

Background: Urinary ATP modulates renal sodium excretion in response to sodium homeostasis.
Results: ATP released through connexin channels in a BKCa channel-sensitive manner inhibits ENaC.
Conclusion: Thus, this ATP is a physiological modulator of sodium excretion.
Significance: Impaired sodium excretion resulting from loss of normal purinergic regulation of ENaC in BK-β4 null mice likely contributes to their elevated blood pressure.
Keywords: Connexin, ENaC, Hypertension, Purinergic Receptor, Sodium Transport, Collecting Duct
Abstract
The epithelial Na+ channel (ENaC) in the aldosterone-sensitive distal nephron (ASDN) is under tonic inhibition by a local purinergic signaling system responding to changes in dietary sodium intake. Normal BKCa channel function is required for flow-sensitive ATP secretion in the ASDN. We tested here whether ATP secreted through connexin channels in a coupled manner with K+ efflux through BKCa channels is required for inhibitory purinergic regulation of ENaC in response to increases in sodium intake. Inhibition of connexin channels relieves purinergic inhibition of ENaC. Deletion of the BK-β4 regulatory subunit, which is required for normal BKCa channel function and flow-sensitive ATP secretion in the ASDN, suppresses increases in urinary ATP in response to increases in sodium intake. As a consequence, ENaC activity, particularly in the presence of high sodium intake, is inappropriately elevated in BK-β4 null mice. ENaC in BK-β4 null mice, however, responds normally to exogenous ATP, indicating that increases in activity do not result from end-organ resistance but rather from lowered urinary ATP. Consistent with this, disruption of purinergic regulation increases ENaC activity in wild type but not BK-β4 null mice. Consequently, sodium excretion is impaired in BK-β4 null mice. These results demonstrate that the ATP secreted in the ASDN in a BKCa channel-dependent manner is physiologically available for purinergic inhibition of ENaC in response to changes in sodium homeostasis. Impaired sodium excretion resulting form loss of normal purinergic regulation of ENaC in BK-β4 null mice likely contributes to their elevated blood pressure.
Introduction
Blood pressure in mammals is set in part by renal sodium excretion. Renal sodium excretion is fine-tuned in the aldosterone-sensitive distal nephron. Here, the activity of the epithelial Na+ channel (ENaC)2 is limiting for sodium reabsorption and, thus, sodium excretion (1–4). ENaC is a heterotrimeric ion channel composed of an α, β, and γ subunit, expressed in the apical membranes of principal cells within the ASDN. The activity of ENaC is under negative-feedback control by the renin-angiotensin-aldosterone system in protection of plasma sodium and blood pressure. Thus, there is a positive relation between the activity of the renin-angiotensin-aldosterone system and ENaC and plasma sodium and blood pressure. The central role of ENaC as an end-effector critical to regulation of blood pressure is apparent when considering that gain-of-function of this ion channel or any of its upstream regulators causes elevations in blood pressure associated with improper decreases in renal sodium excretion, and conversely, loss-of-function causes decreases in blood pressure associated with inappropriate renal sodium and water wasting (1, 2, 5).
Negative-feedback regulation of ENaC by the renin-angiotensin-aldosterone system during control of blood pressure, although clearly important, does not account for all physiological regulation of this key ion channel in the ASDN. Emerging evidence supports that in parallel with the renin-angiotensin-aldosterone system, local control systems intrinsic to the distal nephron also dynamically tune ENaC activity to keep it in balance with moment-to-moment changes in dietary sodium intake and urine flow (6). Such regulation is critical to enable an appropriately graded pressure natriuresis in response to dynamic changes in blood pressure by limiting distal sodium reabsorption to counter increases in filtration and decreases in proximal reabsorption. Increased delivery to the distal nephron favors increased reabsorption here. Thus, this must be mitigated to some extent to enable a strong pressure natriuresis. Local inhibitory control of sodium reabsorption intrinsic to the distal nephron serves this function.
Purinergic signaling intrinsic to the distal nephron is emerging as an important local regulator of sodium excretion and thus ultimately blood pressure (6). Urine flow and/or other surrogates of a positive sodium balance provoke ATP secretion into the urine of the distal nephron (7–11). Connexin (Cx) hemichannels, including Cx30 in mice, function at least in part as conduits for this ATP secretion (7, 10). Potassium efflux through activated calcium-activated big K+ channels (BKCa) facilitates this release by contributing to an electrochemical ATP-K+ efflux-coupled mechanism (9). The activity of BKCa channels is flow-sensitive, possibly providing the mechanism underpinning at least some of the flow-sensitive ATP secretion into the distal nephron (12–15). Importantly, both BKCa channel and Cx30 hemichannel proteins are expressed in the apical membranes of intercalated cells of the ASDN (16, 17). Similar to Cx30, the BKCa channel regulatory subunit BK-β4 is expressed exclusively in the ASDN in intercalated cells (9, 17). This regulatory subunit is required for normal BKCa channel function in intercalated cells and flow-sensitive ATP secretion from these cells (9).
Paracrine-acting ATP in the urine of the distal nephron serves as a ligand for metabotropic G-protein-coupled P2Y2 purinergic receptors expressed in the apical membranes of principal cells (11, 18, 19). Stimulation of P2Y2 receptors activate phospholipase C to metabolize membrane phosphatidylinositol 4,5-bisphosphate (18–20). ENaC activity is dependent on membrane phosphatidylinositol 4,5-bisphosphate levels for a region in the intracellular NH2 terminus of the channel directly binds this phospholipid to relieve the negative-regulatory effects of this region on gating (20, 21). Thus, activation of purinergic signaling by ATP decreases the activity of ENaC in ASDN principal cells by depleting apical membrane phosphatidylinositol 4,5-bisphosphate levels to decrease channel open probability.
The physiological importance of inhibitory purinergic regulation of ENaC in response to changes in sodium balance is emphasized by the findings that genetic deletion of Cx30 and P2Y2 receptors lead to inappropriate sodium retention due to hyperactive ENaC (7, 8, 10, 19, 22). This results in a decrease in sodium excretion particularly in the presence of high dietary sodium intake. Consequently, Cx30 and P2Y2 null mice have elevated blood pressure, particularly in the presence of a positive sodium balance, that is ameliorated by amiloride; an ENaC blocker (7, 8, 22). Such amiloride- and sodium-sensitive increases in blood pressure parallel that observed for gain-of-function mutations of ENaC (1, 2, 5).
Although it is clear that local regulation of ENaC by inhibitory purinergic signaling plays a key role in modulating sodium excretion and, thus, blood pressure in response to changes in sodium intake and that BKCa channels function during flow-sensitive, K+ efflux-coupled ATP release into the ASDN, it is obscure whether ATP released through this mechanism is biologically available for inhibition of ENaC and whether such release constitutes a source of ATP important for conveying information to ENaC about changes in sodium balance. That blood pressure is elevated in BK-β4 null mice is consistent with disruption of inhibitory purinergic regulation of ENaC in these animals (9, 23). The current study tests these ideas. We find that dietary sodium intake-dependent ATP release via Cx hemichannels via a mechanism requiring the normal function of BKCa channels in intercalated cells functions as a strong paracrine inhibitor of the activity of ENaC in principal cells of the ASDN. A consequence of compromising this inhibitory purinergic regulation of ENaC in BK-β4 null mice is that ENaC and thus renal sodium excretion no longer respond appropriately to changes in sodium intake. This inappropriate hyperactivity of ENaC and loss of normal sodium-dependent feedback regulation of the channel likely contribute to the elevations in blood pressure noted in BK-β4 null mice.
EXPERIMENTAL PROCEDURES
Materials
All chemicals and materials were from Sigma unless noted otherwise.
Animals
All animal use and welfare adhered to the NIH Guide for the Care and Use of Laboratory Animals following the protocols reviewed and approved by the Institutional Laboratory Animal Care and Use Committees of the University of Texas Health Science Center at San Antonio and the University of Nebraska Medical Center. Adult (∼25 g, 6–8 weeks old) male C57BL/6J control and BK-β4 null mice were used for experiments. Generation of the BK-β4 null mouse has been described previously (9). Mice were maintained with either a high sodium (2% Na+; TD.92034, Harlan Teklad), regular Na+ (0.32% Na+; TD.7912, Harlan Teklad), or nominally sodium free (Na+ < 0.01%; TD.90228, Harlan Teklad) diet with ad libitum water for 1 week before experimentation.
Isolated, Split-open ASDN Preparation
Isolation of the aldosterone-sensitive distal nephron containing connecting tubule and collecting duct suitable for electrophysiology has been described (10, 11, 18, 19). In brief, mice were sacrificed by CO2 administration followed by cervical dislocation. Kidneys were removed immediately and cut into thin slices (<1 mm) with slices placed into ice-cold physiologic saline solution buffered with HEPES (pH 7.4). The ASDN was identified by the merging of the connecting tubule into the collecting duct and mechanically isolated by micro-dissection using watchmaker forceps under a stereomicroscope. Isolated ASDN was allowed to settle onto a 5 × 5-mm cover glass coated with poly-l-lysine. Cover glass containing ASDN was placed in a perfusion chamber mounted on an inverted Nikon Eclipse TE2000 microscope and superfused with room temperature HEPES-buffered (pH 7.4) saline solution. ASDN were split open with two sharpened micropipettes controlled with different micromanipulators to gain access to the apical membrane of principal cells. Isolated, split-open ASDN were used for patch clamp analysis within 2 h after sacrifice.
Patch Clamp Electrophysiology
ENaC activity in principal cells of murine ASDN was quantified in cell-attached patches of the apical membrane made under voltage-clamp conditions (−Vp = −60 mV) using standard procedures (10, 11, 18, 19). For the current experiments, typical bath and pipette solutions were 150 mm NaCl, 5 mm KCl, 1 mm CaCl2, 2 mm MgCl2, 5 mm glucose, and 10 mm HEPES (pH 7.4) and 140 mm LiCl, 2 mm MgCl2, and 10 mm HEPES (pH 7.4), respectively. Gap-free single channel current data from gigaohm seals (recording pipette resistance 7–8 megaohms) were acquired (and subsequently analyzed) with an Axopatch 200B (Axon Instruments) or EPC-9 (HEKA Instruments Inc.) patch clamp amplifier interfaced via a Digidata 1322A (Axon Instr.) to a PC running the pClamp 9.2 suite of software (Axon Instr.). Currents were low pass-filtered at 100 Hz with an eight-pole Bessel filter (Warner Instruments). Unitary current (i) was determined from all-point amplitude histograms fitted with single- or multi-Gaussian curves using the standard 50% threshold criterion to differentiate between events. Channel activity, defined as NPo, was calculated using the standard equation, NPo = (t1 + 2t2 + … +ntn), where N and Po are the number of ENaC in a patch and the mean open probability of these channels, respectively, and tn is the fractional open time spent at each of the observed current levels. Po was calculated by dividing NPo by the number of active channels within a patch as defined by all-point amplitude histograms.
Measurement of Urinary Sodium and ATP
Urinary ATP was quantified using a luciferase bioluminescence assay (ATP Determination kit A22066; Molecular Probes, Eugene OR) following the manufacturer's instructions. For these experiments, urine was collected directly from the bladder immediately after sacrifice. Sodium excretion was quantified in mice maintained in mouse metabolic cages (Nalgene) following a standard protocol (24). In brief, animals were allowed to acclimate to the metabolic cage for 3 days. Mice were then maintained with either high, regular, or nominally Na+-free diets for a week with urine samples collected and analyzed at the end of this feeding regimen. Urine samples were collected several times over the course of the final day to prevent possible food or fecal contamination. Urinary sodium concentration was measured with a flame photometer (Jenway, model PFP 7).
Statistical Analysis and Data Presentation
Data are reported as the mean ± S.E. Unpaired data were compared with a two-sided unpaired Student's t test or a one-way analysis of variance with a Dunnett's post test (comparing to the nominally Na+ free diet). Paired data were compared with a paired t test. The criterion for significance was p ≤ 0.05. For presentation, slow base-line drifts were corrected, and current data from some patches were software-filtered at 50 Hz.
RESULTS
Local ATP Release via Cx Hemichannels Results in Tonic Inhibition of ENaC in the ASDN
Carbenoxolone inhibits connexin hemichannels (25, 26). Local secretion of ATP through connexin channels is thought to inhibit ENaC in the ASDN (7, 10). The results in Fig. 1 are consistent with this. The representative current trace (Fig. 1A) and summary graph of Po (Fig. 1B) for ENaC before and after treatment demonstrated that the addition of carbenoxolone significantly increases ENaC activity.
FIGURE 1.
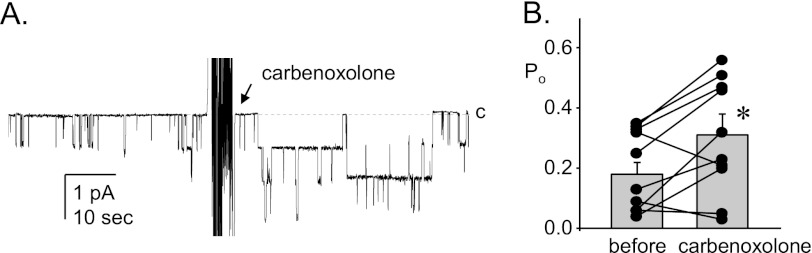
Inhibition of connexin channels relieves tonic inhibition of ENaC. A, shown is a representative gap-free current trace from a cell-attached patch made on the apical membrane of a principal cell in an isolated split-open murine ASDN from a wild type mouse before and after the addition of 25 μm carbenoxolone. This seal contains at least three ENaC. The closed state is denoted with a dashed line and c. Inward current is downward. The holding potential for this patch was −Vp = −60 mV. B, shown is a summary graph of ENaC Po before and after carbenoxolone. Data are from experiments identical to that in A. *, significantly greater compared with before carbenoxolone.
Disruption of BKCa Channel Function Decreases Urinary ATP
Increases in dietary sodium intake increase urinary ATP, likely as a result of increased urine flow (9–11). BKca channels sense flow (12–15) and are believed to mediate flow-sensitive potassium-coupled ATP secretion in the ASDN (9). As shown in Fig. 2, increases in dietary sodium intake increase urinary ATP. The positive relation between elevated sodium intake and urinary ATP is significantly muted in BK-β4 null mice, which lack a critical BKCa channel subunit that is restrictively expressed in the kidney in intercalated cells of the ASDN (17).
FIGURE 2.

ATP secretion is decreased in BK-β4 null mice. Summary graph of urinary ATP concentration for wild type (gray; n = 11, 10, and 13 for <0.01, 0.32, and 2.0% Na+, respectively) and BK-β4 null (black; n = 8, 12, and 11) mice maintained with nominally free, regular, and high sodium diets for at least 1 week before experimentation. *, significant increase versus <0.01% Na+ diet. **, significantly less compared with wild type on the same diet.
ENaC Activity Is Elevated in the ASDN of BK-β4 Null Mice
A disruption in ATP secretion in the ASDN is expected to result in an increase in ENaC activity due to relief from inhibitory purinergic signaling (11, 19). This should be particularly notable under conditions, such as elevated dietary sodium intake, where ATP secretion into the ASDN and, thus, ENaC inhibition is maximal. Fig. 3 shows representative current traces for ENaC in the ASDN from wild type (Fig. 3A) and BK-β4 null (Fig. 3B) mice maintained with a nominally sodium-free and high sodium regimen. Also shown in this figure (see also Table 1) are the corresponding summary graphs of Po (Fig. 3C) and NPo (Fig. 3D) for ENaC in wild type and BK-β4 null mice maintained with a sodium free, regular, and high sodium diet. As expected (27), the Po and activity of ENaC in wild type mice is inversely related to dietary sodium intake. In contrast, the Po of ENaC in BK-β4 null mice is independent of dietary sodium intake with activity elevated compared with that in wild type, particularly so with the high sodium feeding regimen.
FIGURE 3.

ENaC activity is elevated in BK-β4 null mice. Shown are representative gap-free current traces of ENaC from wild type (A) and BK-β4 null (B) mice maintained with nominally sodium-free (top) and high sodium (bottom) diets. Holding potentials were −60 mV. Closed states are noted with dashed lines and c. Summary graphs of Po (C) and NPo (D) for ENaC in wild type (gray; n = 42, 36, 29) and BK-β4 null (black; n = 10, 36, and 14) mice were maintained with nominally free, regular, and high sodium diets for at least 1 week before experimentation. *, shown is a significant decrease compared with <0.01% Na+. **, significantly greater compared with wild type with identical conditions.
TABLE 1.
The effect of dietary Na+ on ENaC activity in BK-β4−/− mice
| % Dietary Na+ | NPo | N | Po | f a | |
|---|---|---|---|---|---|
| Wild type | <0.01 | 1.4 ± 0.18 | 2.9 ± 0.30 | 0.45 ± 0.03 | 0.46 (42/93) |
| 0.32 | 0.79 ± 0.18b | 2.4 ± 0.31 | 0.26 ± 0.03b | 0.46 (36/79) | |
| 2.0 | 0.35 ± 0.09b | 1.6 ± 0.18b | 0.16 ± 0.03b | 0.32 (29/88) | |
| BK-β4−/− | <0.01 | 2.0 ± 0.47 | 4.5 ± 0.81c | 0.43 ± 0.05 | 0.50 (10/20) |
| 0.32 | 1.0 ± 0.13b | 2.6 ± 0.25b | 0.40 ± 0.03c | 0.50 (36/72) | |
| 2.0 | 1.2 ± 0.23b,c | 3.3 ± 0.53c | 0.33 ± 0.04c | 0.47 (14/30) |
a f = frequency (patches with at least one active channel/total number of viable seals for that condition).
b Significant decrease vs. <0.01 Na+.
c Significantly greater compared to wild type under identical conditions.
ENaC in BK-β4 Null Mice Remains Sensitive to Exogenous ATP
The above results are consistent with elevations in dietary sodium intake increasing urinary ATP levels through a BKCa channel-dependent secretory mechanism, resulting in inhibition of ENaC. Such inhibition is absent in BK-β4 null mice, which lack normal BKCa channel function in the ASDN (9, 23). To exclude end-organ resistance to ATP signaling as a mechanism in these mutant mice, we tested whether ENaC in BK-β4 null mice is responsive to exogenous ATP. As shown in Fig. 4 in the representative current trace (Fig.4, A and B) and summary graph of Po (Fig. 4C) for ENaC from BK-β4 null mice, exogenous ATP decreases ENaC activity even in the absence of functional BKCa channels. Metabolism of ATP with hexokinase combined with blockade of purinergic receptors with suramin reverses the effects of exogenous ATP on ENaC in these mutant mice. Such findings are consistent with compromised ATP secretion rather than end-organ resistance being causative for disruption of purinergic inhibition of ENaC in BK-β4 null mice.
FIGURE 4.

ENaC in BK-β4 null mice retains sensitivity to exogenous ATP. A, shown is a representative gap-free current trace for ENaC from a BK-β4 null mouse before (1) and after 10 μm ATP (2) and then 100 μm suramin plus 10 units/ml hexokinase and 5 mm glucose (3). Holding potential is −60 mV. The closed state is noted with dashed lines and c. B, shown are areas before (1) and after (2) ATP and suramin plus hexokinase (3) shown at expanded time scales. C, shown is a summary graph of ENaC Po before and after ATP and suramin (Sur) plus hexokinase (Hexo). *, significantly less than before. **, significantly greater compared with ATP.
Suppressed Tonic Inhibition by ATP in BK-β4 Null Mice Results in ENaC Being Less Responsive to Changes in Dietary Sodium Intake
If sodium-dependent ATP levels in the urine are decreased in BK-β4 null mice, then ENaC activity should be elevated, particularly so in the presence of elevated sodium intake and blockade of inhibitory purinergic signaling should have little effect on ENaC activity in mutant mice as compared with having a notable effect in wild type mice. Recall that ENaC activity is elevated compared with that in wild type in BK-β4 null mice when both are maintained on a high sodium diet (see Fig. 3 and Table 1). As shown in the summary graph of Po in Fig. 5A, blockade of inhibitory purinergic signaling increases the activity of ENaC in ASDN from wild type mice, but has little effect on the already elevated activity of ENaC in the ASDN from BK-β4 null mice. This compromise in inhibitory purinergic regulation results, as shown in Fig. 5B, in ENaC being less able to respond to changes in dietary sodium intake in BK-β4 null mice due to disruption of normal feedback control. Disruption of feedback control is indicated by an increase in fractional ENaC activity: the quotient of ENaC activity in mice maintained with a high sodium diet versus that with a nominally sodium free diet (10, 11). This loss in BK-β4 null mice of normal feedback regulation of ENaC in response to changes in dietary sodium intake by local inhibitory purinergic signaling is akin to that observed previously in P2Y2 receptor and Cx30 knock-out mice (10, 11, 19).
FIGURE 5.

Suppressed tonic ATP inhibition of ENaC in BK-β4 null mice. A, shown is a summary graph of ENaC Po in control (gray; n = 10, 10) and BK-β4 null (black; n = 21, 11) mice in the absence (filled bars) and presence (striped bars) of suramin plus hexokinase and glucose. *, significantly greater compared with wild type under identical conditions. **, significantly greater compared with the control condition. B, shown is a summary graph of fractional ENaC activity (NPo high sodium diet/NPo nominally sodium free diet) in wild type, BK-β4 null, P2Y2 receptor null, and Cx30 null mice. Data for P2Y2 receptor null and Cx30 null mice are shown for comparison purposes only and were previously published in Refs. 10 and 11.
Disruption of Normal Purinergic Inhibition of ENaC Impairs Excretion of a Sodium Load in BK-β4 Null Mice
If ENaC activity is inappropriately elevated in BK-β4 null mice particularly during high sodium intake because these animals lack local inhibitory purinergic regulation of the channel, then BK-β4 null mice should have a decreased ability to excrete a sodium load during a positive sodium balance. As shown in the summary graph in Fig. 6A of urinary sodium excretion in wild type and BK-β4 null mice maintained with nominally sodium free, regular, and high sodium diets, the ability to excrete sodium during elevated sodium intake is significantly impaired in mutant mice. Sodium intake for wild type and BK-β4 null mice, as shown in Fig. 6B, increased as a function of percent sodium in the diet but did not significantly differ when wild type and mutant mice were maintained with the same diet.
FIGURE 6.

Renal sodium excretion is impaired in BK-β4 null mice. A, shown is a summary graph of renal sodium excretion (UNaV) in wild type (gray; n = 10, 17, and 11) and BK-β4 null (black; n = 9, 6, and 6) mice maintained with nominally free, regular, and high sodium diets for 7 days. The excretion rate shown here is that for the final day of treatment. B, shown is a summary graph of sodium intake in wild type (gray; n ≥ 6) and BK-β4 null (black; n ≥ 6) mice maintained with nominally free, regular, and high sodium diets for 7 days. Average daily sodium intake shown here is for the final day of treatment. *, significant increase versus <0.01% Na+. **, significantly less compared with wild type on the same diet.
DISCUSSION
The current results demonstrate that paracrine-acting ATP secreted through connexin hemichannels in the apical membrane of intercalated cells via an electrochemical-coupled mechanism also involving K+ efflux through BKCa channels is biologically available for inhibition of ENaC in the apical membrane of principal cells in response to changes in sodium intake. Fig. 7 shows an illustration of how this local inhibitory purinergic system is thought to work. In this system BKCa channels likely function as flow sensors promoting ATP secretion. Disruption of normal BKCa channel function in intercalated cells of BK-β4 null mice, akin to disruption of Cx30 channels in these cells and disruption of the P2Y2 receptor in principal cells, results in elevated blood pressure (6, 7, 9, 22, 23). These elevations of blood pressure arise from a common final mechanism, hyperactivation of ENaC that is no longer responsive to negative purinergic regulation, causing inappropriately decreased sodium excretion. In this sense any disruption of the inhibitory purinergic signaling system intrinsic to the ASDN that controls ENaC activity in response to changes in sodium balance leads to elevated blood pressure mirroring that caused by gain-of-function mutations in the channel itself (1, 5).
FIGURE 7.

Illustration of how the local inhibitory purinergic system intrinsic to the distal nephron facilitates sodium excretion. Increases in urine flow resulting from a positive sodium balance activate luminal BKCa channels in intercalated cells (IC). K+ secretion through this channel facilitates ATP efflux into the urine through luminal connexin 30 hemichannels. This urinary ATP stimulates luminal P2Y2 receptors in principal cells (PC) to decrease the activity of ENaC. Decreased ENaC activity lessens sodium reabsorption across the ASDN favoring renal sodium excretion.
The Mechanism of ATP Release from Intercalated Cells
The current results demonstrating that the connexin channel inhibitor, carbenoxolone (25, 26), increases the open probability of ENaC in the isolated, split-open ASDN identifies these hemichannel proteins as physiologically important negative regulators of the channel. The consequence of connexin channel function rather than the protein itself is the negative regulatory factor; i.e. ATP released through connexin hemichannels inhibits ENaC (10). Cx30, which is expressed exclusively in the murine ADSN in intercalated cells (16), functions as a conduit for dietary sodium intake-sensitive ATP release where increases in sodium intake promote increased ATP release into the urine via these hemichannels (7, 10). This sensitivity to sodium intake reflects the fact that Cx30 hemichannels function during flow-sensitive ATP release (7).
Disruption of normal BKCa channel function in intercalated cells by deletion of the BK-β4 regulatory subunit, as shown here and previously (9), lessens ATP secretion muting the positive relation between increases in sodium intake and increased urinary ATP. This observation is similar to our previous finding where deletion of Cx30 abolishes sodium intake-sensitive ATP secretion (10). The compromise of ATP secretion in mice lacking Cx30 or BK-β4 results from loss of a common mechanism; ATP is secreted from intercalated cells through electrochemical coupling with secretion of K+, where connexin hemichannels and BKCa channels serve as conduits for the efflux of the prior and latter, respectively.
BKCa channels in the ASDN respond to flow with an increase in activity (12–15). This property enables them to make an important contribution to flow-sensitive potassium secretion in the distal nephron. This also positions BKCa channels expressed in intercalated cells to function as flow sensors modulating ATP secretion in response to changes in sodium intake. Although not yet definitive, such a possibility is consistent with the current observation that deletion of the BK-β4 regulatory subunit compromises the strong positive relation between urinary ATP and dietary sodium intake and the prior observation that BK-β4 siRNA abolishes flow-sensitive ATP release from cultured MDCK cells (9). Further linking flow-sensitive BKCa channel activity and dependent ATP secretion with the function of connexin hemichannels are the observations that carbenoxolone, similar to BK-β4 siRNA, abolishes flow-sensitive ATP release form MDCK cells (9) and that sodium intake- and flow-sensitive urinary ATP is decreased in BK-β4 and Cx30 null mice (7, 9, 10).
Thus, the current results, in agreement with earlier findings, provides compelling evidence that ATP released from intercalated cells via connexin hemichannels in a manner coupled to K+ efflux from these cells through BKCa channels is biologically available for inhibitory regulation of ENaC in principal cells. That ATP secreted through connexin hemichannels arises from intercalated cells and the final effector, ENaC, is in principal cells makes this regulation paracrine. Moreover, that connexin-mediated ATP secretion is sensitive to sodium intake and flow because it is coupled to the function of flow-sensitive BKCa channels enables this local paracrine signaling system to regulate the activity of ENaC in a sodium intake-sensitive manner. This has important implications for this source of ATP during physiological and pathological control of renal sodium excretion and blood pressure.
The Consequences of ATP Release from Intercalated Cells
The physiological importance of paracrine-acting ATP released from intercalated cells is apparent when considering that blood pressure in animals with compromised Cx30, BKCa channel, and P2Y2 receptor function, akin to those with ENaC gain-of-function, is elevated due to inappropriate sodium retention (1, 7–9, 17, 22). As we demonstrate here, the activity of ENaC is elevated in BK-β4 null mice due to lowered ATP secretion. ENaC activity is elevated in BK-β4 null mice, in particular, during high sodium intake. This represents inappropriate regulation of the channel. Normally, the activity of ENaC is inversely related to sodium intake (27). This allows appropriate pressure natriuresis responses in protection of blood pressure for it mitigates any increase in distal sodium reabsorption mandated by increased delivery (6). When this is compromised, animals retain sodium inappropriately, resulting in elevated blood pressure. Such is the case for BK-β4 null mice (9, 23). Similarly, loss-of-function of Cx30 and P2Y2 receptors, both of which disrupt inhibitory purinergic regulation of ENaC in response to changes in sodium intake, result in inappropriate decreases in sodium excretion during a positive sodium balance, causing elevations in blood pressure (7, 8, 11, 19, 22).
Currently it is unclear if changes in sweat and/or gastrointestinal sodium excretion compensate for decreases in renal sodium excretion in BK-β4 null mice. Nevertheless, these mutant mice have a compromised ability to excrete a sodium load through their kidneys and have increased blood pressure.
The inappropriately decreased sodium excretion observed in BK-β4 null mice is not a manifestation of end-organ resistance. Rather it reflects loss of normal feedback inhibition of ENaC by compromise of the local purinergic signaling system that no longer is able to secret the inhibitory factor, ATP, in response to elevations in urine flow. The outcome of disrupting sodium intake-sensitive ATP secretion in BK-β4 and Cx30 null mice is identical to that in the end-organ resistance states of P2Y2 receptor null mice and mice harboring gain-of-function mutations in ENaC. This conclusion emphasizes the physiological importance to sodium balance and control of blood pressure of appropriate regulation of ENaC by the paracrine-acting, inhibitory purinergic control system intrinsic to the ASDN. As demonstrated here, ATP released (via connexin hemichannels) in an electrochemical-coupled manner with K+ efflux through BKCa channels in intercalated cells is a key component of this regulatory system.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK059594 (to J. D. S.) and R01 DK71014 (to S. C. S.). This work was also supported by American Heart Association Fellowship 10POST3200019 (to V. B.).
- ENaC
- epithelial Na+ channel
- ASDN
- aldosterone-sensitive distal nephron
- Cx
- connexin.
REFERENCES
- 1. Rossier B. C., Pradervand S., Schild L., Hummler E. (2002) Epithelial sodium channel and the control of sodium balance. Interaction between genetic and environmental factors. Annu. Rev. Physiol 64, 877–897 [DOI] [PubMed] [Google Scholar]
- 2. Bonny O., Hummler E. (2000) Dysfunction of epithelial sodium transport. from human to mouse. Kidney Int. 57, 1313–1318 [DOI] [PubMed] [Google Scholar]
- 3. Garty H., Palmer L. G. (1997) Epithelial sodium channels. Function, structure, and regulation. Physiol. Rev. 77, 359–396 [DOI] [PubMed] [Google Scholar]
- 4. Kellenberger S., Schild L. (2002) Epithelial sodium channel/degenerin family of ion channels. A variety of functions for a shared structure. Physiol. Rev. 82, 735–767 [DOI] [PubMed] [Google Scholar]
- 5. Lifton R. P., Gharavi A. G., Geller D. S. (2001) Molecular mechanisms of human hypertension. Cell 104, 545–556 [DOI] [PubMed] [Google Scholar]
- 6. Toney G. M., Vallon V., Stockand J. D. (2012) Intrinsic control of sodium excretion in the distal nephron by inhibitory purinergic regulation of the epithelial Na+ channel. Curr. Opin. Nephrol Hypertens. 21, 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sipos A., Vargas S. L., Toma I., Hanner F., Willecke K., Peti-Peterdi J. (2009) Connexin 30 deficiency impairs renal tubular ATP release and pressure natriuresis. J. Am. Soc. Nephrol. 20, 1724–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rieg T., Gerasimova M., Boyer J. L., Insel P. A., Vallon V. (2011) P2Y2 receptor activation decreases blood pressure and increases renal Na+ excretion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R510–R518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Holtzclaw J. D., Cornelius R. J., Hatcher L. I., Sansom S. C. (2011) Coupled ATP and potassium efflux from intercalated cells. Am. J. Physiol. Renal Physiol.. 300, F1319–F1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mironova E., Peti-Peterdi J., Bugaj V., Stockand J. D. (2011) Diminished paracrine regulation of the epithelial Na+ channel by purinergic signaling in mice lacking connexin 30. J. Biol. Chem. 286, 1054–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stockand J. D., Mironova E., Bugaj V., Rieg T., Insel P. A., Vallon V., Peti-Peterdi J., Pochynyuk O. (2010) Purinergic inhibition of ENaC produces aldosterone escape. J. Am. Soc. Nephrol. 21, 1903–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rieg T., Vallon V., Sausbier M., Sausbier U., Kaissling B., Ruth P., Osswald H. (2007) The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int. 72, 566–573 [DOI] [PubMed] [Google Scholar]
- 13. Liu W., Morimoto T., Woda C., Kleyman T. R., Satlin L. M. (2007) Ca2+ dependence of flow-stimulated K+ secretion in the mammalian cortical collecting duct. Am. J. Physiol. Renal Physiol. 293, F227–F235 [DOI] [PubMed] [Google Scholar]
- 14. Pluznick J. L., Wei P., Grimm P. R., Sansom S. C. (2005) BK-β1 subunit. Immunolocalization in the mammalian connecting tubule and its role in the kaliuretic response to volume expansion. Am. J. Physiol. Renal Physiol. 288, F846–F854 [DOI] [PubMed] [Google Scholar]
- 15. Taniguchi J., Imai M. (1998) Flow-dependent activation of maxi K+ channels in apical membrane of rabbit connecting tubule. J. Membr. Biol. 164, 35–45 [DOI] [PubMed] [Google Scholar]
- 16. McCulloch F., Chambrey R., Eladari D., Peti-Peterdi J. (2005) Localization of connexin 30 in the luminal membrane of cells in the distal nephron. Am. J. Physiol. Renal Physiol. 289, F1304–F1312 [DOI] [PubMed] [Google Scholar]
- 17. Holtzclaw J. D., Grimm P. R., Sansom S. C. (2010) Intercalated cell BK-α/β4 channels modulate sodium and potassium handling during potassium adaptation. J. Am. Soc. Nephrol. 21, 634–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pochynyuk O., Bugaj V., Rieg T., Insel P. A., Mironova E., Vallon V., Stockand J. D. (2008) Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. J. Biol. Chem. 283, 36599–36607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pochynyuk O., Rieg T., Bugaj V., Schroth J., Fridman A., Boss G. R., Insel P. A., Stockand J. D., Vallon V. (2010) Dietary Na+ inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y2 receptor tone. FASEB J. 24, 2056–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pochynyuk O., Bugaj V., Vandewalle A., Stockand J. D. (2008) Purinergic control of apical plasma membrane PI(4,5)P2 levels sets ENaC activity in principal cells. Am. J. Physiol. Renal Physiol. 294, F38–F46 [DOI] [PubMed] [Google Scholar]
- 21. Pochynyuk O., Tong Q., Medina J., Vandewalle A., Staruschenko A., Bugaj V., Stockand J. D. (2007) Molecular determinants of PI(4,5)P2 and PI(3,4,5)P3 regulation of the epithelial Na+ channel. J. Gen. Physiol. 130, 399–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rieg T., Bundey R. A., Chen Y., Deschenes G., Junger W., Insel P. A., Vallon V. (2007) Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB J. 21, 3717–3726 [DOI] [PubMed] [Google Scholar]
- 23. Holtzclaw J. D., Grimm P. R., Sansom S. C. (2011) Role of BK channels in hypertension and potassium secretion. Curr. Opin. Nephrol. Hypertens. 20, 512–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grimm P. R., Irsik D. L., Settles D. C., Holtzclaw J. D., Sansom S. C. (2009) Hypertension of Kcnmb1−/− is linked to deficient K+ secretion and aldosteronism. Proc. Natl. Acad. Sci. U.S.A. 106, 11800–11805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anselmi F., Hernandez V. H., Crispino G., Seydel A., Ortolano S., Roper S. D., Kessaris N., Richardson W., Rickheit G., Filippov M. A., Monyer H., Mammano F. (2008) ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc. Natl. Acad. Sci. U.S.A. 105, 18770–18775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Vuyst E., Decrock E., De Bock M., Yamasaki H., Naus C. C., Evans W. H., Leybaert L. (2007) Connexin hemichannels and gap junction channels are differentially influenced by lipopolysaccharide and basic fibroblast growth factor. Mol. Biol. Cell 18, 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vallon V., Hummler E., Rieg T., Pochynyuk O., Bugaj V., Schroth J., Dechenes G., Rossier B., Cunard R., Stockand J. (2009) Thiazolidinedione-induced fluid retention is independent of collecting duct αENaC activity. J. Am. Soc. Nephrol. 20, 721–729 [DOI] [PMC free article] [PubMed] [Google Scholar]