarized in Table 2. Inhibitor 18, containing a known 7,6,5 tricyclic moiety as P2 ligand showed a low nanomolar activity towards BACE 1 (Ki = 7.3 nM). This inhibitor is substantially less potent than inhibitor 5 however, the ratio of cell inhibitory to enzyme inhibitory efficacy was improved significantly (3 vs >58), indicating better cell permeability for compound 18. Inhibitor 19 with a sterically more demanding propyl group in the P1′- region has shown around 18-fold improvement in the potency (entries 1 and 2). Inhibitor 20 with a dimethyl substituted indole derivative as the P2 ligand resulted in >10-fold potency enhancement over unsubstituted inhibitor 19. This inhibitor exhibited a cellular IC50 value of 15 nM. The ligand was also designed especially to halt the possibility of retro-Michael reaction of the P2-α,α-unsubstituted sultam functionality in inhibitors 18 and 19.27 Inhibitors 21 and 22, designed by replacing the two methyl groups of P2 ligand in 20 with cyclopropyl group, were also exhibited impressive potency.
Table 2.
BACE 1 inhibitory and cellular activity of inhibitors 18–22.
| Entry | Inhibitor | Ki (nM) | IC50 (nM)a |
|---|---|---|---|
| 1 | 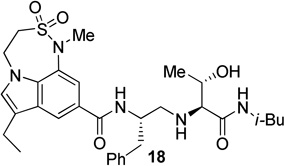 |
7.3 | 22 |
| 2 | 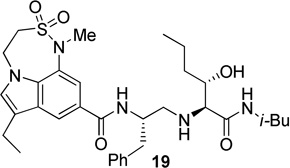 |
0.4 | --- |
| 3 | 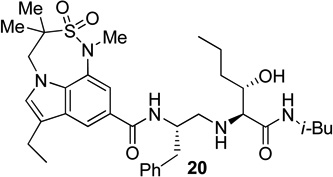 |
0.036 | 15 |
| 4 | 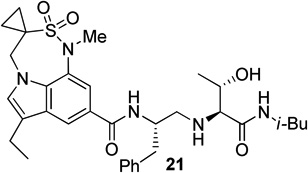 |
7.2 | --- |
| 5 |  |
0.47 | 14 |
IC50 was determined in neuroblastoma cells. GRL-8234 exhibited Ki = 1.8 nM, IC50 = 2.5 nM in this assay.
We then evaluated the potencies of selected inhibitors against recombinant BACE 2 and human cathepsin D and the results are shown in Table 3. Interestingly, inhibitor 5 displayed very impressive selectivity against BACE 2 (BACE 2, Ki = 120 nM, selectivity >7000 fold) and cathepsin D (Ki = 4.3 PM, selectivity >250,000 fold) as well. In comparison, deoxyinhibitor 4 has shown a BACE 2 Ki of 1450 nM (selectivity >50 fold) and cathepsin D Ki of 8264 nM (selectivity >300 fold). This result suggested that the allothreonine hydroxyl group on the P1′-side chain is critical to selectivity and potency of inhibitor 5. Inhibitor 18 has also exhibited good selectivity (over 970 fold selective) towards BACE 1 over cathepsin D (entry 3). Inhibitor 19 with a butyl side chain has shown improvement in cathepsin D selectivity (entry 4). Inhibitor 20 displayed good selectivity against BACE 2 and excellent selectivity against cathepsin D (entry 5). Inhibitor 22 has also shown a >4200-fold selectivity over cathepsin D (entry 6).
Table 3.
Selectivity studies of BACE 1 inhibitors against BACE 2 & cathepsin D
| Entry | Inhibitor | BACE 1 Ki(nM) |
BACE 2 Ki(nM |
CD Ki(nM) |
BACE 2/BACE 1 (Selectivity) |
CD/BACE1 (Selectivity) |
|---|---|---|---|---|---|---|
| 1 | 4 | 27.12 | 1450 | 8264 | >55 | >300 |
| 2 | 5 | 0.017 | 120 | 4300 | >7000 | >250,000 |
| 3 | 18 | 7.3 | --- | 7100 | --- | >970 |
| 4 | 19 | 0.4 | --- | 690 | --- | >1700 |
| 5 | 20 | 0.036 | 11 | 530 | >300 | >14,700 |
| 6 | 22 | 0.47 | --- | 2000 | --- | >4,200 |
Conclusion
In conclusion, we have designed, synthesized and examined the biological activity of isophthalamide based BACE 1 inhibitors containing various functional groups in the prime region. Selectivity of inhibitors 4 and 5 against BACE 2 and cathepsin D were also examined. Inhibitor 5 with an allothreonine moiety exhibited superior potency and exceptionally high selectivity when compared to inhibitors with a threonine moiety or an ethylglycine (homoalanine) moiety. Inhibitors with N-isobutyl-N-methylamide and P2′ reduced amide groups on the prime side lost their efficacy. These results clearly demonstrate the significance of prime region of the inhibitors on the potency and selectivity. The X-ray structure of 5-bond β-secretase also showed the presence of effective hydrogen bond between the prime side of inhibitor and Thr72, Gly34, and Tyr 198 residues of BACE 1. Based upon this molecular insight, we have further designed and synthesized inhibitors by replacing the isophthalamide moiety with conformationally constrained 7,6,5-tricyclicindole moieties and by keeping the allothreonine moiety intact. These inhibitors have also shown very good potency and selectivity against cathepsin D. These results further support the significance of hydrogen bonding interactions in the prime region for the potency and selectivity. The combination of active site interactions along with the Tyr-198 may be responsible for the observed selectivity. This molecular insight may aid further design of selectivity against other aspartic acid proteases. Further investigations into the origin of selectivity are in progress.
Experimental Section
General
All anhydrous solvents were obtained according to the following procedures: diethyl ether and tetrahydrofuran (THF) were distilled from sodium/benzophenone under argon; dichloromethane from calcium hydride. Other solvents were used without purification. All moisture-sensitive reactions were carried out in flame-dried flasks under argon atmosphere. Reactions were monitored by thin layer chromatography (TLC) using Silicycle 60A-F254 silica gel pre-coated plates. Flash column chromatography was performed using Silicycle 230–400 mesh silica gel. Yields refer to chromatographically and spectroscopically pure compounds. 1H NMR and 13C NMR spectra were recorded on a Varian Inova-300 (300 MHz and 75 MHz, respectively), Bruker Avance ARX- 400 (400 MHz and 100 MHz), and Bruker Avance DRX-500 (500 MHz and 125 MHz). High and low resolution mass spectra were carried out by the Mass Spectroscopy Center at Purdue University. The purity of all test compounds was determined by HRMS and HPLC analysis. All test compounds showed ≥95% purity.
Synthesis of compound 12a
To a mixture of (S)-2-[(tert-butoxycarbonyl)amino]butanoic acid 10a (2.3 mmol, 0.47 g) and iPr2NEt (2.76 mmol, 0.48 mL) in CH2Cl2 (12 mL), HOBt.H2O (2.76 mmol, 0.37 g), isobutyl amine (2.76 mmol, 0.27 mL) and EDC.HCl (2.76 mmol, 0.53 g) were added simultaneously at 23 °C and the resulting mixture was stirred for 17 h at 23 °C. Reaction mixture was quenched with saturated aqueous NaHCO3 solution and extracted with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (1–3% MeOH/CH2Cl2) to furnish (S)-tert-butyl [1-(isobutylamino)-1-oxobutan-2-yl]carbamate in 86% yields (0.51 g). 1H NMR (400 MHz, CDCl3) δ 6.24 (brs, 1H), 5.08 (br, 1H), 4.07-3.89 (m, 1H), 3.18-2.96 (m, 2H), 1.92-1.70 (m, 2H), 1.62 (hept, J = 7.3 Hz, 1H), 1.43 (s, 9H), 0.93 (t, J = 7.4 Hz, 3H), 0.89 (d, J = 6.8 Hz, 6H).
To a solution of (S)-tert-butyl [1-(isobutylamino)-1-oxobutan-2-yl]carbamate (1.97 mmol, 0.51 g) in CH2Cl2 (9 mL), trifluoroacetic acid (3 mL) was added at 0 °C and the resulting mixture was stirred for 1.5 h at 23 °C. Excess trifluoroacetic acid and CH2Cl2 were removed under reduced pressure and the resulting residue was purified by silica gel column chromatography [2–5% (5%NH3/MeOH)/CH2Cl2] to furnish (S)-2-amino-N-isobutylbutanamide in 96% yield (0.298 g).
To a solution of the above (S)-2-amino-N-isobutylbutanamide (1.87 mmol, 0.296 g) and (S)-tert-butyl (1-oxo-3-phenylpropan-2-yl)carbamate 11 (prepared from the corresponding Weinreb amide (2 mmol) following a similar literature procedure)7 in CH2Cl2 (20 mL), Na(OAc)3BH (2.62 mmol, 0.55 g) was added at 0 °C. The resulting mixture was stirred for 1 h at 0 °C and 15 h at 23 °C. Reaction mixture was quenched with saturated aqueous NaHCO3 solution and extracted with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (1–3% MeOH/CH2Cl2) to furnish the corresponding adduct 12a in 79% yields (0.58 g). 1H NMR (400 MHz, CDCl3) δ 7.34-7.26 (m, 2H), 7.25-7.11 (m, 4H), 4.50 (br, 1H), 3.95 (brs, 1H), 3.10-2.96 (m, 2H), 2.92 (dd, J = 7.3, 5.0 Hz, 1H), 2.86-2.69 (m, 2H), 2.62 (dd, J = 11.9, 4.3 Hz, 1H), 2.52 (dd, J = 12.0, 7.9 Hz, 1H), 1.80-1.63 (m, 2H), 1.63-1.50 (m, 1H), 1.41 (s, 9H), 0.92 (t, J = 7.5 Hz, 3H), 0.85 (d, J = 6.5 Hz, 6H).
Synthesis of inhibitor 4
To a solution of 12a (0.72 mmol, 0.282 g) in CH2Cl2 (9 mL), trifluoroacetic acid (3 mL) was added at 0 °C. After stirring the reaction mixture for 1 h at 23 °C, CH2Cl2 and trifluoroacetic acid were removed under reduced pressure. The resulting residue was purified by silica gel column chromatography [2–6% (5%NH3/MeOH)/CH2Cl2] to furnish the corresponding amine in 93% yield (0.195 g).
To a solution of the above amine (0.125 mmol, 36.4 mg) in CH2Cl2 (10 mL), iPr2NEt (0.03 mL), HOBt.H2O (0.16 mmol, 21.6 mg), (R)-3-(N-methylmethylsulfonamido)-5-[(1-phenylethyl)-carbamoyl]benzoic acid 13 (0.16 mmol, 60.2 mg) and EDC.HCl (0.16 mmol, 30.7 mg) were added simultaneously at 23 °C and the resulting mixture was stirred for 14 h at the same temperature. Reaction mixture was quenched with saturated aqueous NaHCO3 solution and extracted with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (1–3% MeOH/CH2Cl2) to furnish the inhibitor 4 in 73% yields (59.6 mg). 1H NMR (400 MHz, CDCl3) δ 8.26 (s, 1H), 8.03 (s, 1H), 7.98 (s, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.39-7.34 (m, 2H), 7.33-7.24 (m, 5H), 7.24-7.18 (m, 3H), 6.71 (t, J = 6.2 Hz, 1H), 5.31 (p, J = 7.5 Hz, 1H), 4.40-4.26 (m, 1H), 3.29 (s, 3H), 3.04-2.91 (m, 3H), 2.88-2.83 (m, 1H), 2.81 (s, 3H), 2.81-2.73 (m, 2H), 2.55 (dd, J = 12.3, 4.1 Hz, 1H), 1.75-1.58 (m, 2H), 1.57 (d, J = 7.0 Hz, 3H), 1.55-1.48 (m, 1H), 0.89 (t, J = 7.4 Hz, 3H), 0.77 (d, J = 6.7 Hz, 3H), 0.76 (d, J = 6.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.15, 165.23, 164.53, 143.06, 142.23, 137.64, 135.65, 129.1, 128.62, 128.58, 127.93, 127.87, 127.33, 126.64, 126.23, 123.39, 65.20, 51.68, 50.24, 49.56, 46.41, 38.55, 37.84, 35.52, 28.36, 26.96, 21.60, 19.98, 19.95, 10.33. HRMS-ESI (m/z): [M+H]+ calcd for C35H48N5O5S 650.3376, found 650.3370.
Synthesis of compound 12b
Tert-butyl [(2S, 3S)-3-hydroxy-1-(isobutylamino)-1-oxobutan-2-yl]carbamate was synthesized in 71% yield by coupling of (2S, 3S)-2-[(tert-butoxycarbonyl)amino]-3-hydroxybutanoic acid 10b with isobutyl amine in the presence of EDC, HOBt and iPr2NEt as described for (S)-tert-butyl [1-(isobutylamino)-1-oxobutan-2-yl]carbamate. 1H NMR (400 MHz, CDCl3) δ 6.47 (brs, 1H), 5.53 (brs, 1H), 4.10-3.75 (m, 3H), 3.23-3.09 (m, 1H), 3.06-2.92 (m, 1H), 1.78 (hept, J = 6.7 Hz, 1H), 1.45 (s, 9H), 1.29 (d, J = 6.1 Hz, 3H), 0.91 (d, J = 6.5 Hz, 6H).
Compound 12b was synthesized in 52% yield (for two steps) from tert-butyl [(2S, 3S)-3-hydroxy-1-(isobutylamino)-1-oxobutan-2-yl]carbamate following the procedure described for the synthesis of compound 12a. 1H NMR (400 MHz, CDCl3) δ 7.35-7.27 (m, 3H), 7.26-7.19 (m, 1H), 7.17 (d, J = 7.4 Hz, 2H), 4.53 (d, J = 8.9 Hz, 1H), 4.05-3.86 (m, 2H), 3.39 (brs, 1H), 3.13-3.00 (m, 2H), 2.99 (d, J = 5.7 Hz, 1H), 2.87-2.71 (m, 2H), 2.67 (dd, J = 12.1, 4.4 Hz, 1H), 2.58 (dd, J = 12.1, 7.6 Hz, 1H), 1.73 (hept, J = 6.7 Hz, 1H), 1.41 (s, 9H), 1.18 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 7.1 Hz, 6H).
Synthesis of inhibitor 5
Inhibitor 5 has been prepared (yield 71%, over 2 steps) from 12b by boc deprotection followed by coupling with (R)-3-(N-methylmethylsulfonamido)-5-[(1-phenylethyl)carbamoyl]benzoic acid 13 following the similar reaction procedure described for the inhibitor 4. 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 1H), 7.93 (s, 1H), 7.88 (s, 1H), 7.39-7.27 (m, 6H), 7.26-7.14 (m, 6H), 5.27 (p, J = 7.3 Hz, 1H), 4.48-4.30 (m, 1H), 4.04-3.94 (m, 1H), 3.24 (s, 3H), 3.09 (d, J = 4.3 Hz, 1H), 3.02-2.88 (m, 3H), 2.88-2.77 (m, 1H), 2.80 (s, 3H), 2.73 (dd, J = 12.4, 3.7 Hz, 1H) 2.64 (dd, J = 12.3, 7.7 Hz, 1H), 1.64 (hept, J = 6.7 Hz 1H), 1.56 (d, J = 6.9 Hz, 3H), 1.04 (d, J = 6.2 Hz, 3H), 0.80 (d, J = 6.8 Hz, 3H), 0.80 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 172.37, 166.01, 164.75, 142.91, 142.03, 137.54, 135.90, 135.77, 129.16, 128.65, 128.59, 127.89, 127.62, 127.43, 126.65, 126.23, 123.87, 68.21, 68.06, 52.16, 51.60, 49.66, 46.40, 39.00, 37.83, 35.56, 28.34, 21.65, 20.06, 18.21. HRMS-ESI (m/z): [M+H]+ calcd for C35H48N5O6S 666.3325, found 666.3321.
Synthesis of compound 12c
Tert-butyl [(2S, 3R)-3-hydroxy-1-(isobutylamino)-1-oxobutan-2-yl]carbamate has been synthesized (yield 91%) by coupling of (2S, 3R)-2-[(tert-butoxycarbonyl)amino]-3-hydroxybutanoic acid 10c with isobutyl amine in the presence of iPr2NEt, HOBt and EDC as described for (S)-tert-butyl [1-(isobutylamino)-1-oxobutan-2-yl]carbamate. 1H NMR (400 MHz, CDCl3) δ 6.71 (brs, 1H), 5.52 (d, J = 7.6 Hz, 1H), 4.45-4.30 (m, 1H), 3.97 (dd, J = 8.2, 1.9 Hz, 1H), 3.22-3.08 (m, 1H), 3.07-2.92 (m, 1H), 1.77 (heptet, J = 6.7 Hz, 1H), 1.45 (s, 9H), 1.18 (d, J = 6.4 Hz, 3H), 0.90 (d, J = 6.9 Hz, 6H).
Compound 12c was prepared from tert-butyl [(2S, 3R)-3-hydroxy-1-(isobutylamino)-1-oxobutan-2-yl]carbamate following the procedure described for the synthesis of compound 12a (yield 49% over 2 steps). 1H NMR (400 MHz, CDCl3) δ 7.37-7.26 (m, 2H), 7.25-7.13 (m, 4H), 4.59 (d, J = 8.5 Hz, 1H), 4.04-3.75 (m, 2H), 3.15-2.96 (m, 2H), 2.94 (d, J = 5.2 Hz, 1H), 2.80 (d, J = 6.2 Hz, 2H), 2.70-2.55 (m, 2H), 1.92 (br, 1H), 1.74 (hept, J = 6.7 Hz, 1H), 1.40 (s, 9H), 1.18 (d, J = 6.3 Hz, 3H), 0.87 (d, J = 6.4 Hz, 6H).
Synthesis of inhibitor 6
Inhibitor 6 has been prepared (yield 75%, over 2 steps) from 12c by boc deprotection followed by coupling with (R)-3-(N-methylmethylsulfonamido)-5-[(1-phenylethyl)carbamoyl]benzoic acid 13 following the similar reaction procedure described for the inhibitor 4. 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 7.92 (s, 1H), 7.87 (s, 1H), 7.48 (d, J = 7.7 Hz, 1H), 7.38-7.27 (m, 5H), 7.26-7.15 (m, 5H), 7.08 (t, J = 5.9 Hz, 1H), 5.27 (p, J = 7.1 Hz, 1H), 4.42-4.27 (m, 1H), 3.79 (p, J = 6.3 Hz, 1H), 3.22 (s, 3H), 3.01-2.87 (m, 4H), 2.84 (dd, J = 13.7, 7.7 Hz, 1H), 2.78 (s, 3H), 2.71-2.59 (m, 2H), 1.69-1.59 (m, 1H), 1.56 (d, J = 7.0 Hz, 3H), 1.14 (d, J = 6.2 Hz, 3H), 0.79 (d, J = 6.4 Hz, 3H), 0.78 (d, J = 6.6 Hz, 3H).13C NMR (100 MHz, CDCl3) δ 172.78, 165.82, 164.81, 143.03, 142.05, 137.58, 135.88, 135.77, 129.14, 128.61, 127.78, 127.64, 127.39, 126.64, 126.23, 123.80, 69.13, 68.38, 52.04, 50.90, 49.68, 46.54, 38.59, 37.79, 35.56, 28.33, 21.68, 20.03, 19.68. HRMS-ESI (m/z): [M+H]+ calcd for C35H48N5O6S 666.3325, found 666.3335.
Synthesis of compound 12d
To a mixture of (2S, 3S)-2-[(tert-butoxycarbonyl)amino]-3-hydroxybutanoic acid (10b) (1 mmol, 0.22 g) and iPr2NEt (1.2 mmol, 0.21 mL) in CH2Cl2 (5 mL), were added HOBt.H2O (1.2 mmol, 0.162 g), N-isobutyl-N-methylamine (1.2 mmol, 0.14 mL) and EDC.HCl (1.2 mmol, 0.23 g) at 23 °C and the resulting reaction mixture was stirred for 15 h at 23 °C. Reaction mixture was quenched with saturated aqueous NaHCO3 solution and extracted with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (1-3%MeOH/CH2Cl2) to obtain tert-butyl {(2S, 3S)-3-hydroxy-1-[isobutyl(methyl)amino]-1-oxobutan-2-yl}carbamate.
To a solution of the above tert-butyl {(2S, 3S)-3-hydroxy-1-isobutyl(methyl)amino]-1-oxobutan-2-yl}carbamate in CH2Cl2 (6 mL), was added trifluoroacetic acid (2 mL) at 0 °C. The resulting reaction mixture was warmed to 23 °C and stirred for 1.5 h at 23 °C. Reaction mixture was concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography [2–5% (5%NH3/MeOH)/CH2Cl2] to furnish (2S, 3S)-2-amino-3-hydroxy-N-isobutyl-N-methyl-butanamide in 49% (93.2 mg) yields over two steps. 1H NMR (400 MHz, CDCl3) δ 3.89-3.76 (m, 1H), 3.67 & 3.64 (two doublets, J = 4.7 Hz, 4.6 Hz, 1H), 3.36 (dd, J = 13.2, 8.0 Hz, 0.5H), 3.19 (dd, J = 14.4, 8.3 Hz, 0.5H), 3.10 (dd, J = 14.3, 7.0 Hz, 0.5H), 3.05 (s, 1.7H), 3.03-2.98 (m, 0.5H), 2.91 (s, 1.3H), 2.51 (brs, 3H), 2.00-1.87 (m, 1H), 1.10 (two doublets, J = 6.3, 6.3 Hz, 3H), 0.93-0.81 (m, 6H).
Compound 12d was synthesized from (2S, 3S)-2-amino-3-hydroxy-N-isobutyl-N-methyl-butanamide by reductive amination with aldehyde 11 following a similar procedure described for the synthesis of compound 12a (yield 30%). 1H NMR (300 MHz, CDCl3) δ 7.33 – 7.10 (m, 5H), 4.80-4.64 (m, 1H), 3.95-3.78 (m, 2H), 3.43 (dd, J = 11.8, 4.4 Hz, 1H), 3.35 – 3.09 (m, 2H), 3.06-2.96 (m, 2.5H), 2.91 (s, 1.5H), 2.84 (br, 2H), 2.72 – 2.59 (m, 1H), 2.48-2.33 (m, 1H), 2.01-1.87 (m, 1H), 1.39 (s, 9H), 1.10 & 1.06 (two doublets, J = 6.4, 6.4 Hz, 3H), 0.90 & 0.86 (two doublets, J = 6.7, 6.3 Hz, 6H).
Synthesis of inhibitor 7
Inhibitor 7 was synthesized from compound 12d by boc removal using trifluoroacetic acid followed by coupling of the resulting amine with the known acid 13 using EDC, HOBt, and iPr2NEt following the similar procedure described for the synthesis of inhibitor 4 (yield 61%, over two steps). 1H NMR (400 MHz, CDCl3) δ 8.21 & 8.17 (two singlets, 1H), 8.07 – 7.92 (m, 2H), 7.45 – 7.30 (m, 4H), 7.30 – 7.15 (m, 6H), 7.07 (t, J = 7.8 Hz, 1H), 5.40-5.26 (m, 1H), 4.34 – 4.19 (m, 1H), 3.97-3.82 (m, 1H), 3.55 & 3.53 (two doublets, J = 4.2, 4.2 Hz, 1H), 3.33 (s, 3H), 3.23 – 3.03 (m, 2.5H), 3.01 (s, 2H), 2.95 – 2.87 (m, 1H), 2.85 & 2.84 (two singlets, 3H), 2.77 (s, 1H), 2.73 – 2.57 (m, 2.5H), 1.96-1.78 (m, 1H), 1.60 (d, J = 6.9 Hz, 3H), 1.13 & 1.10 (two doublets, J = 6.4, 6.5 Hz, 3H), 0.89 & 0.84 (two doublets, J = 6.6, 6.5 Hz, 2.7H), 0.79 & 0.75 (two doublets, J = 6.7, 6.7 Hz, 3.3H). HRMS-ESI (m/z): [M+H]+ calcd for C36H50N5O6S 680.3482, found 680.3478.
Synthesis of (2S, 3S)-2-[(tert-butoxycarbonyl)amino]-3-methoxybutanoic acid (15)
To a solution of (2S, 3S)-methyl 2-[(tert-butoxycarbonyl)amino]-3-hydroxybutanoate (14) (2.14 mmol, 0.5 g) in CH3CN (21 mL), were added Ag2O (10.7 mmol, 2.48 g) and MeI (21.4 mmol, 1.3 mL) at 23 °C and the resulting reaction mixture was stirred for 8 days at 23 °C. Reaction mixture was filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (20–25% EtOAc/hexanes) to furnish (2S, 3S)-methyl 2-[(tert-butoxycarbonyl)amino]-3-methoxybutanoate in 55% yields (0.29 g). 1H NMR (400 MHz, CDCl3) δ 5.27 (d, J = 9.3 Hz, 1H), 4.41 (dd, J = 8.6, 3.6 Hz, 1H), 3.74 (s, 3H), 3.66-3.55 (m, 1H), 3.34 (s, 3H), 1.43 (s, 9H), 1.18 (d, J = 6.4 Hz, 3H).
To a solution of (2S, 3S)-methyl 2-[(tert-butoxycarbonyl)amino]-3-methoxybutanoate (0.86 mmol, 0.213 g) in THF (4 mL) and H2O (2 mL), was added LiOH.H2O (5.16 mmol, 0.216 g) at 23 °C. The resulting reaction mixture was stirred for 5 h at 23 °C. Reaction mixture was diluted with H2O and diethyl ether. Organic layer was separated and the aqueous layer was carefully acidified with 2N HCl and extracted with ethyl acetate. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to provide crude (2S, 3S)-2-[(tert-butoxycarbonyl)amino]-3-methoxybutanoic acid (15). This crude acid was used in the coupling reaction with-out any further purification.
Synthesis of compound 12e
Synthesis of tert-butyl [(2S, 3S)-1-(isobutylamino)-3-methoxy-1-oxobutan-2-yl]carbamate has been carried out by coupling of the above crude acid 15 with isobutyl amine using EDC, HOBt, iPr2NEt following the procedure described for the synthesis of (S)-tert-butyl [1-(isobutylamino)-1-oxobutan-2-yl]carbamate (yield 74%, over two steps). 1H NMR (300 MHz, CDCl3) δ 6.22 (brs, 1H), 5.15 (brs, 1H), 4.10 (dd, J = 7.7, 6.8 Hz, 1H), 3.59 (p, J = 6.4 Hz, 1H), 3.34 (s, 3H), 3.21-3.10 (m, 1H), 3.09 – 2.98 (m, 1H), 1.77 (hept, J = 6.7 Hz, 1H), 1.44 (s, 9H), 1.18 (d, J = 6.3 Hz, 3H), 0.91 (d, J = 6.8 Hz, 6H).
To a solution of tert-butyl [(2S, 3S)-1-(isobutylamino)-3-methoxy-1-oxobutan-2-yl]-carbamate (0.624 mmol, 0.18 g) in CH2Cl2 (6 mL), was added trifluoroacetic acid (2 mL) at 0 °C and the resulting mixture was stirred for 1.5 h at 23 °C. Trifluoroacetic acid and CH2Cl2 were removed under reduced pressure and the resulting residue was diluted with EtOAc and basified with saturated aqueous NaHCO3 solution. Organic layer was separated and the aqueous layer was extracted several times with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to obtain crude (2S, 3S)-2-amino-N-isobutyl-3-methoxybutanamide in 88% yield.
To a solution of the above crude amine (0.55 mmol, 0.104 g) and (S)-tert-butyl (1-oxo-3-phenylpropan-2-yl)carbamate 11 [prepared from the corresponding Weinreb amide (0.55 mmol) following the similar literature procedure]7 in CH2Cl2 (6 mL), Na(OAc)3BH (0.825 mmol, 0.175 g) was added at 0 °C. The resulting mixture was stirred for 1 h at 0 °C and 15 h at 23 °C. Reaction mixture was quenched with saturated aqueous NaHCO3 solution and extracted with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (20–45% EtOAc/hexanes) to furnish the corresponding adduct 12e in 67% yield (0.155 g). 1H NMR (400 MHz, CDCl3) δ 7.35 (brs, 1H), 7.29 – 7.21 (m, 2H), 7.21-7.10 (m, 3H), 4.64 (d, J = 8.2 Hz, 1H), 3.94 (brs, 1H), 3.77 – 3.68 (m, 1H), 3.29 (s, 3H), 3.27 (d, J = 4.0 Hz, 1H), 3.00 (t, J = 6.4 Hz, 2H), 2.82 (dd, J = 12.9, 5.7 Hz, 1H), 2.69 (dd, J = 13.6, 7.7 Hz, 1H), 2.58 – 2.46 (m, 2H), 1.68 (hept, J = 6.7 Hz, 1H), 1.39 (s, 9H), 1.00 (d, J = 6.4 Hz, 3H), 0.83 (d, J = 6.6 Hz, 6H).
Synthesis of inhibitor 8
To a solution of 12e (0.073 mmol, 30.8 mg) in CH2Cl2 (3 mL), was added trifluoroacetic acid (1 mL) at 0 °C. The resulting reaction mixture was warmed to 23 °C and stirred for 1.5 h at 23 °C. Reaction mixture was concentrated under reduced pressure and the resulting residue was used in the next step without any further purification.
To a solution of the above residue in CH2Cl2 (5 mL), were added iPr2NEt (0.1 mL), HOBt.H2O (0.14 mmol, 18.9 mg), acid 13 (0.07 mmol, 26.3 mg), EDC.HCl (0.14 mmol, 26.8 mg) simultaneously at 23 °C. The resulting reaction mixture was stirred for 17 h at 23 °C. Reaction mixture was quenched with saturated aqueous NaHCO3 solution and extracted with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (2%MeOH/CH2Cl2) to obtain the inhibitor 8 in 67% yields (32.1 mg). 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 8.02 (s, 1H), 7.96 (s, 1H), 7.42 – 7.29 (m, 5H), 7.28-7.25 (m, 2H), 7.22 (d, J = 7.4 Hz, 3H), 7.14-7.02 (m, 3H), 5.32 (p, J = 7.2 Hz, 1H), 4.48-4.36 (m, 1H), 3.73-3.61 (m, 1H), 3.33 (s, 3H), 3.30 (s, 3H), 3.27 (d, J = 4.2 Hz, 1H), 3.04-2.94 (m, 3H), 2.90-2.80 (m, 4H), 2.76 – 2.65 (m, 2H), 1.69-1.61 (m, 1H), 1.61 (d, J = 6.9 Hz, 3H), 1.04 (d, J = 6.4 Hz, 3H), 0.80 (d, J = 6.4 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 171.84, 165.41, 164.41, 142.87, 142.20, 137.42, 135.67, 135.64, 129.11, 128.60, 128.57, 127.80, 127.73, 127.38, 126.63, 126.18, 123.41, 65.95, 56.52, 51.99, 51.19, 49.53, 46.33, 38.73, 37.83, 35.43, 28.32, 21.63, 20.00, 14.62. HRMS-ESI (m/z): [M+H]+ calcd for C36H50N5O6S 680.3482, found 680.3488.
Synthesis of tert-butyl {(2R, 3S)-3-[(tert-butyldimethylsilyl)oxy]-1-(isobutylamino)butan-2-yl}carbamate (16)
To a solution of (2S, 3S)-methyl 2-[(tert-butoxycarbonyl)amino]-3-hydroxybutanoate (14) (2.2 mmol, 0.513 g) in CH2Cl2 (11 mL), were added Et3N (3.3 mmol, 0.46 mL), and TBSOTf (2.86 mmol, 0.66 mL) at 0 °C. The resulting reaction mixture was warmed to 23 °C and stirred for 2 h at the same temperature. Reaction mixture was diluted with H2O and CH2Cl2. Organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (10% EtOAc/hexanes) to obtain (2S, 3S)-methyl 2-[(tert-butoxycarbonyl)-amino]-3-[(tert-butyldimethylsilyl)oxy]butanoate in 88% yields (0.67 g). 1H NMR (400 MHz, CDCl3) δ 5.25 (d, J = 8.4 Hz, 1H), 4.16 (dd, J = 8.3, 3.7 Hz, 1H), 4.05 – 3.96 (m, 1H), 3.66 (s, 3H), 1.36 (s, 9H), 1.16 (d, J = 6.3 Hz, 3H), 0.79 (s, 9H), −0.03 (s, 6H).
To a solution of (2S, 3S)-methyl 2-[(tert-butoxycarbonyl)amino]-3-[(tert-butyldimethyl-silyl)oxy]butanoate (0.288 mmol, 0.1 g) in Et2O (3 mL), was added DiBAL-H (0.63 mmol, 0.63 mL, 1M solution in CH2Cl2) at −78 °C and the resulting mixture was stirred for 1.5 h at −78 °C. Reaction mixture was quenched with 0.5 mL EtOAc and diluted with Et2O and saturated aqueous sodium potassium tartrate. After stirring for 0.5 h, organic layer was separated and the aqueous layer was extracted with Et2O. The combine extracts were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was used in the next step without any further purification.
To a solution of the above aldehyde in CH2Cl2 (6 mL), were added isobutyl amine (0.864 mmol, 0.086 mL) and Na(OAc)3BH (0.576 mmol, 0.122 g) at 0 °C and the resulting reaction mixture was stirred for 1.5 h at 0 °C & 15 h at 23 °C. Reaction mixture was quenched with saturated aqueous NaHCO3 solution and extracted with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography [3% (5%NH3/MeOH)/CH2Cl2] to obtain (tert-butyl {(2R, 3S)-3-[(tert-butyldimethylsilyl)oxy]-1-(isobutylamino)butan-2-yl}-carbamate (16) in 75% yields (80.7 mg). 1H NMR (400 MHz, CDCl3) δ 5.36 (d, J = 7.9 Hz, 1H), 4.11-3.98 (m, 1H), 3.57-3.42 (m, 1H), 2.90 (dd, J = 12.2, 5.3 Hz, 1H), 2.62 (dd, J = 12.2, 4.3 Hz, 1H), 2.44-2.30 (m, 2H), 1.72 (hept, J = 6.7 Hz, 1H), 1.43 (s, 9H), 1.14 (d, J = 6.4 Hz, 3H), 0.92-0.84 (m, 15H), 0.04 (s, 3H), 0.03 (s, 3H).
Synthesis of compound 17
Compound 17 was synthesized by Boc removal of tert-butyl {(2R, 3S)-3-[(tert-butyldimethylsilyl)oxy]-1-(isobutylamino)butan-2-yl}carbamate (16) using trifluoroacetic acid followed by reductive amination with known aldehyde 11 following the procedure described for the synthesis of 12e. 1H NMR (400 MHz, CDCl3) δ 7.31-7.23 (m, 2H), 7.23-7.15 (m, 3H), 5.20 (br, 1H), 3.94 – 3.75 (m, 2H), 2.95-2.81 (m, 1H), 2.79 – 2.67 (m, 2H), 2.58 (dd, J = 12.4, 5.0 Hz, 2H), 2.50-2.43 (m, 2H), 2.38 (qd, J = 11.5, 6.9 Hz, 2H), 1.74 (hept, J = 6.7 Hz, 1H), 1.40 (s, 9H), 1.10 (d, J = 6.3 Hz, 3H), 0.90 (d, J = 6.6 Hz, 6H), 0.87 (s, 9H), 0.04 (s, 3H), 0.02 (s, 3H).
Synthesis of inhibitor 9
Boc removal of compound 17 using trifluoroacetic acid followed by coupling of the resulting amine with known acid 13 using EDC, HOBt and iPr2NEt following the procedure described for the synthesis of inhibitor 8 afforded the corresponding amide in 70% yields. 1H NMR (400 MHz, CDCl3) δ 8.20 (s, 1H), 8.00 (s, 1H), 7.94 (d, J = 7.8 Hz, 1H), 7.92 (s, 1H), 7.66 (d, J = 7.6 Hz, 1H), 7.40 (d, J = 7.5 Hz, 2H), 7.33 (t, J = 7.5 Hz, 2H), 7.29 – 7.19 (m, 5H), 7.20-7.12 (m, 1H), 5.29 (p, J = 7.1 Hz, 1H), 4.20 – 4.06 (m, 1H), 4.06 – 3.93 (m, 1H), 3.28 (s, 3H), 3.20 (dd, J = 12.4, 8.2 Hz, 1H), 3.10 – 2.98 (m, 2H), 2.86 (dd, J = 13.6, 6.4 Hz, 1H), 2.82-2.76 (m, 4H), 2.76 – 2.67 (m, 3H), 2.60 (dd, J = 12.4, 7.1 Hz, 1H), 1.81 (hept, J = 6.8 Hz, 1H), 1.59 (d, J = 7.0 Hz, 3H), 1.11 (d, J = 6.4 Hz, 3H), 0.86 (s, 9H), 0.75 (d, J = 6.7 Hz, 3H), 0.75 (d, J = 6.7 Hz, 3H), 0.07 (s, 3H), 0.05 (s, 3H).
To a solution of the above amide (0.104 mmol, 79.7 mg) in THF (10 mL), was added TBAF (1.25 mmol, 1.25 mL, 1M solution in THF) at 0 °C and the resulting reaction mixture was stirred for 18 h at 23 °C. Solvent was removed under reduced pressure and the resulting residue was purified by silica gel column chromatography [3–5% (5%NH3/MeOH)/CH2Cl2] to obtain inhibitor 9 in 65% (43.9 mg) yields. 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 8.01 – 7.93 (m, 2H), 7.40 (d, J = 7.3 Hz, 2H), 7.37 – 7.24 (m, 7H), 7.24 – 7.16 (m, 3H), 5.31 (p, J = 7.3 Hz, 1H), 4.37-4.25 (m, 1H), 3.93-3.83 (m, 1H), 3.31 (s, 3H), 3.04 (dd, J = 13.5, 5.8 Hz, 1H), 2.81 (s, 3H), 2.80 – 2.67 (m, 5H), 2.44 – 2.27 (m, 3H), 1.70-1.54 (m, 4H), 1.13 (d, J = 6.8 Hz, 3H), 0.81 (d, J = 6.5 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 165.78, 164.64, 143.13, 142.13, 137.87, 135.80, 129.24, 128.63, 128.53, 128.16, 127.46, 127.37, 126.53, 126.34, 123.78, 68.48, 60.61, 57.62, 51.94, 49.74, 49.48, 49.30, 38.67, 37.88, 35.46, 27.73, 21.78, 20.41, 20.38, 20.34. HRMS-ESI (m/z): [M+H]+ calcd for C35H50N5O5S 652.3533, found 652.3543.
Synthesis of inhibitor 18
Inhibitor 18 was synthesized from compound 12b by Boc removal using trifluoroacetic acid followed by coupling of the resulting amine with known acid 23 using EDC, HOBt, and iPr2NEt following the similar procedure described for the synthesis of inhibitor 4 (yield 68%, over two steps). 1H NMR (400 MHz, CDCl3) δ 7.85 (s, 1H), 7.47 (s, 1H), 7.38-7.28 (m, 3H), 7.24-7.19 (m, 2H), 6.84 (s, 1H), 6.47 (d, J = 8.0 Hz, 1H), 4.62-4.36 (m, 3H), 4.02 (p, J = 6.0 Hz, 1H), 3.90-3.81 (m, 2H), 3.47 (s, 3H), 3.12 (d, J = 5.0 Hz, 1H), 3.10-2.93 (m, 4H), 2.84 (dd, J = 12.2, 4.7 Hz, 1H), 2.77 (dd, J = 11.1, 6.8 Hz, 1H), 2.72 (q, J = 7.5 Hz, 2H), 1.76-1.65 (m, 1H), 1.30 (t, J = 7.5 Hz, 3H), 1.15 (d, J = 6.4 Hz, 3H), 0.85 (d, J = 6.7 Hz, 3H), 0.84 (d, J = 6.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 172.75, 167.90, 137.52, 133.84, 130.72, 129.28, 128.65, 127.72, 127.23, 126.75, 125.78, 120.26, 118.03, 117.60, 68.32, 67.48, 56.77, 52.30, 51.39, 46.41, 43.60, 39.66, 39.03, 28.42, 20.11, 18.95, 17.93, 14.25. HRMS-ESI (m/z): [M+H]+ calcd for C31H44N5O5S 598.3063, found 598.3060.
Synthesis of compound 25
To a solution of 24 (0.43 mmol, 0.145 g) in DMF (4 mL), was added NaH (1.72 mmol, 68.8 mg, 60% NaH in mineral oil) at 23 °C and the resulting mixture was stirred for 15 min at the same temperature. Iodo methane (1.72 mmol, 0.11 mL) was added to the reaction mixture and stirring was continued for further 2.5 h at 23 °C. Reaction mixture was quenched with methanol and then diluted with EtOAc. The resulting solution was washed with H2O & brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (30% EtOAc/hexanes) to obtain the corresponding α,α-dimethylated adduct in 51% (76.8 mg) yields. 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 1.0 Hz, 1H), 7.74 (s, 1H), 6.81 (s, 1H), 4.14 (s, 2H), 3.93 (s, 3H), 3.55 (s, 3H), 2.76 (q, J = 7.5 Hz, 2H), 1.57 (s, 6H), 1.32 (t, J = 7.5 Hz, 3H).
A mixture of the above ester (0.219 mmol, 76.7 mg) and NaOH (20 mmol, 10 mL, 2N NaOH) in EtOH (10 mL) and THF (10 mL) was stirred for 2.5 days at 23 °C. Solvent was removed under reduced pressure and the resulting mixture was diluted with H2O and diethyl ether. Organic layer was separated, aqueous layer was acidified with aqueous 1N HCl and extracted with ethyl acetate. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to furnish the corresponding crude acid (25) in 72% yields (53.2 mg) which was used directly in the coupling reaction without any further purification. LRMS-ESI (m/z): [M+Na]+ 359.19
Synthesis of 1-(benzyloxymethyl)cyclopropanesulfonyl chloride (27)
Solutions of BuLi [17.2 mmol, 6.9 mL (2.5 M in hexanes) in 15 mL THF] and butyl 3-chloro-1-propanesulfonate (26) (16.3 mmol, 3.5 g in 15 mL THF) were added at the same time via cannula to an oven dried flask containing THF (100 mL) at −78 °C and the resulting mixture was stirred for 5–10 min at −78 °C and 30 min at 0 °C. Reaction mixture was cooled back to −78 °C and a solution of BuLi [19.6 mmol, 7.8 mL (2.5 M in hexanes)] was added to this mixture. After stirring for 15 min at −78 °C, BOMCl (19.6 mmol, 2.7 mL) was added to the reaction flask and stirring was continued for further 2 h at −78 °C and 3 h at 23 °C. Reaction mixture was quenched with H2O and THF was removed under reduced pressure. The resulting mixture was diluted with CH2Cl2 and H2O. Organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (8–12% EtOAc/hexanes) to furnish butyl 1-(benzyloxymethyl)cyclopropanesulfonate in 80% yields (3.9 g). 1H NMR (300 MHz, CDCl3) δ 7.40-7.25 (m, 5H), 4.55 (s, 2H), 4.23 (t, J = 6.6 Hz, 2H), 3.79 (s, 2H), 1.73-1.58 (m, 2H), 1.48 (ABq, J = 6.9, 5.0 Hz, 2H), 1.44-1.30 (m, 2H), 1.11 (ABq, J = 7.0, 5.1 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 137.41, 128.33, 127.78, 127.61, 73.09, 70.66, 69.84, 37.81, 31.04, 18.55, 13.41, 10.72.
To a mixture of butyl 1-(benzyloxymethyl)cyclopropanesulfonate (13 mmol, 3.88 g) in DME (40 mL) and H2O (40 mL), KSCN (13.65 mmol, 1.33 g) was added at 23 °C and the resulting reaction mixture was refluxed for 15 h. Reaction mixture was cooled to 23 °C and diluted with H2O and ethyl acetate. Organic layer was separated and the aqueous layer was concentrated under reduced pressure to provide the crude potassium 1-(benzyloxymethyl)cyclopropanesulfonate which was used directly in the next step without additional purification.
A mixture of potassium 1-(benzyloxymethyl)cyclopropanesulfonate in SOCl2 (35 mL) and DMF (3.5 mL) was refluxed for 1.5 h and excess SOCl2 was removed under reduced pressure. Water was added carefully to the resulting mixture and extracted with ethyl acetate. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (5–10% EtOAc/hexanes) to obtain 1-(benzyloxymethyl)cyclopropanesulfonyl chloride 27 in 91% yields (3.1 g) (over 2 steps). 1H NMR (300 MHz, CDCl3) δ 7.46-7.28 (m, 5H), 4.63 (s, 2H), 4.01 (s, 2H), 1.85-1.77 (m, 2H), 1.46-1.37 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 136.99, 128.41, 127.89, 127.61, 73.29, 68.38, 52.39, 14.03.
Synthesis of sulfonamide 29
To a mixture of amine 28 (3.66 mmol, 0.8 g), pyridine (11 mmol, 0.89 mL) and DMAP (0.73 mmol, 89.2 mg) in CH2Cl2 at 0°C, was added a solution of 1-(benzyloxymethyl)-cyclopropanesulfonyl chloride (27) (3.84 mmol, 1 g in 5 mL CH2Cl2) and the resulting mixture was stirred for 44 h at 23 °C. Reaction mixture was diluted with CH2Cl2, washed with aqueous 1N HCl, brine and dried over anhydrous Na2SO4. Dichloromethane solution was filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (20–30% EtOAc/hexanes) to afford the corresponding sulfonamide 29 in 74% yields (1.2 g). 1H NMR (400 MHz, CDCl3) δ 9.38 (s, 1H), 8.25 (s, 1H), 7.87 (s, 1H), 7.49 (d, J = 7.1 Hz, 2H), 7.42 (t, J = 7.4 Hz, 2H), 7.39-7.33 (m, 1H), 6.79 (s, 1H), 6.69 (s, 1H), 4.73 (s, 2H), 3.91 (s, 3H), 3.90 (s, 2H), 2.75 (q, J = 7.5 Hz, 2H), 1.30 (t, J = 7.5 Hz, 3H), 1.11-0.99 (m, 2H), 0.77-0.65 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 167.62, 136.35, 135.01, 128.91, 128.63, 128.44, 122.14, 121.20, 120.85, 120.76, 120.61, 120.36, 74.15, 73.10, 51.84, 38.91, 18.09, 14.25, 10.25.
Synthesis of 7,6,5-tricyclic indole derivative 30
To a solution of sulfonamide 29 (2.7 mmol, 1.19 g) in MeOH (75 mL) and AcOH (25 mL), 10% Pd/C (0.2 g) was added under argon. Argon ballon was now replaced with H2 ballon and the resulting mixture was stirred for 16 h at 23 °C. Reaction mixture was filtered through celite and washed with MeOH. Solvent was removed under reduced pressure and the resulting residue was diluted with toluene and concentrated under reduced pressure to furnish the corresponding alcohol in 95% yield.
To a mixture of the above alcohol (0.6 mmol, 0.21 g) and Et3N (0.9 mmol, 0.12 mL) in CH2Cl2 (25 mL) at 0°C, was added methanesulfonyl chloride (0.63 mmol, 0.049 mL) and the resulting mixture was stirred for 2 h at 23 °C. Reaction mixture was diluted with CH2Cl2, washed with aqueous 1N HCl, brine and dried over anhydrous Na2SO4. Dichloromethane solution was filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (5–15% diethyl ether/CH2Cl2) to afford the corresponding mesylate in 46% (70% BRSM) yields (0.12 g, 69 mg of alcohol was recovered). 1H NMR (400 MHz, CDCl3) δ 9.24 (s, 1H), 8.26 (s, 1H), 7.87 (d, J = 1.0 Hz, 1H), 7.41 (s, 1H), 7.07 (s, 1H), 4.58 (s, 2H), 3.93 (s, 3H), 3.16 (s, 3H), 2.79 (q, J = 7.5 Hz, 2H), 1.38-1.28 (m, 5H), 1.06-0.97 (m, 2H).
To a solution of the above mesylate (0.69 mmol, 0.297g) in DMF (30 mL), was added NaH (2.76 mmol, 0.11 g, 60% dispersion in mineral oil) at 23 °C. After stirring the mixture for 3 h at 23 °C, iodo methane (3.5 mmol, 0.22 mL) was added to the flask and stirring was continued for further 1 h at 23 °C. Reaction mixture was carefully quenched with MeOH, diluted with ethyl acetate and washed with aqueous 1N HCl. Ethyl acetate solution was filtered and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (35–40% EtOAc/hexanes) to afford the corresponding 7, 6, 5-tricyclic indole derivative in 87% yields (0.21 g). 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 1.1 Hz, 1H), 7.83 (d, J = 1.0 Hz, 1H), 6.78 (s, 1H), 4.32 (s, 2H), 3.93 (s, 3H), 3.48 (s, 3H), 2.77 (q, J = 7.5 Hz, 2H), 1.53 (t, J = 6.6 Hz, 2H), 1.31 (t, J = 7.5 Hz, 3H), 1.03-0.95 (m, 2H).
A mixture of the above 7, 6, 5-tricyclic indole derivative (0.2 mmol, 69.7 mg) and NaOH (20 mmol, 0.8 g in 10 mL H2O) in EtOH (5 mL) and THF (10 mL) was stirred for 4 days at 23 °C. Solvent was removed under reduced pressure and the resulting mixture was diluted with H2O and diethyl ether. Organic layer was separated, aqueous layer was acidified with aqueous 1N HCl and extracted with ethyl acetate. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to furnish the corresponding crude acid (30) in 75% yields (50 mg) which was used directly in the coupling reaction without any further purification. LRMS-ESI (m/z): [M+Na]+ 357.26
Synthesis of (2R, 3S)-ethyl 3-hydroxy-2-{[(4-nitrophenyl)sulfonyl]oxy}hexanoate (32)
To a solution of (E)-ethyl hex-2-enoate (31) (12.6 mmol, 1.79 g) in tBuOH (30 mL) and H2O (30 mL), were added AD-mixα (17.6 g) and MeSO2NH2 (15.1 mmol, 1.44 g) at −1 °C and the resulting reaction mixture was stirred for 7 days at −1 °C. Reaction mixture was quenched with saturated aqueous Na2S2O3 solution and extracted with ethyl acetate. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (20–35% EtOAc/hexanes) to furnish (2R, 3S)-ethyl 2,3-dihydroxyhexanoate in 60% yields (1.34 g). 1H NMR (300 MHz, CDCl3) δ 4.29 (q, J = 7.2 Hz, 2H), 4.10-4.04 (m, 1H), 3.96-3.84 (m, 1H), 3.07 (d, J = 5.2 Hz, 1H), 1.90 (d, J = 7.7 Hz, 1H), 1.68-1.38 (m, 4H), 1.32 (t, J = 7.1 Hz, 3H), 0.96 (t, J = 7.2 Hz, 3H).
To a solution of (2R, 3S)-ethyl 2,3-dihydroxyhexanoate (6.7 mmol, 1.18 g) in CH2Cl2 (30 mL), were added Et3N (10 mmol, 1.39 mL) and 4-nitrobenzenesulfonyl chloride (6.7 mmol, 1.48 g) at 0 °C. The resulting reaction mixture was stirred for 24 h at 23 °C. Reaction mixture was diluted with H2O and CH2Cl2. Organic layer was separated and the aqueous layer was extracted with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (10–25% EtOAc/hexanes) to obtain (2R, 3S)-ethyl 3-hydroxy-2-{[(4-nitrophenyl)sulfonyl]oxy}hexanoate (32) in 54 % yields (1.3 g). 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 8.8 Hz, 2H), 8.16 (d, J = 8.9 Hz, 2H), 4.96 (d, J = 2.9 Hz, 1H), 4.15 (q, J = 7.1 Hz, 2H), 4.12-4.04 (m, 1H), 2.07 (brs, 1H), 1.62 – 1.44 (m, 3H), 1.43 – 1.30 (m, 1H), 1.21 (t, J = 7.1 Hz, 3H), 0.92 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 166.74, 150.75, 141.85, 129.45, 124.18, 80.97, 71.23, 62.30, 34.98, 18.47, 13.89, 13.63.
Synthesis of (2S, 3S)-ethyl 2-[(tert-butoxycarbonyl)amino]-3-hydroxyhexanoate (33)
To a solution of (2R, 3S)-ethyl 3-hydroxy-2-{[(4-nitrophenyl)sulfonyl]oxy}hexanoate (32) (3.6 mmol, 1.3 g) in DMF (10 mL), NaN3 (5.76 mmol, 0.374 g) was added at 23 °C and the resulting mixture was heated at 55 °C for 15 h. Reaction mixture was cooled to 23 °C, diluted with ethyl acetate and the resulting solution was washed with H2O, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (15% EtOAc/hexanes) to obtain (2S, 3S)-ethyl 2-azido-3-hydroxyhexanoate in 83% yields (0.6 g). 1H NMR (400 MHz, CDCl3) δ 4.29 (qd, J = 7.2, 1.3 Hz, 2H), 3.94 (s, 2H), 2.32 (brs, 1H), 1.59-1.45 (m, 3H), 1.44-1.37 (m, 1H), 1.33 (t, J = 7.2 Hz, 3H), 0.94 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 168.92, 71.56, 66.15, 61.99, 35.03, 18.52, 14.06, 13.74.
To a mixture of (2S, 3S)-ethyl 2-azido-3-hydroxyhexanoate (2.98 mmol, 0.6 g), and (Boc)2O (4.47 mmol, 0.975 g) in EtOH (15 mL) was added 10% Pd-C (0.15 g) at 23 °C under argon. Argon balloon was replaced with H2 balloon and the reaction mixture was stirred for 15 h under H2 atmosphere. Reaction mixture was filtered through celite, washed with ethyl acetate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (10–25% EtOAc/hexanes) to obtain (2S, 3S)-ethyl 2-[(tert-butoxycarbonyl)amino]-3-hydroxyhexanoate (33) in 96% yields (0.79 g). 1H NMR (300 MHz, CDCl3) δ 5.52 (d, J = 7.2 Hz, 1H), 4.42 – 4.28 (m, 1H), 4.27 - 4.13 (m, 2H), 3.94 – 3.80 (m, 1H), 3.03 (brs, 1H), 1.59 – 1.30 (m, 13H), 1.26 (t, J = 7.1 Hz, 3H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 170.68, 155.98, 80.20, 72.67, 61.48, 58.34, 35.29, 28.18, 18.86, 14.06, 13.82. LRMS-ESI (m/z): [M+Na]+ 298.26
Synthesis of tert-butyl [(2S, 3S)-3-hydroxy-1-(isobutylamino)-1-oxohexan-2-yl]carbamate (34a)
To a solution of (2S, 3S)-ethyl 2-[(tert-butoxycarbonyl)amino]-3-hydroxyhexanoate (33) (0.67 mmol, 0.18 g) in THF (6 mL) and H2O (3 mL), was added LiOH.H2O (3.45 mmol, 0.145 g). The resulting reaction mixture was stirred for 12 h at 23 °C. Reaction mixture was diluted with H2O and ethyl acetate. Organic layer was separated and the aqueous layer was acidified with 1N HCl and extracted with ethyl acetate. The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was used in the coupling reaction with-out any further purification.
Synthesis of tert-butyl [(2S, 3S)-3-hydroxy-1-(isobutylamino)-1-oxohexan-2-yl]carbamate (34a) has been carried out by coupling of (2S, 3S)-2-[(tert-butoxycarbonyl)amino]-3-hydroxyhexanoic acid with isobutyl amine using EDC, HOBt and iPr2NEt following the similar procedure described for the synthesis of (S)-tert-butyl [1-(isobutylamino)-1-oxobutan-2-yl]carbamate (yield 60%, over two steps). 1H NMR (400 MHz, CDCl3) δ 6.51 (brs, 1H), 5.62 (d, J = 7.2 Hz, 1H), 3.98-3.82 (m, 2H), 3.69 (brs, 1H), 3.21-3.08 (m, 1H), 3.06-2.92 (m, 1H), 1.77 (hept, J = 6.7 Hz, 1H), 1.63-1.46 (m, 3H), 1.44 (s, 9H), 1.41 – 1.32 (m, 1H), 0.97-0.83 (m, 9H).
Synthesis of compound 35a
Compound 35a was synthesized from tert-butyl [(2S, 3S)-3-hydroxy-1-(isobutylamino)-1-oxohexan-2-yl]carbamate (34a) by Boc removal using trifluoroacetic acid followed by reductive amination of the resulting amine with aldehyde 11 following the procedure described for the synthesis of compound 12a (yield 49%, over two steps). 1H NMR (400 MHz, CDCl3) δ 7.28 (t, J = 7.2 Hz, 2H), 7.22 (d, J = 7.0 Hz, 1H), 7.19-7.13 (m, 2H), 4.63 (d, J = 7.4 Hz, 1H), 3.92 (brs, 1H), 3.84 – 3.74 (m, 1H), 3.12-2.94 (m, 3H), 2.86-2.71 (m, 2H), 2.64 (dd, J = 12.1, 4.6 Hz, 1H), 2.55 (dd, J = 12.0, 7.4 Hz, 1H), 1.71 (hept, J = 6.7 Hz, 1H), 1.58-1.21 (m, 13H), 0.90 (t, J = 7.0 Hz, 3H), 0.86 (d, J = 6.6 Hz, 6H).
Synthesis of inhibitor 19
Inhibitor 19 has been synthesized from compound 35a by Boc removal using trifluoroacetic acid followed by coupling of the resulting amine with known acid 23 using EDC, HOBt, and iPr2NEt following the similar procedure described for the synthesis of inhibitor 8 (yield 50%). 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 1.0 Hz, 1H), 7.46 (s, 1H), 7.35 – 7.28 (m, 2H), 7.26-7.19 (m, 3H), 6.84 (s, 1H), 6.41 (d, J = 8.2 Hz, 1H), 4.54 – 4.44 (m, 3H), 3.89 – 3.76 (m, 3H), 3.47 (s, 3H), 3.13 (d, J = 4.9 Hz, 1H), 3.11 – 2.99 (m, 2H), 2.97 (d, J = 6.8 Hz, 2H), 2.83 (dd, J = 12.3, 4.8 Hz, 1H), 2.79-2.68 (m, 3H), 1.71 (hept, J = 6.7 Hz, 1H), 1.54 – 1.33 (m, 4H), 1.30 (t, J = 7.5 Hz, 3H), 0.91 – 0.80 (m, 9H). 13C NMR (100 MHz, CDCl3) δ 172.89, 167.85, 137.51, 133.83, 130.72, 129.30, 128.67, 127.72, 127.26, 126.78, 125.83, 120.25, 117.93, 117.56, 72.00, 66.43, 56.81, 51.98, 51.20, 46.44, 43.62, 39.61, 38.94, 35.26, 28.42, 20.10, 18.98, 17.94, 14.26, 13.94. HRMS-ESI (m/z): [M+H]+ calcd for C33H48N5O5S 626.3376, found 626.3369.
Synthesis of inhibitor 20
Inhibitor 20 has been synthesized from compound 35a by Boc removal using trifluoroacetic acid followed by coupling of the resulting amine with acid 25 using EDC, HOBt, and iPr2NEt following the similar procedure described for the synthesis of inhibitor 8 (yield 52%). 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 0.8 Hz, 1H), 7.40 (s, 1H), 7.34 – 7.28 (m, 2H), 7.26-7.18 (m, 3H), 6.80 (s, 1H), 6.39 (d, J = 8.2 Hz, 1H), 4.54-4.40 (m, 1H), 4.11 (s, 2H), 3.80 (p, J = 4.2 Hz, 1H), 3.50 (s, 3H), 3.13 (d, J = 4.8 Hz, 1H), 3.11 – 2.99 (m, 2H), 2.97 (d, J = 6.8 Hz, 2H), 2.82 (dd, J = 12.3, 5.0 Hz, 1H), 2.79 – 2.64 (m, 3H), 1.70 (hept, J = 6.7 Hz, 1H), 1.56 (s, 6H), 1.53 – 1.33 (m, 4H), 1.30 (t, J = 7.5 Hz, 3H), 0.93 – 0.79 (m, 9H). 13C NMR (125 MHz, CDCl3) δ 172.59, 167.99, 137.38, 134.28, 130.53, 129.22, 128.81, 128.59, 127.37, 126.83, 126.71, 120.29, 116.36, 114.86, 71.83, 66.29, 65.68, 56.50, 51.89, 51.03, 46.38, 39.40, 38.81, 35.15, 28.33, 22.49, 22.46, 20.03, 19.71, 18.90, 17.88, 14.02, 13.87. HRMS-ESI (m/z): [M+H]+ calcd for C35H52N5O5S 654.3689, found 654.3696.
Synthesis of Inhibitor 21
Inhibitor 21 was synthesized (yield 65%, over 2 steps) from 12b by Boc deprotection followed by coupling with 7,6,5 tricyclic indole derivative 30 as described for the inhibitor 4. 1H NMR (400 MHz, CDCl3) δ 7.88 (s, 1H), 7.47 (s, 1H), 7.36 (t, J = 6.0 Hz, 1H), 7.33-7.27 (m, 2H), 7.26-7.17 (m, 3H), 6.77 (s, 1H), 6.55 (d, J = 8.2 Hz, 1H), 4.56-4.43 (m, 1H), 4.27 (s, 2H), 4.03 (p, J = 6.2 Hz, 1H), 3.41 (s, 3H), 3.12 (d, J = 5.1 Hz, 1H), 3.08-2.99 (m, 2H), 2.95 (dd, J = 6.5, 4.4 Hz, 2H), 2.82 (dd, J = 12.2, 4.7 Hz, 1H), 2.78-2.65 (m, 3H), 1.69 (hept, J = 6.6 Hz, 1H), 1.52 (t, J = 6.3 Hz, 2H), 1.28 (t, J = 7.5 Hz, 3H), 1.13 (d, J = 6.4 Hz, 3H), 1.00 (t, J = 6.4 Hz, 2H), 0.83 (d, J = 6.7 Hz, 3H), 0.82 (d, J = 6.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 172.75, 167.89, 137.56, 134.44, 130.80, 129.26, 128.62, 128.14, 127.09, 126.71, 126.13, 120.33, 118.15, 117.90, 68.26, 67.53, 52.30, 52.23, 51.39, 46.39, 43.05, 39.77, 39.02, 28.40, 20.09, 18.86, 17.94, 14.19, 12.67. HRMS-ESI (m/z): [M+H]+ calcd for C33H46N5O5S 624.3220, found 624.3223.
Synthesis of compound 35b
(2S, 3S)-2-Amino-3-hydroxy-N-isopropylhexanamide was synthesized from (2S, 3S)-2-[(tert-butoxycarbonyl)amino]-3-hydroxyhexanoic acid by coupling with isopropyl amine using EDC, HOBt, and iPr2NEt followed by boc removal using trifluoroacetic acid following the procedure described for the synthesis of (2S, 3S)-2-amino-3-hydroxy-N-isobutyl-N-methylbutanamide (yield 79% over two steps). 1H NMR (400 MHz, CDCl3) δ 7.22 (br, 1H), 4.14-3.92 (m, 1H), 3.81 – 3.67 (m, 1H), 3.22 (d, J = 6.2 Hz, 1H), 2.40 (brs, 2H), 1.62 – 1.24 (m, 4H), 1.18 – 1.08 (m, 6H), 0.91 (t, J = 6.9 Hz, 3H).
Compound 35b was prepared from (2S, 3S)-2-amino-3-hydroxy-N-isopropylhexanamide by reductive amination with aldehyde 11 following the procedure described for the synthesis of compound 12a (yield 81%). 1H NMR (400 MHz, CDCl3) δ 7.28 (t, J = 7.4 Hz, 2H), 7.22 (d, J = 7.3 Hz, 1H), 7.20-7.13 (m, 2H), 7.00 (br, 1H), 4.62 (d, J = 8.1 Hz, 1H), 4.08 – 3.85 (m, 2H), 3.83 – 3.72 (m, 1H), 2.98 (d, J = 5.3 Hz, 1H), 2.82 (dd, J = 13.7, 6.5 Hz, 1H), 2.75 (dd, J = 13.3, 7.4 Hz, 1H), 2.60 (dd, J = 12.2, 4.7 Hz, 1H), 2.54 (dd, J = 12.1, 7.3 Hz, 1H), 1.59 – 1.20 (m, 13H), 1.11 (d, J = 6.5 Hz, 3H), 1.06 (d, J = 6.6 Hz, 3H), 0.90 (t, J = 7.0 Hz, 3H).
Synthesis of inhibitor 22
Inhibitor 22 was synthesized by Boc removal of 35b using trifluoroacetic acid followed by coupling of the resulting amine with the acid 30 using EDC, HOBt, and iPr2NEt following the similar procedure described for the synthesis of inhibitor 4 (yield 65% over two steps). 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 1.1 Hz, 1H), 7.46 (d, J = 1.0 Hz, 1H), 7.36 – 7.28 (m, 2H), 7.28 – 7.20 (m, 3H), 6.97 (d, J = 8.2 Hz, 1H), 6.78 (s, 1H), 6.38 (d, J = 8.2 Hz, 1H), 4.54 – 4.42 (m, 1H), 4.28 (ABq, J = 20.6, 14.9 Hz, 2H), 4.08 – 3.95 (m, 1H), 3.84-3.75 (m, 1H), 3.44 (s, 3H), 3.09 (d, J = 4.9 Hz, 1H), 2.98 (d, J = 6.8 Hz, 2H), 2.81 (dd, J = 12.3, 4.9 Hz, 1H), 2.77 – 2.69 (m, 3H), 1.56-1.33 (m, 5H), 1.33-1.24 (m, 4H), 1.07 (t, J = 6.9 Hz, 6H), 1.01 – 0.96 (m, 2H), 0.87 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 171.67, 167.77, 137.47, 134.36, 130.68, 129.20, 128.58, 128.06, 126.97, 126.69, 126.05, 120.23, 118.05, 117.83, 71.81, 66.05, 52.15, 51.63, 50.99, 43.01, 40.92, 39.60, 38.81, 35.05, 22.46, 18.90, 17.89, 14.15, 13.86, 12.64. HRMS-ESI (m/z): [M+H]+ calcd for C34H48N5O5S 638.3376, found 638.3371.
Determination of X-ray Structure of β-secretase-Inhibitor 5 Complex
Expression and purification of recombinant human beta-secretase, crystal growing, inhibitor soaking of the crystal and diffraction data collection were as previously described.8 The structure was determined by molecular replacement implemented with the program AMoRe using the the C molecule of previously determined memapsin 2 structure (PDB ID: 1FKN) as a search model8 with removed inhibitor and water molecules. Rotation and translation functions followed by the rigid-body refinement with data from 15 Å to 3.5 Å resolution in space group P21 gave unambiguous solutions for the four memapsin 2 molecules in the asymmetric unit. A random selection of 7% of reflections (9028 reflections) was set aside as the test set for cross-validation during the refinement. The refined model has well-defined electron density for the inhibitor and its corresponding structure was built into the active site. The four molecules in the crystallographic asymmetric unit have essentially identical structures. The crystal form was determined to be monoclinic with a resolution of 2.0 Å. The unit cell parameters are a = 86.4Å, b = 130.3 Å, c = 88.4 Å, β = 97.5. The coordinates and structure factors of the β-secretase and 5 complex have been deposited in Protein Data Bank28 with pdb id: 4GID.
Supplementary Material
Acknowledgements
Financial support by the National Institutes of Health (AG 18933) is gratefully acknowledged. We thank Mr. Cuthbert D. Martyr for his help with the MS data.
Abbreviations
- AD
Alzheimer’s Disease
- BACE 1
Beta-site APP Cleaving Enzyme 1
- BACE 2
Beta-site APP Cleaving Enzyme 2
- APP
β-Amyloid Precursor Protein
- CD
Cathepsin D
Footnotes
The PDB accession code for 5-bound β-secretase X-ray structure is 4GID.
Supporting Information: HPLC, and HRMS data of inhibitors 4–9 and 18–22; crystallographic data collection and refinement statistics for inhibitor 5. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Goedert M, Spillantini MG. A Century of Alzheimer's Disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ, Schenk D. Alzheimer's Disease: Molecular Understanding Predicts Amyloid-Based Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 3.Citron M. Alzheimer's Disease: Strategies for Disease Modification. Nat. Rev. Drug Discovery. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 4.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Aβ Causes the Onset of Early Alzheimer’s Disease-Related Cognitive Deficits in Transgenic Mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh AK, Brindisi M, Tang J. Developing β-Secretase Inhibitors for Treatment of Alzheimer’s Disease. J. Neurochem. 2012;120:71–83. doi: 10.1111/j.1471-4159.2011.07476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang J, Hong L, Ghosh AK. The Discovery of β-Secretase and Development Toward a Clinical Inhibitor for AD: An Exciting Academic Collaboration. In: Ghosh AK, editor. Aspartic Acid Proteases as Therapeutic Targets. Vol. 45. Wiley-VCH; 2010. pp. 413–440. [Google Scholar]
- 7.Ghosh AK, Shin D, Downs D, Koelsch G, Lin X, Ermolieff J, Tang J. Design of Potent Inhibitors for Human Brain Memapsin 2 (β-Secretase) J. Am. Chem. Soc. 2000;122:3522–3523. doi: 10.1021/ja000300g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong L, Koelsch G, Lin X, Wu S, Terzyan S, Ghosh AK, Zhang XC, Tang J. Structure of the Protease Domain of Memapsin 2 (β-Secretase) Complexed with Inhibitor. Science. 2000;290:150–153. doi: 10.1126/science.290.5489.150. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni S, Xu X, Miller HB, Reddy DS, Weerasena V, Turner R, Chang W, Koelsch G, Tang J. Potent Memapsin 2 (β-Secretase) Inhibitors: Design, Synthesis, Protein-Ligand X-ray Structure, and in vivo Evaluation. Bioorg. Med. Chem. Lett. 2008;18:1031–1036. doi: 10.1016/j.bmcl.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang W, Huang X, Downs D, Cirrito JR, Koelsch G, Holtzman DM, Ghosh AK, Tang J. β-Secretase Inhibitor GRL-8234 Rescues Age-related Cognitive Decline in APP Transgenic Mice. FASEB J. 2011;25:775–784. doi: 10.1096/fj.10-167213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner RT, Loy JA, Nguyen C, Devasamudram T, Ghosh AK, Koelsch G, Tang J. Specificity of Memapsin 1 and Its Implications on the Design of Memapsin 2 (β-Secretase) Inhibitor Selectivity. Biochemistry. 2002;41:8742–8746. doi: 10.1021/bi025926t. [DOI] [PubMed] [Google Scholar]
- 12.Diment S, Leech MS, Stahl PD. Cathepsin D is Membrane-Associated in Macrophage Endosomes. J. Biol. Chem. 1988;263:6901–6907. [PubMed] [Google Scholar]
- 13.(a) Koike M, Nakanishi H, Saftig P, Ezaki J, Isahara K, Ohsawa Y, Schulz-Schaeffer W, Watanabe T, Waguri S, Kametaka S, Shibata M, Yamamoto K, Kominami E, Peters C, Figura K, Uchiyama Y. Cathepsin D Deficiency Induces Lysosomal Storage with Ceroid Lipofuscin in Mouse CNS Neurons. J. Neurosci. 2000;20:6898–6906. doi: 10.1523/JNEUROSCI.20-18-06898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saftig P, Hetman M, Schmahl W, Weber K, Heine L, Mossmann H, Koster A, Hess B, Evers M, von Figura K. EMBO J. 1995;14:3599–3608. doi: 10.1002/j.1460-2075.1995.tb00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh AK, Kumaragurubaran N, Hong L, Lei H, Hussain KA, Liu C-F, Devasamudram T, Weerasena V, Turner R, Koelsch G, Bilcer G, Tang J. Design, Synthesis and X-ray Structure of Protein–Ligand Complexes: Important Insight into Selectivity of Memapsin 2 (β-Secretase) Inhibitors. J. Am. Chem. Soc. 2006;128:5310–5311. doi: 10.1021/ja058636j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Labby KJ, Xue F, Kraus JM, Ji H, Mataka J, Li H, Martasek P, Roman LJ, Poulos TL, Silverman RB. Intramolecular Hydrogen Bonding: A Potential Strategy for More Bioavailable Inhibitors of Neuronal Nitric Oxide Synthase. Bioorg. Med. Chem. 2012;20:2435–2443. doi: 10.1016/j.bmc.2012.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kuhn B, Mohr P, Stahl M. Intramolecular Hydrogen Bonding in Medicinal Chemistry. J. Med. Chem. 2010;53:2601–2611. doi: 10.1021/jm100087s. [DOI] [PubMed] [Google Scholar]
- 16.(a) Iserloh U, Cumming JN. Peptidomimetic BACE1 Inhibitors for Treatment of Alzheimer's Disease: Design and Evolution. In: Ghosh AK, editor. Aspartic Acid Proteases as Therapeutic Targets. Vol. 45. Wiley-VCH; 2010. pp. 441–479. [Google Scholar]; (b) Coburn CA, Stachel SJ, Jones KG, Steele TG, Rush DM, DiMuzio J, Pietrak BL, Lai M-T, Huang Q, Lineberger J, Jin L, Munshi S, Holloway MK, Espeseth A, Simon A, Hazuda D, Graham SL, Vacca JP. BACE-1 Inhibition by a Series of ψ[CH2NH] Reduced Amide Isosteres. Bioorg. Med. Chem. Lett. 2006;16:3635–3638. doi: 10.1016/j.bmcl.2006.04.076. [DOI] [PubMed] [Google Scholar]
- 17.Velmourougane G, Harbut MB, Dalal S, McGowan S, Oellig CA, Meinhardt N, Whisstock JC, Klemba M, Greenbaum DC. Synthesis of New (−)-Bestatin-Based Inhibitor Libraries Reveals a Novel Binding Mode in the S1 Pocket of the Essential Malaria M1 Metalloaminopeptidase. J. Med. Chem. 2011;54:1655–1666. doi: 10.1021/jm101227t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni SS, Xu X, Chang W, Weerasena V, Turner R, Koelsch G, Bilcer G, Tang J. Design, Synthesis, and X-ray Structure of Potent Memapsin 2 (β-Secretase) Inhibitors with Isophthalamide Derivatives as the P2-P3-Ligands. J. Med. Chem. 2007;50:2399–2407. doi: 10.1021/jm061338s. [DOI] [PubMed] [Google Scholar]
- 19.Andurkar SV, Stables JP, Kohn H. Synthesis and Anticonvulsant Activities of (R)-(O)-Methylserine Derivatives. Tetrahedron: Asymmetry. 1998;9:3841–3854. [Google Scholar]
- 20.Charrier N, Clarke B, Cutler L, Demont E, Dingwall C, Dunsdon R, East P, Hawkins J, Howes C, Hussain I, Jeffrey P, Maile G, Matico R, Mosley J, Naylor A, O’Brien A, Redshaw S, Rowland P, Soleil V, Smith KJ, Sweitzer S, Theobald P, Vesey D, Walter DS, Wayne G. Second Generation of Hydroxyethylamine BACE-1 Inhibitors: Optimizing Potency and Oral Bioavailability. J. Med. Chem. 2008;51:3313–3317. doi: 10.1021/jm800138h. [DOI] [PubMed] [Google Scholar]
- 21.Hubert DE, Sally R, Simon WD. Tricyclic Indole Derivatives and Their Use in the Treatment of Alzheimer’s Disease. WO 2004/094430 A1 [Google Scholar]
- 22.Charrier N, Demont E, Dunsdon R, Maile G, Naylor A, O'Brien A, Redshaw S, Theobald P, Vesey D, Walter D. Synthesis of Indoles: Efficient Functionalisation of the 7-Position. Synthesis. 2006:3467–3477. [Google Scholar]
- 23.Fleming PR, Sharpless KB. Selective Transformations of Threo-2,3-dihydroxy Esters. J. Org. Chem. 1991;56:2869–2875. [Google Scholar]
- 24.Memapsin 2 inhibition was measured using recombinant enzyme produced from E. coli expression. A fluorogenic substrate Arg-Glu(EDANS)-Glu-Val-Asn-Leu-Asp-Ala-Glu-Phe-Lys (Dabcyl)-Arg was used with 0.47 PM of the enzyme in 0.1 M Na-acetate + 5% dimethylsulfoxide, pH 4.5 at 37 °C. The excitation wavelength was 350 nm and the emission wavelength was 490 nm. For details, please see: Ermolieff J, Loy JA, Koelsch G, Tang J. Proteolytic Activation of Recombinant Pro-memapsin 2 (Pro-β-secretase) Studied with New Fluorogenic Substrates. Biochemistry. 2000;39:12450–12456. doi: 10.1021/bi001494f.
- 25.Chang WP, Koelsch G, Wong S, Downs D, Da H, Weerasena V, Gordon B, Devasamudram T, Bilcer G, Ghosh AK, Tang J. In vivo Inhibition of Aβ Production by Memapsin 2 (β-Secretase) Inhibitors. J. Neurochem. 2004;89:1409–1416. doi: 10.1111/j.1471-4159.2004.02452.x. [DOI] [PubMed] [Google Scholar]
- 26.Patel S, Vuillard L, Cleasby A, Murray CW, Yon J. Apo and Inhibitor Complex Structures of BACE (β-Secretase) J. Mol. Biol. 2004;343:407–416. doi: 10.1016/j.jmb.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 27.Charrier N, Clarke B, Cutler L, Demont E, Dingwall C, Dunsdon R, Hawkins J, Howes C, Hubbard J, Hussain I, Maile G, Matico R, Mosley J, Naylor A, O’Brien A, Redshaw S, Rowland P, Soleil V, Smith KJ, Sweitzer S, Theobald P, Vesey D, Walter DS, Wayne G. Second Generation of BACE-1 Inhibitors Part 3: Towards Non-hydroxyethylamine Transition State Mimetics. Bioorg. Med. Chem. Lett. 2009;19:3674–3678. doi: 10.1016/j.bmcl.2009.03.149. [DOI] [PubMed] [Google Scholar]
- 28.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.