

Abstract
Background:
Amplification of aurora kinase A (AK-A) overrides the mitotic spindle assembly checkpoint, inducing resistance to taxanes. RNA interference targeting AK-A in human pancreatic cancer cell lines enhanced taxane chemosensitivity. In this study, a novel AK-A inhibitor, CYC3, was investigated in pancreatic cancer cell lines, in combination with paclitaxel.
Methods:
Western blot, flow cytometry and immunostaining were used to investigate the specificity of CYC3. Sulforhodamine B staining, time-lapse microscopy and colony-formation assays were employed to evaluate the cytotoxic effect of CYC3 and paclitaxel. Human colony-forming unit of granulocyte and macrophage (CFU-GM) cells were used to compare the effect in tumour and normal tissue.
Results:
CYC3 was shown to be a specific AK-A inhibitor. Three nanomolar paclitaxel (growth inhibition 50% (GI50) 3 nℳ in PANC-1, 5.1 nℳ in MIA PaCa-2) in combination with 1 μℳ CYC3 (GI50 1.1 μℳ in MIA PaCa2 and 2 μℳ in PANC-1) was synergistic in inhibiting pancreatic cell growth and causing mitotic arrest, achieving similar effects to 10-fold higher concentrations of paclitaxel (30 nℳ). In CFU-GM cells, the effect of the combination was simply additive, displaying significantly less myelotoxicity compared with high concentrations of paclitaxel (30 nℳ; 60–70% vs 100% inhibition).
Conclusion:
The combination of lower doses of paclitaxel and CYC3 merits further investigation with the potential for an improved therapeutic index in vivo.
Keywords: pancreatic cancer, paclitaxel, CYC3, mitotic inhibitor, combination treatment
Aurora kinases (AKs) are a family of serine/threonine kinases that are important for mitosis. In mammals, there are three members in this family, AK-A, AK-B and AK-C (Gautschi et al, 2008). Both AK-A and AK-B are overexpressed in a number of cancers, including breast, lung, bladder and pancreas (Gautschi et al, 2008). Given their association with cancer, both AK-A and AK-B have become targets for cancer therapy (Lok et al, 2010). Various selective or non-selective inhibitors of AKs have been developed and tested as potential cancer therapeutics in clinical or preclinical studies (Cheung et al, 2009; Kitzen et al, 2010). The majority of these compounds are pan-aurora inhibitors. Those drugs under evaluation for use in solid tumours that are reported to be specific for AK-A include ENMD-2076, which also has VEGFR inhibitor activity. It showed promising anti-tumour activity, especially in ovarian cancer in a phase I trial (Diamond et al, 2011), but it has also recently been shown to inhibit AK-B at concentrations that are cytotoxic (Wang et al, 2010b), so it is not an AK-A-specific inhibitor in vivo. MK5108 has more than 200-fold selectivity for AK-A over AK-B and appears to be AK-A-specific in cells and in xenograft tumours (Shimomura et al, 2010a). MK5108 is being tested in a phase I trial in advanced solid tumours with and without docetaxel (Minton et al, 2010). Another AK-A-specific inhibitor, MLN8054, was discontinued because of benzodiazepine-like effects (Dees et al, 2010; Macarulla et al, 2010), and has been replaced by MLN8237 (alisertib; Manfredi et al, 2011b). MLN8237 is now in phase II trials in lung, breast, head and neck, and gastrointestinal cancers (Sharma et al, 2011). To our knowledge, no AK-A-specific inhibitor is being investigated in clinical trials in pancreatic cancer.
Pancreatic ductal adenocarcinoma is a particularly lethal cancer with a cumulative 5-year survival rate of less than 5% (Warshaw and Fernandez-del Castillo, 1992). It is resistant to most current forms of cytotoxic chemotherapy and ionising radiation (Li et al, 2010). Overexpression of AK-A has been shown in pancreatic cancer cell lines and tissues (Li et al, 2003; Rojanala et al, 2004). Of relevance to our studies, amplification of AK-A has been reported to induce resistance to taxanes (Anand et al, 2003). Knockdown of AK-A by RNA interference in pancreatic cancer cell lines not only led to growth inhibition, cell cycle arrest and apoptosis (Hata et al, 2005b; Warner et al, 2006), but also enhanced the taxane chemosensitivity in these cells (Hata et al, 2005b). Therefore, co-treatment of an AK-A-specific inhibitor with paclitaxel may be an intriguing drug combination for pancreatic cancer.
Bone marrow (BM) is very sensitive to many anti-proliferative agents (Negro et al, 2001). A clinical side effect of paclitaxel treatment is neutropenia, caused by damage to the neutrophil progenitor cells in BM (Rowinsky et al, 1993). A concern in considering combinations of AK inhibitors and taxanes is that in the phase I clinical trials of several different dual inhibitors for both AK-A and AK-B, neutropenia is one of the major side effects observed (Cheung et al, 2009). In pre-clinical studies to assess potential myelotoxicity, the colony-forming unit of granulocyte and macrophage (CFU-GM) assay has been optimised and validated to predict potential drug myelotoxicity in vitro (Parent-Massin, 2001), but to our knowledge this has not been used to test AK-A inhibitors.
In this report, an AK-A-specific inhibitor CYC3 from Cyclacel Ltd has been tested alone and in combination with paclitaxel in pancreatic cancer cell lines. To distinguish additivity from synergy, we employed growth inhibition assays (by sulforhodamine B (SRB) staining) and mathematical modelling to search for real synergistic combinations. Later, we confirmed the synergy by time-lapse microscopy and colony-formation assays. In addition, we investigated the potential myelotoxicity of the synergistic combination identified using a CFU-GM assay with human BM cells.
Materials and methods
Cell culture
PANC-1 and MIA PaCa-2 (human pancreatic carcinoma) cells obtained from the European Collection of Cell Cultures (ECACC; Health Protection Agency, Salisbury, UK) were verified by STR genotyping and tested negative for mycoplasma. They were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum at 37 °C and 5% CO2. Paclitaxel (catalogue number 1097) was obtained from Tocris Bioscience (Bristol, UK). Paclitaxel and CYC3 were dissolved in dimethylsulphoxide (DMSO) and then diluted in culture medium to a final concentration of 0.2% DMSO.
Sulforhodamine assay
Cells were seeded in 96-well plates at concentrations of 3000 PANC-1 cells per well or 2000 MIA PaCa-2 cells per well. Twenty-four hours later, cells were treated with drugs for 72 h. Then, cells were fixed with trichloroacetic acid and stained with SRB (Skehan et al, 1990). Fluorescence was quantified using an Infinite 200 PRO plate reader (Tecan, Reading, UK) at a wavelength of 545 nm. Drug-treated wells were compared with solvent control wells, and the concentration of drug that resulted in 50% of the cell growth of the solvent control was designated as the GI50 concentration, calculated using Graphpad PRISM 5 (Graphpad Software, La Jolla, CA, USA). At least three biological replicates were performed for each assay.
Synergy calculation
Ninety-six-well plates were treated with a dilution series of each drug in an 8 × 8 checkerboard pattern of combinations. After SRB staining to obtain the growth inhibition data, we used software that we developed to identify synergistic drug combinations (SynergySurface, www.synergysurface.com; Figure 3). The single agent inhibition values were used to calculate a drug combination surface under the assumption of an additive effect. To obtain this additive surface, different models of additivity can be used (Bliss, Loewe and the Highest Single Agent). Details of the three models and their relative advantages can be found in previous publications (Jonker et al, 2005; Fitzgerald et al, 2006a); we used the Bliss model in these studies because it assumes independent mechanisms of action of the two drugs. Regions of synergy are then detected by comparing obtained data from a combination with the calculated additive effect. This is done by subtracting the calculated additive inhibition values from the measured inhibition to obtain the final difference values. In the final synergy surface, positive values hence indicate synergy regions, whereas negative difference values identify antagonistic effects.
Time-lapse microscopy
Cells were seeded in 96-well plates, and 24 h later cells were exposed to drug and imaged every 3 h by time-lapse microscopy using an Incucyte (Essen Bioscience, Welwyn Garden City, UK) for 72 h. The cell confluence was calculated using the Incucyte software (2010A, Rev2).
Cancer cell colony-formation assay
A total of 20 000 cells were seeded in 6-well plates, and 24 h later cells were exposed to drugs for 72 h, then counted. Equal numbers of cells (1000, 2500 and 5000) from each sample were then seeded in 6 cm plates and left to grow for 7 days. After that, cells were fixed with 70% methanol and stained with 5% Giemsa (Sigma, Gillingham, UK). Colony numbers were calculated using ImageQuanTL (GE Healthcare, Little Chalfont, UK).
Immunostaining
A total of 5000 cells were seeded in 8-well chamber slides (Ibidi, München, Germany) and treated with drugs 24 h later. After treatment, cells were fixed with 4% paraformaldehyde in PBS, permeabilised with 0.3% Triton X-100 in PBS, then stained with antibodies and counterstained with DAPI. The primary antibodies were anti-α-tubulin (T5168, Sigma) and anti-p-AK-A (T288; 3079, Cell Signalling Technologies, Danvers, MA, USA); secondary antibodies were anti-rabbit IgG Alexa 647 (4414, Cell Signalling Technologies) and anti-mouse IgG Alexa 488 (4408, Cell Signalling Technologies). The confocal image was taken using a Leica IR Laser microscope (Milton Keynes, UK). The image quantification was done using Icys research imaging cytometer (Compucyte, Westwood, MA, USA) and analysed as described in http://www.compucyte.com/cellcycle.htm.
Western blot
For a typical experiment, proteins were extracted from cells using RIPA buffer (50 mℳ Tris HCl pH 8.0, 150 mℳ NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) and equal amounts (measured using Bradford assay reagent (Biorad, Hemel Hempstead, UK)) were separated by SDS-PAGE using Novex Bris-Tris 4-12% gels (Invitrogen, Paisley, UK) in MOPs buffer (Invitrogen) and then transferred to nitrocellulose membrane (Invitrogen). After blocking with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE, USA), blots were stained with primary antibodies from Cell Signalling Technologies: anti-phospho AK-A/B/C (2914), anti-AK-A (3092S), anti-cleaved PARP (5625S); from Abcam (Cambridge, UK): anti-β-actin (ab6276), anti-AK-B (ab2254); and from Millipore (Watford, UK): anti-p-H3 S10 (06-570). Secondary antibodies from LiCOR Biosciences were IRDye 680- or 800CW-conjugated goat anti-mouse or anti-rabbit IgGs (926–32210, 926–32211, 926–32220, 926–32221). The blots were imaged and quantified using the Odyssey Infrared Imaging System (LI-COR Biosciences). To detect phospho-aurora A in PANC-1 and MiaPaCa-2 cells, it was necessary to first increase the level of aurora A expression by arresting the cells in mitosis with nocodazole, because the level of aurora A was too low in asynchronous cell populations.
Flow cytometry
In a typical experiment, cells were harvested by 0.05% trypsin, retaining the culture media containing floating cells. Cells were fixed with 70% ethanol, stained with propidium iodide solution (0.1% v/v Triton-X-100, 20 μg ml−1 propidium iodide, 0.2 mg ml−1 ribonuclease A in PBS) and analysed using a BD FACSCalibur flow cytometer (Becton Dickinson, Oxford, UK). Machine configuration details and representative plots are shown in the Supplementary Methods). The cell cycle distribution of the samples was determined using FlowJo version 7.6.1 (Tree Star, Inc., Ashland, OR, USA).
LC-MS/MS analysis of paclitaxel
Cell pellets obtained after washing in PBS were fixed in ice-cold methanol (70% v/v) by vigorous vortexing, followed by incubation at −80 °C for 1 h. The suspension was sonicated, and the cell extract from untreated cells was used to prepare calibration standards. An aliquot of the cell suspension (200 μl) was added to 200 μl of ice-cold acetonitrile (100% v/v) containing paclitaxel-[2H5] internal standard. After centrifugation, the supernatant was evaporated to dryness in a speedvac. For media, 25 μl of the supernatant was processed in the same way as an aliquot of the cell suspension. The residue was reconstituted in acetonitrile (50% v/v) and 20 μl was injected into a Dionex-Ultimate 3000 LC with autosampler using an Acquity UPLC HSS T3 50 mm × 2.1 mm i.d., 1.8 μm column (Waters, Elstree, UK). The mobile phase was (A) 0.1% acetic acid: (acetonitrile : methanol, (1 : 1)) 70 : 30 and (B) 0.1% acetic acid: (acetonitrile : methanol, (1 : 1)) 10 : 90. The gradient, at a flow rate of 225 μl min−1, was 25% B for 0.2 min, increased to 100% over 0.2 min and maintained at 100% for 2.8 min, decreased to 25% over 0.2 min and maintained at 25% for 2.1 min to give a total run time of 5.5 min. The LC-MS/MS was performed on an Applied Biosystems Sciex 4000 Q-trap mass spectrometer (Applied Biosystems, Carlsbad, CA, USA) equipped with a turbo-ionspray source and data was acquired using Analyst 1.4.2 software (Applied Biosystems). The mass spectrometry parameters were set essentially as described by Zhang et al (2011). Quantitation of the internal standard was done by multiple-reaction monitoring of the transition 881.4–308.1, with all other parameters identical to those used for paclitaxel.
Colony-forming unit of granulocyte and macrophage assay frozen
Human BM mononuclear cells and methylcellulose-based culture medium (MethoCult H4025 optimum without EPO) were purchased from Stem Cell Technologies (Grenoble, France). The cells were counted and suspended in MethoCult medium with or without drugs, and then 2 × 104 cells were plated in 35 mm petri dishes and cultured for 14 days as described in the manufacturer’s manual, catalogue number 28404. Colonies (aggregates with more than 30 cells) were counted manually using a Nikon TS100 microscope (Nikon, Surrey, UK); IC50 and IC90 were calculated using Graphpad PRISM 5.
Kinase assays
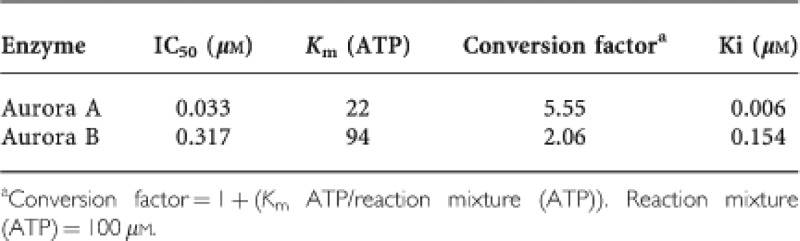
The IC50 values for purified proteins were determined as previously described (Wang et al, 2010a). The IC50 values were determined using XLfit software (IDBS, Surrey, UK). Apparent inhibition constants (Ki) were calculated from IC50 values and the appropriate Km (ATP) values for each kinase using the method of Cheng and Prusoff (1973). Recombinant human AK-A and AK-B were purchased from Upstate Discovery (Dundee, UK). Aurora A kinase assays were performed using a 25-μl reaction volume (25 mℳ β-glycerophosphate, 20 m Tris/HCl pH 7.5, 5 mkm EGTA, 1 mℳ DTT, 1 mℳ Na3VO4, 10 μg kemptide (peptide substrate) and recombinant AK-A diluted in 20 mℳ Tris/HCl pH 8, containing 0.5 mg ml−1 BSA, 2.5% glycerol and 0.006% Brij-35. Reactions were started by the addition of 5 μl Mg/ATP mix (15 mℳ MgCl2 and 100 μℳ ATP, with 18.5 kBq γ-32P-ATP per well) and incubated at 30 °C for 30 min before terminating by the addition of 25 μl 75 mℳ H3PO4. Aurora B kinase assays were performed as for aurora A, except that before use aurora B was activated in a separate reaction at 30 °C for 60 min with inner centromeres protein (Upstate, Dundee, UK; Sessa et al, 2005). The conversion of IC50 to Ki values was as follows:
M30 ELISA
Cells (106) were seeded in 10 cm plates. After drug treatment, the culture media were collected and floating cells were removed by centrifugation. Cleaved cytokeratin 18 induced by apoptosis was measured and quantified by M30 Apoptosense ELISA (PEVIVA AB, via Enzo Life Sciences (Exeter, UK) Ltd) according the manufacturer’s manual.
Results
CYC3 is an AK-A-specific inhibitor
CYC3 is a novel small-molecule inhibitor of AK-A. The inhibitor is a pyrimido-diazepine analogue (International Patent Number: WO 2009/040556 A1), which has high selectivity for AK-A in vitro. CYC3 inhibits AK-A at least 25-fold more potently than AK-B (Table 1) and has limited activity against a panel of 65 additional protein kinases (Supplementary Table S1). To evaluate the CYC3 effect in pancreatic cancer cells, we first investigated the ability of CYC3 to inhibit AK-A autophosphorylation on the T288 site (p-AK-A T288) in cells, which is a marker for AK-A kinase activity. In both MIA PaCa-2 and PANC-1 cells, CYC3 suppressed p-AK-A T288 in a dose-dependent manner (Figures 1A and B). In flow cytometry analysis, CYC3 induced the accumulation of G2/M cells in a dose-dependent manner (Figure 1C). There was no observation of significant populations of cells with 8 𝒩 DNA content, which could have indicated endoreduplication, a sign of AK-B inhibition. In confocal microscopy experiments to investigate the effect of CYC3 in a single-cell manner, the p-AK-A T288 localised at the centrosomes as reported (Gautschi et al, 2008) and disappeared with CYC3 incubation (Figure 1D). Analysis of the cell cycle profile of the immunostained CYC3-treated cells using iCys (which can distinguish between cells in G2 and in M phase) revealed an increase in the mitotic cell population (Figure 1E) and a dramatic decrease in the proportion of mitotic cells, which were p-AK-A T288-positive (Figure 1F), consistent with the observed cell cycle arrest and kinase inhibition (Figures 1A–C).
Table 1. Structure and in vitro kinase selectivity of CYC3.

Figure 1.

CYC3 specifically inhibits aurora kinase A activity in cells. (A, B) Cells were treated with 100 nℳ Nocodazole and DMSO, or indicated concentrations of CYC3 for 24 h before collecting. The protein expression levels were determined by western blot and quantified using ImageQuantTL. (C) Flow cytometry analysis of the cell cycle profiles of MIA PaCa-2 and PANC-1 cells upon treatment with CYC3 as indicated. (D–F) Cells were treated with DMSO or indicated concentrations of CYC3 for 24 h before subjecting to immunofluorescent detection of p-AK-A T288 (arrows) and DAPI (scale bar=25 μm). The fluorescent images are presented in D. iCys research imaging cytometer was used to quantify the mitotic cells (E), and mitotic cells with a positive staining of p-AK-A T288 (F).
After confirming that CYC3 functions as an AK-A-specific inhibitor in cells, the effect of CYC3 on cell survival was investigated in growth inhibition assays using SRB staining. CYC3 efficiently inhibited both MIA PaCa-2 and PANC-1 cell proliferation. The 72-h GI50 was 1.1 μℳ for MIA PaCa-2 cells and 2 μℳ for PANC-1 cells (Figure 2A). Next, we used time-lapse microscopy to compare the effect of CYC3 on cell growth over time. It is clear that CYC3 slows down the cell growth in both MIA PaCa-2 (Figure 2B) and PANC-1 (Figure 2C) cells in a dose-dependent manner, with significant inhibition from 1.5 μℳ CYC3 in both cell lines. In addition, increasing concentrations of CYC3 enhanced apoptosis in both MIA PaCa-2 (Figure 2D) and PANC-1 cells (Figure 2E) as measured by PARP cleavage, which is also consistent with previous publications about the cellular effects of AK-A-specific inhibition (Hata et al, 2005b; Warner et al, 2006). Phosphorylated histone H3 at the serine 10 site (p-H3 S10) is a marker of mitosis and AK-B activity (Gautschi et al, 2008). Increasing concentrations of CYC3 enhanced the expression of p-H3 S10 dramatically in PANC-1 cells (Figure 2E), but not in MIA PaCa-2 cells (Figure 2D), consistent with the greater increase in mitotic cells seen in PANC-1 in Figure 1E. Of note, CYC3 does not decrease p-H3 S10 in either cell line, which confirms that at concentrations ⩽3 μℳ; CYC3 does not significantly inhibit AK-B. The anti-proliferative effect of CYC3 was confirmed in six additional cell lines from a variety of cancers, with a mean IC50 at 72 h of 2.3±1 μℳ (Supplementary Table S2).
Figure 2.

CYC3 inhibits cell growth and induces apoptosis. (A) MIA PaCa-2 and PANC-1 cells were treated with CYC3, and the cells remaining after 72 h of treatment were measured by SRB assay. (B, C) Time-lapse microscopic measurement of the cell growth on treatment with CYC3. These data are the mean of two replicates (**means P<0.01 vs DMSO control). (D, E) Cells were treated with CYC3 as indicated for 24 h before collecting. The protein expression levels were determined by western blot.
Synergy between CYC3 and low concentration of paclitaxel
To fully evaluate the combination effects of paclitaxel and CYC3, 8 × 8 concentration combination experiments were performed in MIA PaCa-2 cells using SRB assays at 72 h; investigating concentration ranges of 0.03–30 nℳ of paclitaxel and 0.25–3 μℳ of CYC3. We then used the SynergySurface software to investigate how both drugs interact to inhibit growth in this data set. This approach identified that 1-3 nℳ paclitaxel with 0.25–1.5 μℳ CYC3 inhibits growth more than expected under an additive effect assumption. However, there was no such synergy detected at higher concentrations of either agent (Figure 3A). The Emax (maximum growth inhibition within the area of synergy) was 89±7% growth inhibition at 1 μℳ CYC3 (IC37 as single agent)+3 nℳ paclitaxel (IC34 as single agent). In statistical analysis of the SRB data, the inhibitory effect of the 3 nℳ paclitaxel and 1 μℳ CYC3 combination on MIA PaCa-2 cells is significantly different (P<0.01) from the predicted addictive inhibition (Figure 3B). A similar synergistic region was found in PANC-1 cells, with Emax 70±16% (1 μℳ CYC3, IC13, and 3 nℳ paclitaxel, IC49).
Figure 3.

Evaluation of the effect of paclitaxel and CYC3 in combination. (A) MIA PaCa-2 cells were seeded in 96-well plates and treated with an 8 × 8 combinations of CYC3 and paclitaxel for 72 h. The experimental data (mean of 6 replicates, left), the predicted inhibition by Bliss additivity model based on single-agent data (middle) and the difference value (Data additivity, right) tables are presented. The Synergy spots are in blue in the ‘Data additivity’ table. (B) Statistical analysis of the combination of 3 nℳ paclitaxel and 1 μℳ CYC3 in cells using SRB assays. Error bars represent the s.d. (C) Time-lapse microscopic measurement of the cell growth with CYC3 and paclitaxel as indicated. The predicted growth curve induced by additive suppression with 3 nℳ paclitaxel and 1 μℳ CYC3 is presented. These data are the mean of three replicates. ***Interaction P<0.0001, treatment difference P=0.0007. (D) Cells were incubated with paclitaxel and/or CYC3 for 72 h, then plated in colony-forming assays. The left panels are the photos of the colony plates and the right panels are the quantification of the plates using ImageQuantTL. The predicted colony numbers induced by additive suppression with 3 nℳ paclitaxel and 1 μℳ CYC3 are presented. These data are the mean of six replicates.
To further validate the synergy, time-lapse microscopy was used to evaluate the effect of the combination on cell growth over time (Figure 3C). On the basis of the growth curves of cells treated with either 3 nℳ paclitaxel or 1 μℳ CYC3 alone, an expected additive growth curve of the combination was calculated based on the Bliss Additivity Model. The experimental inhibition achieved using the combination suppressed the cell growth more than expected under the assumption of an additive effect of paclitaxel and CYC3. In MIA PaCa-2 cells, the cell confluence at 72 h in comparison with the initial cell confluence is 266±11%, compared with an expected additive effect of 772% (Figure 3C), whereas in PANC-1 cells it is 236±2% vs 393% (Supplementary Figure S1A), supporting the existence of synergy between these two compounds. As a third test of synergy, a colony-formation assay was also used to evaluate the effect of the combination on cancer cell clonogenic ability (Figure 3D). On the basis of the effects of single agents, the Bliss additivity model was used to calculate the expected additive combination effect on colony formation. We detected a much greater inhibition of colony formation using the combination (3.6±1.4% of control) than expected for using an additive combination (41.4% of control) in the MIA PaCa-2 (Figure 3D) and PANC-1 cells (13.2±6.5% of control vs predicted 39.1%, Supplementary Figure S1B), which further confirms the synergistic interaction of 3 nℳ paclitaxel and 1 μℳ CYC3 for inhibiting cell proliferation.
Myelotoxicity of the combination treatment using CYC3 and paclitaxel
A key question is if the combination will provide a better therapeutic window (i.e., lower myelotoxicity) when compared with the high-concentration single-agent activity of paclitaxel.
The potential myelotoxicity of the combination of 3 nℳ paclitaxel and 1 μℳ CYC3 was compared with that seen with 30 nℳ paclitaxel, using the CFU-GM assay with human BM cells. Consistent with other reports (Kurtzberg et al, 2009), paclitaxel had a very steep dose response in colony inhibition from 3 to 10 nℳ, suggesting there may be a threshold for paclitaxel toxicity in these progenitor cells (Figure 4A). In contrast, CYC3 demonstrated a shallow dose-dependent increase in toxicity (Figure 4B). The Bliss additivity model was used to calculate an additive combination effect on CFU-GM colony formation. The experimental colony inhibitory effect of 3 nℳ paclitaxel with 1 μℳ CYC3 combination (40.3±2.7% of control) was similar to the calculated additive inhibition (43.9 of control), whereas 30 nℳ paclitaxel treatment completely abolished all the colonies (Figure 4C). Thus, the combination of CYC3 and 3 nℳ paclitaxel was only additive in terms of toxicity to CFU-GM, whereas it was synergistic in toxicity to pancreatic cancer cells.
Figure 4.

Effect of the combination of CYC3 and paclitaxel in CFU-GM assays. CFU-GM assays using human BM cells treated with paclitaxel (A), CYC3 (B) and the combination of paclitaxel and CYC3 (C). The predicted colony number induced by additive suppression with 3 nℳ paclitaxel and 1 μℳ CYC3 is presented in (C). These data are the mean of three replicates.
Mechanism of the synergy
Next, the mechanism underlying the synergy was explored further. The LC-MS spectrometry was used to investigate the cellular and media concentration of paclitaxel with or without CYC3 co-treatment in PANC-1 cells. When CYC3 was present, the cellular paclitaxel level was not significantly different from that observed in paclitaxel treatment alone (Figure 5A), suggesting CYC3 does not enhance the cellular uptake of paclitaxel.
Figure 5.

Investigation of the mechanism of the synergy between CYC3 and paclitaxel. (A) 0.5 × 106 PANC-1 cells were seeded in 6-cm dishes with 3 ml DMEM media for 24 h before being treated with 3 nℳ paclitaxel±1 μℳ CYC3 for 3 h before collecting. The amount of paclitaxel in cells and media were measured by LC-MS. (B) Flow cytometry analysis of the cell cycle profiles of cells upon treatments as indicated. (C, D) MIA PaCa-2 (C) and PANC-1 (D) cells were treated as indicated for 24 and 48 h before collecting. The protein expression levels were determined by western blot. (E, F) MIA PaCa-2 (E) and PANC-1 (F) cells were treated as indicated for 24, 48 and 72 h. The secreted caspases-cleaved keratin 18 in the media was measured by M30 Apoptosense ELISA (Enzo Life Sciences).
The cell cycle arrest and apoptosis induction effects of the combination treatments were also investigated. Both 30 nℳ paclitaxel and the combination of 3 nℳ paclitaxel with 1 μℳ CYC3 caused significant G2/M arrest in PANC-1 cells (Figure 5B), which is accompanied by an increase in p-H3 S10 phosphorylation (Figure 5D). Although in MIA PaCa-2 cells the induction of G2/M cell cycle arrest and p-H3 S10 phosphorylation by the same combination was less, there was an accompanying increase in the sub-G1 population, suggestive of apoptosis (Figures 5B and C). Apoptosis was induced sooner in MIA PaCa-2 cells (24 h) than in PANC-1 (48 h), as measured by PARP cleavage (Figures 5C and D). Apoptosis was confirmed by the detection of cleaved cytokeratin in the medium by M30 ELISA (Figure 5E and F, apparent at 48 h in MIA PaCa-2 vs 72 h in PANC-1). Thus, MIA PaCa-2 cells respond to the CYC3/paclitaxel combination with less stable arrest in mitosis and earlier apoptosis than in Panc-1, but in both cells the combination induces effective growth inhibition when measured at 72 h.
Discussion
CYC3 shows a 25-fold differential between the in vitro activities against purified AK-A (IC50 6 nℳ) and AK-B (IC50 154 nℳ). In comparison, MK-5108 (which was in clinical development) had an IC50 of 0.064 nℳ against AK-A and 14.1 nℳ against AK-B (Shimomura et al, 2010b), and MLN8237 has an IC50 of 1.2 nℳ against AK-A and 396.5 nℳ against AK-B (Manfredi et al, 2011a). In this study, we have demonstrated that the AK-A inhibitor CYC3 specifically inhibits AK-A activity in vitro in pancreatic cancer cells, arresting cells at mitosis, suppressing cell growth and inducing apoptosis.
We then investigated the activity of CYC3 in combination with paclitaxel. Many drug combination assays use the combination-index isobologram method, which is based on the median effect principle developed by Chou and Talalay (1984), but this method tests fixed dose ratios of the two drugs, and we wished to investigate the full interaction surface across a broad range of concentrations of both drugs. The method developed by Chou and Talalay (1984) uses a line-fitting technique, but modern advances in numerical nonlinear solvers can determine the expected combined effect for any combination of inhibitor concentrations (Prichard and Shipman, 1990; Fitzgerald et al, 2006b). We chose to use a checkerboard design to investigate 8 × 8 dose combinations in a 96-well plate format. The relative proliferation associated with different drug concentrations was determined using the SRB assay (Vichai and Kirtikara, 2006). We then developed a customed software, which automatically analyses the resulting combination data for synergistic effects, applying mathematical models to compare the predicted effect with the experimental data, using methods similar to those used by Prichard and Shipman (1990), and Prichard et al (1991). The mathematical models included in the software include the two most commonly used models for calculating the expected dose–response relationships from single-agent data: the Loewe additivity and Bliss independence. The Loewe additivity model assumes that two inhibitors act through a similar mechanism and, therefore, the resulting effect can be described by different equipotent dose ratios (Chou, 2006). The Loewe additivity model can describe the trivial situation that both agents are actually the same drug (Jonker et al, 2005), but to apply this mathematical method both agents have to show a typical dose–response relationship as single agents. In contrast, the Bliss independence model assumes that both drugs modulate different mechanisms (Fitzgerald et al, 2006a). The Bliss independence model can be used on any data set, which describes a combination effect regardless of the shape of the single-agent dose–response curves, and this is the model we used in these studies. As the software is able to automatically analyse raw data output from plate readers, it allows us to test a large number of plates and concentration combinations more efficiently than other available software that requires pre-processing of the derived data (CompuSyn). This approach generates a 3D surface, which can be interrogated to identify regions of interaction.
Using the software to compare the experimental data with additivity predictions identified areas of synergy when CYC3 was combined with a low concentration of paclitaxel (3 nℳ).
Our data are consistent with that of Hata et al (2005a) who showed in MIA PaCa-2 and PANC-1 cells that siRNA knockdown of AK-A enhanced cytotoxicity by 10 nℳ paclitaxel. Previous reports of the interaction between AK-A-specific inhibitors and taxanes in other cell types appear to be consistent. MK-5108 was shown to synergise with docetaxel to inhibit HeLa-S3 xenograft tumour growth (Shimomura et al, 2010b), and VE-465 was reported to synergise with paclitaxel to induce apoptosis in paclitaxel-resistant and -sensitive ovarian cancer cells (Scharer et al, 2008). In contrast, Wysong et al (2009) showed that inhibition of AK-A by MLN8054 abrogated the mitotic arrest induced by paclitaxel in colorectal and lung cancer cell lines by allowing mitotic slippage, because AK-A is required for spindle assembly checkpoint maintenance. However, these authors did not report the ultimate cell fate beyond 24 h, so this is not necessarily contradictory to the synergistic cytotoxicity of the taxane/AK-A inhibitor combination. Also, the paclitaxel used in their study was 100 nℳ, much higher than the synergistic 3-nℳ concentration we identified in our study. Indeed, in the experiments we report above, at high concentrations of paclitaxel (30 nℳ), no synergy was observed. This highlights the importance of investigating wide ranges of concentrations of both agents, as described in this paper, to generate a surface of interaction, which can then be interrogated using modelling approaches.
By measuring the paclitaxel concentration in cells and in media, it was shown that CYC3 did not alter the uptake of paclitaxel. P-glycoprotein (P-gp, Mdr-1) is reported to be involved in drug resistance to paclitaxel by pumping paclitaxel out of the cells (Ueda et al, 1987a, 1987b). Our result is consistent with a report in breast cancer cells showing AK-A inhibition does not influence the expression and function of P-gp (Li et al, 2011), and suggests that a molecular mechanism underlies the synergy between paclitaxel and CYC3.
It is likely that the combination of 3 nℳ paclitaxel and 1 μℳ CYC3 synergise to induce mitotic arrest and subsequent cell death. This hypothesis is consistent with the observations in PANC-1 cells, but the combination-induced mitotic arrest in MIA PaCa-2 cells was less obvious. However, the combination induced apoptosis sooner in MIA PaCa-2 than in PANC-1 cells. Therefore, a possible explanation for this cell line discrepancy may be that MIA PaCa-2 cells are more vulnerable to mitotic stress and cannot tolerate arrest in mitosis for as long as PANC-1 cells. Indeed MIA PaCa-2 and PANC-1 cells also displayed the same differential response to mitotic arrest by exposure to higher (30 nℳ) paclitaxel, and different cancer cell lines are known to vary in their response to prolonged exposure to anti-mitotic drugs (Gascoigne and Taylor, 2008). The molecular mechanisms underlying this cell line difference are not clear. Further investigations are needed, which may shed light on potential biomarkers for better responses to CYC3 alone and in combination with paclitaxel.
Having identified the areas of synergy, it was important to assess whether this might impact on the therapeutic index, when using combination strategies. Although inhibiting synergistically the growth and clonogenic ability of the cancer cells, the combination of 3 nℳ paclitaxel and 1 μℳ CYC3 did not display synergistic toxicity towards CFU-GM human BM cells. Thus, there was a differential response between pancreatic cancer cells and human BM cells to the drug combination.
Of note, the combination of 3 nℳ paclitaxel and 1 μℳ CYC3 achieved a similar magnitude of cytotoxicity as treatment with higher (30 nℳ) paclitaxel as a single agent in the cancer cell lines, but the combination was significantly less toxic than 30 nℳ paclitaxel in CFU-GM cells. These differences may reflect differences in the molecular action of paclitaxel at different concentrations; 10 nℳ paclitaxel has been shown to induce transient mitotic arrest followed by mitotic slippage in some cell lines, whereas 30 nℳ paclitaxel induced longer mitotic arrest without slippage (Riffell et al, 2009); these differences may be modulated by CYC3 in a different way in cancer cells with multiple genetic abnormalities than in normal CFU-GM cells. The mechanism of the difference in response of the cancer and normal cells warrants further investigation. These data suggest that the combination of CYC3 and low-dose paclitaxel could be associated with less myelotoxicity than higher doses of paclitaxel and yet be equally efficacious. This suggests that clinical trials of AKis with full-dose taxanes may fail because the taxane dose is too high. We plan to exploit this opportunity to ‘resurrect’ the AKi approach in pre-clinical and clinical trials, in combination with paclitaxel, using more rational, science-led dosing schedules.
Acknowledgments
We thank the core facilities in Cancer Research UK Cambridge Research Institute for the technical support, Ashok Venkitaraman (MRC/Hutchison Research Centre), Bob Jackson (Pharmacometrics), Masashi Narita and Mahito Sadaie (CRUK CRI) for the helpful discussion on the project, and Christina Ortmann (Department of Haematology, University of Cambridge) for the help to set up the CFU-GM assay. This work was funded by Cancer Research UK.
Footnotes
Supplementary Information accompanies the paper on British Journal of Cancer website (http://www.nature.com/bjc)
Avril Robertson and Daniella Zheleva are employees of Cyclacel Ltd. Cyclacel provided the compound, but the laboratory costs were funded by Cancer Research UK.
Supplementary Material
References
- Anand S, Penrhyn-Lowe S, Venkitaraman AR (2003) AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell 3(1): 51–62 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22(23): 3099–3108 [DOI] [PubMed] [Google Scholar]
- Cheung CH, Coumar MS, Hsieh HP, Chang JY (2009) Aurora kinase inhibitors in preclinical and clinical testing. Expert Opin Investig Drugs 18(4): 379–398 [DOI] [PubMed] [Google Scholar]
- Chou TC (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58(3): 621–681 [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55 [DOI] [PubMed] [Google Scholar]
- Dees EC, Infante JR, Cohen RB, O'Neil BH, Jones S, von Mehren M, Danaee H, Lee Y, Ecsedy J, Manfredi M, Galvin K, Stringer B, Liu H, Eton O, Fingert H, Burris H (2010) Phase 1 study of MLN8054, a selective inhibitor of Aurora A kinase in patients with advanced solid tumors. Cancer Chemother Pharmacol 67(4): 945–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JR, Bastos BR, Hansen RJ, Gustafson DL, Eckhardt SG, Kwak EL, Pandya SS, Fletcher GC, Pitts TM, Kulikowski GN, Morrow M, Arnott J, Bray MR, Sidor C, Messersmith W, Shapiro GI (2011) Phase I safety, pharmacokinetic, and pharmacodynamic study of ENMD-2076, a novel angiogenic and Aurora kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 17(4): 849–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK (2006a) Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol 2(9): 458–466 [DOI] [PubMed] [Google Scholar]
- Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK (2006b) Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol 2(9): 458–466 [DOI] [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS (2008) Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14(2): 111–122 [DOI] [PubMed] [Google Scholar]
- Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN, Gandara DR (2008) Aurora kinases as anticancer drug targets. Clin Cancer Res 14(6): 1639–1648 [DOI] [PubMed] [Google Scholar]
- Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, Marumoto T, Saya H, Horii A (2005a) RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res 65(7): 2899–2905 [DOI] [PubMed] [Google Scholar]
- Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, Marumoto T, Saya H, Horii A (2005b) RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res 65(7): 2899–2905 [DOI] [PubMed] [Google Scholar]
- Jonker DM, Visser SA, van der Graaf PH, Voskuyl RA, Danhof M (2005) Towards a mechanism-based analysis of pharmacodynamic drug-drug interactions in vivo. Pharmacol Ther 106(1): 1–18 [DOI] [PubMed] [Google Scholar]
- Kitzen JJ, de Jonge MJ, Verweij J (2010) Aurora kinase inhibitors. Crit Rev Oncol Hematol 73(2): 99–110 [DOI] [PubMed] [Google Scholar]
- Kurtzberg LS, Roth SD, Bagley RG, Rouleau C, Yao M, Crawford JL, Krumbholz RD, Schmid SM, Teicher BA (2009) Bone marrow CFU-GM and human tumor xenograft efficacy of three tubulin binding agents. Cancer Chemother Pharmacol 64(5): 1029–1038 [DOI] [PubMed] [Google Scholar]
- Li D, Zhu J, Firozi PF, Abbruzzese JL, Evans DB, Cleary K, Friess H, Sen S (2003) Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res 9(3): 991–997 [PubMed] [Google Scholar]
- Li J, Wientjes MG, Au JL (2010) Pancreatic cancer: pathobiology, treatment options, and drug delivery. AAPS J 12(2): 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tang K, Zhang H, Zhang Y, Zhou W, Chen X (2011) Function of Aurora kinase A in Taxol-resistant breast cancer and its correlation with P-gp. Mol Med Report 4(4): 739–746 [DOI] [PubMed] [Google Scholar]
- Lok W, Klein RQ, Saif MW (2010) Aurora kinase inhibitors as anti-cancer therapy. Anticancer Drugs 21(4): 339–350 [DOI] [PubMed] [Google Scholar]
- Macarulla T, Cervantes A, Elez E, Rodriguez-Braun E, Baselga J, Rosello S, Sala G, Blasco I, Danaee H, Lee Y, Ecsedy J, Shinde V, Chakravarty A, Bowman D, Liu H, Eton O, Fingert H, Tabernero J (2010) Phase I study of the selective Aurora A kinase inhibitor MLN8054 in patients with advanced solid tumors: safety, pharmacokinetics, and pharmacodynamics. Mol Cancer Ther 9(10): 2844–2852 [DOI] [PubMed] [Google Scholar]
- Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ, Wysong DR, Janowick DA, Hyer ML, Leroy PJ, Gershman RE, Silva MD, Germanos MS, Bolen JB, Claiborne CF, Sells TB (2011a) Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res 17(24): 7614–7624 [DOI] [PubMed] [Google Scholar]
- Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ, Wysong DR, Janowick DA, Hyer ML, Leroy PJ, Gershman RE, Silva MD, Germanos MS, Bolen JB, Claiborne CF, Sells TB (2011b) Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res 17(24): 7614–7624 [DOI] [PubMed] [Google Scholar]
- Minton SE, LoRusso P, Lockhart AC, Saif MW, Krishnamurthi SS, Pickett-Gies CA, Wang Y, Guan S, Roben E, Stein MN (2010) A phase I study of MK-5108, an oral aurora A kinase inhibitor, in both monotherapy and in combination with docetaxel in patients with advanced solid tumors. J Clin Oncol 28, Suppl)abstr e13026 [Google Scholar]
- Negro GD, Bonato M, Gribaldo L (2001) In vitro bone marrow granulocyte-macrophage progenitor cultures in the assessment of hematotoxic potential of the new drugs. Cell Biol Toxicol 17(2): 95–105 [DOI] [PubMed] [Google Scholar]
- Parent-Massin D (2001) Relevance of clonogenic assays in hematotoxicology. Cell Biol Toxicol 17(2): 87–94 [DOI] [PubMed] [Google Scholar]
- Prichard MN, Prichard LE, Baguley WA, Nassiri MR, Shipman C (1991) Three-dimensional analysis of the synergistic cytotoxicity of ganciclovir and zidovudine. Antimicrob Agents Chemother 35(6): 1060–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN, Shipman C (1990) A three-dimensional model to analyze drug-drug interactions. Antiviral Res 14(4–5): 181–205 [DOI] [PubMed] [Google Scholar]
- Riffell JL, Zimmerman C, Khong A, McHardy LM, Roberge M (2009) Effects of chemical manipulation of mitotic arrest and slippage on cancer cell survival and proliferation. Cell Cycle 8(18): 3025–3038 [PubMed] [Google Scholar]
- Rojanala S, Han H, Munoz RM, Browne W, Nagle R, Von Hoff DD, Bearss DJ (2004) The mitotic serine threonine kinase, Aurora-2, is a potential target for drug development in human pancreatic cancer. Mol Cancer Ther 3(4): 451–457 [PubMed] [Google Scholar]
- Rowinsky EK, Eisenhauer EA, Chaudhry V, Arbuck SG, Donehower RC (1993) Clinical toxicities encountered with paclitaxel (Taxol). Semin Oncol 20(4 Suppl 3): 1–15 [PubMed] [Google Scholar]
- Scharer CD, Laycock N, Osunkoya AO, Logani S, McDonald JF, Benigno BB, Moreno CS (2008) Aurora kinase inhibitors synergize with paclitaxel to induce apoptosis in ovarian cancer cells. J Transl Med 6: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa F, Mapelli M, Ciferri C, Tarricone C, Areces LB, Schneider TR, Stukenberg PT, Musacchio A (2005) Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell 18(3): 379–391 [DOI] [PubMed] [Google Scholar]
- Sharma S, Kurzrock R, Gouw L, Hong DS, Jones K, Zhou X, Shi H, Fingert H, Falchook GS (2011) Phase I dose-escalation study of the investigational Aurora A kinase (AAK) inhibitor MLN8237 as an enteric-coated tablet (ECT) formulation in patients with nonhematologic malignancies. J Clin Oncol 29, Suppl)abstr 3094 [Google Scholar]
- Shimomura T, Hasako S, Nakatsuru Y, Mita T, Ichikawa K, Kodera T, Sakai T, Nambu T, Miyamoto M, Takahashi I, Miki S, Kawanishi N, Ohkubo M, Kotani H, Iwasawa Y (2010a) MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther 9(1): 157–166 [DOI] [PubMed] [Google Scholar]
- Shimomura T, Hasako S, Nakatsuru Y, Mita T, Ichikawa K, Kodera T, Sakai T, Nambu T, Miyamoto M, Takahashi I, Miki S, Kawanishi N, Ohkubo M, Kotani H, Iwasawa Y (2010b) MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther 9(1): 157–166 [DOI] [PubMed] [Google Scholar]
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82(13): 1107–1112 [DOI] [PubMed] [Google Scholar]
- Ueda K, Cardarelli C, Gottesman MM, Pastan I (1987a) Expression of a full-length cDNA for the human ‘MDR1’ gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci USA 84(9): 3004–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K, Clark DP, Chen CJ, Roninson IB, Gottesman MM, Pastan I (1987b) The human multidrug resistance (mdr1) gene. cDNA cloning and transcription initiation. J Biol Chem 262(2): 505–508 [PubMed] [Google Scholar]
- Vichai V, Kirtikara K (2006) Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 1(3): 1112–1116 [DOI] [PubMed] [Google Scholar]
- Wang S, Midgley CA, Scaerou F, Grabarek JB, Griffiths G, Jackson W, Kontopidis G, McClue SJ, McInnes C, Meades C, Mezna M, Plater A, Stuart I, Thomas MP, Wood G, Clarke RG, Blake DG, Zheleva DI, Lane DP, Jackson RC, Glover DM, Fischer PM (2010a) Discovery of N-phenyl-4-(thiazol-5-yl)pyrimidin-2-amine aurora kinase inhibitors. J Med Chem 53(11): 4367–4378 [DOI] [PubMed] [Google Scholar]
- Wang X, Sinn AL, Pollok K, Sandusky G, Zhang S, Chen L, Liang J, Crean CD, Suvannasankha A, Abonour R, Sidor C, Bray MR, Farag SS (2010b) Preclinical activity of a novel multiple tyrosine kinase and aurora kinase inhibitor, ENMD-2076, against multiple myeloma. Br J Haematol 150(3): 313–325 [DOI] [PubMed] [Google Scholar]
- Warner SL, Munoz RM, Stafford P, Koller E, Hurley LH, Von Hoff DD, Han H (2006) Comparing Aurora A and Aurora B as molecular targets for growth inhibition of pancreatic cancer cells. Mol Cancer Ther 5(10): 2450–2458 [DOI] [PubMed] [Google Scholar]
- Warshaw AL, Fernandez-del Castillo C (1992) Pancreatic carcinoma. N Engl J Med 326(7): 455–465 [DOI] [PubMed] [Google Scholar]
- Wysong DR, Chakravarty A, Hoar K, Ecsedy JA (2009) The inhibition of Aurora A abrogates the mitotic delay induced by microtubule perturbing agents. Cell Cycle 8(6): 876–888 [DOI] [PubMed] [Google Scholar]
- Zhang W, Dutschman GE, Li X, Cheng YC (2011) Quantitation of paclitaxel and its two major metabolites using a liquid chromatography-electrospray ionization tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 879(22): 2018–2022 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.