

Background: Angiogenesis is an important step in the metastatic cascade of tumors.
Results: MMP-13 itself as well as VEGF-A secretion from fibroblasts promotes angiogenesis. Indeed, MMP-13 is well correlated with blood vessel density in human cancer tissues.
Conclusion: MMP-13 can be a marker for prediction of malignant behaviors and a therapeutic target in cancer.
Significance: This work provides new insights regarding the role of MMP-13 in tumor angiogenesis.
Keywords: Cancer, Cancer Biology, Endothelial Cell, Metalloprotease, Metastasis, Tumor Metastases, Tumor Microenvironment, Vascular Endothelial Growth Factor (VEGF)
Abstract
Matrix metalloproteinases (MMPs) are extracellular zinc-dependent endopeptidases involved in the degradation and remodeling of extracellular matrix in physiological and pathological processes. MMPs also have a role in cell proliferation, migration, differentiation, angiogenesis, and apoptosis. We previously identified cancer invasion-related factors by comparing the gene expression profiles between parent and the highly invasive clone of cancer cells. Matrix metalloproteinase-13 (MMP-13) was identified as a common up-regulated gene by cancer invasion-related factors. Although MMP-13 slightly promoted tumor invasion, we found that MMP-13 was involved in tumor angiogenesis. Conditioned medium from MMP-13-overexpressing cells promoted capillary formation of immortalized human umbilical vein endothelial cells. Furthermore, treatment with recombinant MMP-13 protein enhanced capillary tube formation both in vitro and in vivo. MMP-13-promoted capillary tube formation was mediated by activation of focal adhesion kinase and ERK. Interestingly, MMP-13 promoted the secretion of VEGF-A from fibroblasts and endothelial cells. By immunohistochemical analysis, we found a possible correlation between MMP-13 expression and the number of blood vessels in human cancer cases. In summary, these findings suggest that MMP-13 may directly and indirectly promote tumor angiogenesis.
Introduction
The process of metastasis consists of sequential and selective steps, including proliferation, induction of angiogenesis, detachment, motility, invasion into circulation, aggregation and survival in the circulation, cell arrest in distant capillary beds and extravasation into organ parenchyma (1, 2). Induction of angiogenesis is considered one of the important steps in the metastatic cascade of tumors. It is widely accepted that tumor growth and metastasis are angiogenesis-dependent, and hence, blocking angiogenesis could be a strategy to arrest tumor growth (3). The “angiogenic switch” is “off” when the effect of proangiogenic molecules is balanced by that of anti-angiogenic molecules, and is “on” when the net balance is tipped in favor of angiogenesis (4, 5). Pro- and anti-angiogenic molecules can be emanated from cancer cells, endothelial cells, stromal cells, blood, and the extracellular matrix (ECM)3 (6). Their relative contribution is likely to change with tumor type and site. It is also likely to change with tumor growth, regression and relapse. However, the interplay between environmental and genetic mechanisms influencing tumor angiogenesis and growth is a complex and largely unresolved matter.
The ECM undergoes significant remodeling during tumor progression and this is mediated largely by the extracellular proteinases, particularly the matrix metalloproteinases (MMPs), and the major source of these is from the stromal cells (7). MMPs represent a family of zinc-dependent proteinases, which are able to degrade ECM components such as collagens and proteoglycans and have a role in normal development and tissue damage in various pathophysiological conditions involving arthritis, wound healing, and tumor development (8). MMPs can be classified into subgroups, including collagenases, stromelysins, gelatinases, and membrane-type MMPs (9). MMPs have been implicated in the promotion of tumor invasion and metastasis for decades (10). It is now evident that MMP function is more complex than initially thought, given that these enzymes do more than degrade physical barriers. MMPs also affect multiple signaling pathways that modulate the biology of the cell in normal physiological processes and in disease. It is now evident that some MMPs such as MMP-1, -2, -3, -7, -9, -14, and -16 can contribute to distinct vascular events in tumors (11). Among them, MMP-9, conveyed by inflammatory cells, has a distinct role in tumor angiogenesis, mainly regulating the bioavailability of VEGF. MMP-9 enables an angiogenic switch by making sequestered VEGF bioavailable for its receptor VEGFR-2 in pancreatic islet tumors (12). In addition, the direct cleavage of matrix-bound VEGF by MMP-3, -7, -9, or -16 results in modified VEGF molecules with altered bioavailability, which changes the vascular patterning of tumors in vivo (13). However, the degradation of ECM components and other extracellular molecules may generate fragments with new bioactivities that inhibit angiogenesis (14). Thus, MMPs have dual functions as inhibiting and promoting angiogenesis, and the effects of MMPs on angiogenesis might be diverse.
It has recently been shown that a repair of bone fracture in MMP-13-deficient mice is delayed, which suggests a critical role of MMP-13 in the process of angiogenesis during the healing of fracture (15). Additionally, chicken MMP-13 was shown to directly contribute to neovascularization, which clearly extends the physiologic role of MMP-13 associated with cartilage and bone resorption to collagen remodeling in the angiogenic process (16). MMP-13 is known as collagenase-3 and is active against a wide variety of ECM components (17). Moreover, high expression of MMP-13 has been related to tumor behavior and prognosis (18). Recently, it has been shown that MMP-13 produced from stromal fibroblasts promotes angiogenesis through increased protein level of VEGF and VEGFR-2 in cancer invasive area (19). Here, we found that MMP-13 produced by cancer cells directly and indirectly promoted tumor angiogenesis.
EXPERIMENTAL PROCEDURES
Reagents
Active form of recombinant human MMP-13, which truncated from the C terminus was obtained from Chondrex, Inc. (Redmond, WA). This recombinant protein was made using the pET vector system in Escherichia coli. Recombinant TGF-β and ERK inhibitor, U0126 was obtained from R&D Systems (Minneapolis, MN). Focal adhesion kinase (FAK) inhibitor, FAK inhibitor 14, was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). MMP-13 inhibitor, CL82198, was obtained from Merck Millipore (Darmstadt, Germany).
Cell Lines and Culture Conditions
Head and neck squamous cell carcinoma (HNSCC) cell lines (HCS2, HSC3, HSC4, Ca-9-22, Ho-1-N-1, and Ho-1-U-1) were provided by the Japanese Collection of Research Bioresources Cell Bank. These cells were maintained in RPMI 1640 (Nacalai tesque, Inc., Kyoto, Japan) supplemented with 10% heat-inactivated FBS (Invitrogen) and 100 units/ml penicillin-streptomycin (Invitrogen) under the condition of 5% CO2 in air at 37 °C. Immortalized human umbilical vein endothelial cells (HUVECs; HuhT1 cells) were used in this study. HuhT1 cells were previously established by transfection with human telomerase reverse transcriptase (20). HuhT1 cells was maintained in HuMedia-EG2 (Kurabo, Okayama, Japan) under the condition of 5% CO2 in air at 37 °C. Normal fibroblasts were obtained from gingival tissues using standard explant techniques (21). The tissues were obtained undergoing routine dental surgery in the Department of Oral Surgery (Hiroshima University Hospital). Normal fibroblasts were maintained in DMEM supplemented with 10% FBS. Only cells between passages three and five were used in this study.
RT-PCR
Using RNeasy mini kit (Qiagen, Hilden, Germany), total RNA from cultures of confluent cells was isolated. These isolates were quantified and their purity was evaluated by spectrophotometer. The cDNA was synthesized from 1 μg of total RNA according to ReverTra Dash (Toyobo Biochemicals, Tokyo, Japan). We used the following primers: human MMP-13, 5′-ttgagctggactcattgtcg-3′ (forward) and 5′-ggagcctctcagtcatggag-3′ (reverse); human glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-tccaccaccctgttgctgta-3′ (forward) and 5′-accacagtccatgccatcac-3′ (reverse). Aliquots of total cDNA were amplified with 1.25 units of rTaq-DNA polymerase (Qiagen), and this amplification was done in a thermal cycler (MyCyler, Bio-Rad, Richmond, CA) for 30 cycles after initial 30 s of denaturation at 94 °C, annealing for 30 s at 60 °C, and extension for 1 min at 72 °C in all primers used. The amplification reaction products were resolved on 1.2% agarose/TAE gels (Nacalai tesque), electrophoresed at 100 mV, and then finally visualized by using ethidium bromide.
Generation of MMP-13-overexpressing Cells
pcDNA3.1-FLAG-MMP-13 expression vector was kindly provided by Dr. Michael Byrne (Harvard Medical School). We transfected MMP-13 into HSC3 cells. Then, G418 (300 μg/ml; Invivogen, San Diego, CA) was added to the culture medium after 48 h of transfection. After 2 weeks of G418 selection, we obtained the stable pool clones. Cell transfections were performed using FuGENE 6HD (Roche Applied Science) according to the manufacturer's instruction. Conditioned media were collected after incubation with RPMI without FBS for 2 days.
Silencing by siRNA
Logarithmically growing HSC4 and Ho-1-N-1 cells were seeded at a density of 105 cells/dish (6 cm) and transfected with 20 nm siRNA by using Oligofectamine® RNAi MAX (Invitrogen), according to the manufacturer's instructions. Forty-eight hours after transfection, the cells were prepared and analyzed by Western blot analysis. At the same time, we changed to new medium, incubated for 48 h, and collected the conditioned medium. The following siRNA oligonucleotides were obtained from B-Bridge International, Inc. (Mountain View, CA): MMP-13, gaugaaaccuggacaaguaTT. A scrambled sequence without significant homology to rat, mouse, or human gene sequences was used as a control.
Western Blot Analysis
Western blotting was carried out as described previously (22). The protein concentrations were measured by Bradford protein assay (Bio-Rad). Twenty μg of protein was subjected to 10% polyacrylamide gel electrophoresis, followed by electroblotting onto a nitrocellulose filter. For detection of the immunocomplex, the ECL Western blotting detection system (Amersham Biosciences) was used. Anti-MMP-13 monoclonal antibody (Fuji Company Industries, Tokyo, Japan), anti-FLAG monoclonal antibody (Sigma), and anti-β-actin monoclonal antibody (Sigma), α-smooth muscle actin (α-SMA), anti-phospho-FAK (Tyr-576/Tyr-577) monoclonal antibody (Cell Signaling Technology, Beverly, MA), anti-phospho-Src (Tyr-416) polyclonal antibody (Cell Signaling Technology), anti-phospho-ERK (Thr-202/Tyr-204) monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA), anti-FAK polyclonal antibody (Cell Signaling Technology), anti-Src polyclonal antibody (Cell Signaling Technology), and anti-ERK monoclonal antibody (Cell Signaling Technology) were used. For detection of phosphorylated proteins, membranes were blocked with 3% milk/TBS-T and incubated with phospho-specific antibodies overnight at 4 °C. After washing in TBS-T, membranes were incubated with specific secondary antibodies, and the proteins were visualized as described previously.
MMP-13 Activity
The MMP-13 activity was determined by a MMP-13 inhibitor assay kit (Chondrex, Inc., Redmond, WA, distributed by IWAI Chemicals Company, Japan, catalog no. 3003). A designate reaction was performed in the 96-well microtiter plate according to the manufacturer's instructions. The assay procedure was separated into two stages. First, diluted recombinant human MMP-13 (10 μg/ml) with dilution buffer B or conditioned medium (from control cells, MMP-13-overexpressing cells or MMP-13 siRNA-treated cells) was activated by adding 5 μl of aminophenylmercuric acetate at 35 °C for 60 min. Second, appropriate amounts of test samples with or without several inhibitors (U0126, FAK inhibitor 14 and CL82198) that diluted by solution B and reaction buffer to the wells were added to adjust the final volume to 160 μl. The reaction was initiated by adding 100 μl of substrate solution to each well. The collagenase reaction was stopped by adding 10 μl of the stop solution to each well after incubating at room temperature for ∼30 min. The reaction fluorescence intensity was determined at λemission = 450 nm and λexcitation = 345 nm with Varioskan Flash (Thermo Scientific). The MMP-13 activity was determined by comparing with a standard response curve using buffer instead of inhibitor in similar conditions. All assays were carried out in three replications.
Migration Assay
Migration activity was measured by the use of a 24-well cell culture insert with 8-μm pores (Falcon Becton Dickinson). The lower compartment contained 0.5 ml of conditioned medium or serum-free medium with or without 100 ng/ml of recombinant MMP-13. After trypsinization, 5 × 104 cells were resuspended in 100 μl of serum-free medium and placed in the upper compartment of the cell culture insert for 4 h. To examine the activity of migration, the cells that had penetrated onto the lower side of the filter were fixed with formalin and stained with hematoxylin. These were assayed three times.
In Vitro Angiogenesis by HUVECs
An angiogenesis assay kit obtained from Kurabo (Osaka, Japan) was used according to the manufacturer's instructions with minor modifications (23). HUVECs were treated with conditioned medium from MMP-13-overexpressing cells or control cells (1:1 mixture with the medium). HUVECs were also treated with different concentrations of recombinant MMP-13 protein (0, 50, 100 and 200 ng/ml). VEGF-A (2 μg/ml) was used as a positive control, and suramin (1 mm) was used as a negative control. We examined three wells/data point in a single experiment. The media were changed every 3 days. After 12 days, the cells were fixed at room temperature with cold 70% ethanol for 30 min. The cells were incubated with the anti-human CD31 antibody for 1 h at 37 °C and further with an alkaline phosphatase-conjugated goat anti-mouse IgG antibody. Visualization was achieved with 5-bromo-4-chloro-3-indolyl phosphate-nitrobluetetrazolium. Capillary tube score was estimated with the Chalkley count method under a bright-field microscope (24).
Rat Aortic Ring Angiogenesis Assay
The effect of samples on angiogenesis was studied by culturing aortic explants in three-dimensional matrix gels according to the protocol described by Bauer et al. (25). Thoracic aorta was excised from 7-week-old male Sprague-Dawley rat, and the fibroadipose tissue was removed. The aorta was sectioned into 1-mm-long cross-sections, rinsed with EBM-2 medium (Lonza, Walkersville, MD), placed on the Matrigel-coated wells, covered with additional 50 μl of Matrigel, and allowed to form a gel for more than 30 min at 37 °C, 5% CO2. Afterward, control was treated with EBM-2 medium only, and the test sample was treated with EBM-2 medium containing recombinant MMP-13 protein. Each medium was added every other day. All assays were performed by using five aortic rings per sample. Aortic rings were photographed on day 15. The area of angiogenic sprouting was calculated using Image-Pro Plus software program (Media Cybernetics). Microvessel densities were reported in square pixels.
VEGF-A Quantification
A fixed number of fibroblasts cultured in medium without FBS were treated with MMP-13 (0, 10 and 50 ng/ml) and/or TGF-β (1 ng/ml) for 24 h. The concentration of VEGF-A in the culture medium was quantified with commercial ELISA kit according to the manufacturers' instructions (Pierce Biotechnology, Rockford, IL).
Tissue Samples
Sixty-seven tissue samples of human HNSCC were retrieved from the Surgical Pathology Registry of University of Peradeniya and Oral and Maxillofacial unit, Kandy Hospital, after being approved by the Ethical Committee of each institution. Informed consent obtained from all patients was verbal for this study, and then signature was obtained from all patients. Sixty-seven Sri Lankan HNSCC cases (42 male, nine female and 16 unknown; average age was 50.2 ± 13.2) were surgically resected from 1998 to 2004 before radiochemotherapy. Clinical information including metastasis was gathered from surgical records of the patients. Tissues were fixed in 10% buffered formalin and embedded in paraffin.
Immunohistochemistry
Tumor tissues were fixed in 10% formalin, embedded in paraffin, and cut into 4-μm thick sections. For immunohistochemical staining, tissue sections were deparaffinized in xylene and rehydrated in descending grades of ethanol. Endogenous peroxidase activity was blocked with methanol containing 0.3% H2O2 for 30 min. Antigen retrieval was done by the microwaving using a citrate phosphate buffer (pH 6.0), and then the sections were incubated with the primary antibody at 4 °C overnight. Immunohistochemical staining was carried out by a monoclonal anti-MMP-13 antibody (Fuji Company Industries, 1:80). For detection of the reaction after incubation with secondary antibodies, we used diaminobenzidine (DAKO, Glostrup, Denmark). The sections were counterstained by hematoxylin and dehydrated in ascending grades of ethanol, and finally, the slides were mounted. By considering the percentage of positive cells and the overall staining intensity, MMP-13 was considered positive if over 10% of the tumor cells showed strong or diffuse staining. If less than 10% of the cells showed weak or no staining, it was considered negative.
Assay for Blood Vessel Density
CD34 is an antigen present in hematopoietic progenitor cells and endothelial cells. Anti-CD34 antibody is a highly sensitive marker for endothelial cell differentiation and has also been studied as a marker for vascular tumors. To investigate the relation between angiogenesis and MMP-13, we stained all HNSCC cases with CD34 endothelial marker (Novocastra Laboratories Ltd., Newcastle, UK) by SABC method. To assess blood vessel density, we performed histomorphometric analysis. Three representative photomicrographs (areas where MMP-13 positivity were detected including invasive front) were taken from each case stained with CD34. First, we went through all the sections stained with MMP-13 and CD34 antibodies. The area was selected by the following criteria: (i) the expression of MMP-13, (ii) the included invasive tumor front, and (iii) the high number of blood vessels. Photographs of those tumors were taken in close proximity to MMP-13-expressing area including the invasive front. For MMP-13-negative cases, three areas from the invasive front were selected. Any positively stained endothelial cell or endothelial cell cluster, with or without a lumen, was considered as a single, countable blood vessel. Stromal area was quantitatively analyzed using digital image (Adobe Photoshop and Scion Image software). From each figure, total counts of blood vessels per stromal area were taken, and the average was calculated. The results were then compared with MMP-13 expression.
Statistical Analysis
A p value < 0.05 was required for assessing the significance. Correlation between variables was estimated using Fisher's exact test, and for correlation between MMP-13 expression and blood vessel density, a Welch test was used.
RESULTS
MMP-13 Promotes Angiogenesis
We previously identified periostin, interferon-induced transmembrane protein 1 (IFITM1), and Wnt-5b as cancer invasion-related factors by comparing the gene expression profiles between parent and highly invasive clone of a cancer cell line (22). MMP-13 was identified as a common up-regulated molecule by comparing the gene expression profiles between control cells and periostin-overexpressing cells, control cells and IFITM1-overexpressing cells, and control cells and Wnt-5b-overexpressing cells (Fig. 1A) (26). It is known that MMP-13 is highly expressed in various tumors and is related to tumor behavior and prognosis (18). To know the role of MMP-13 in cancer development, we generated MMP-13-overexpressing cancer cells. Expression of MMP-13 mRNA was examined in six head and neck cancer cell lines (Fig. 1B). Among six cell lines, HSC2 and HSC3 cells showed lower expression of MMP-13 mRNA. Expression level of MMP-13 in these cells was lower than that in other cancer cells. Therefore, we transfected a FLAG-MMP-13 plasmid into HSC3 cells. Then, we obtained four stable clones and one stable pool clone of MMP-13-overexpressing cells (Fig. 1C). All stable clones highly expressed ectopic MMP-13 (Fig. 1C). In further experiments, clone 1 was used. By using MMP-13-overexpressing cells, we examined the role of MMP-13 in cell growth and invasion. MMP-13 overexpression did not affect cell proliferation and slightly promoted the invasion of HNSCC cells (data not shown). We also confirmed that conditioned medium from MMP-13-overexpressing cells had a higher protease activity than that from control cells (Fig. 1D).
FIGURE 1.

Identification of MMP-13. A, schema shows the strategy to identify MMP-13. Periostin, IFITM1, and Wnt-5b are identified as the invasion-related molecules by comparing the gene expression profile between the parent (MSCC-1 cells) and a highly invasive clone (MSCC-Inv1 cells). To identify common up-regulated genes, we compared the gene expression profiles of control versus periostin-overexpressing cells, control versus IFITM1-overexpressing cells, and control versus Wnt-5b-overexpressing cells. MMP-13 is commonly up-regulated among periostin-, IFITM1-, and Wnt-5b-overexpressing cells. B, expression of MMP-13 in HNSCC cell lines. Expression of MMP-13 mRNA in six HNSCC cell lines: HSC2, HSC3, HSC4, Ca-9-22, Ho-1-N-1, and Ho-1-U-1 was examined by RT-PCR. GAPDH was used as a loading control. C, generation of MMP-13-overexpressing cells. pcDNA3.1-FLAG-MMP13 was transfected into HSC3 cells. After selection, we obtained four stable clones and one stable pool clone of MMP-13-overexpressing cells. Ectopic expression of MMP-13 was examined by immunoblotting with an anti-FLAG antibody. In further experiments, clone 1 was used. D, MMP-13 ability was determined by a MMP-13 inhibitor assay kit as described under “Experimental Procedures.” Conditioned medium was collected from control HSC3, MMP-13-overexpressing HSC3, control Ho-1-N-1, MMP-13 siRNA-treated Ho-1-N-1, control HSC4, and MMP-13 siRNA-treated HSC4 cells. The reaction was initiated by adding 100 μl of substrate solution, and the reaction fluorescence intensity was determined at λemission = 450 nm and λexcitation = 345 nm. The graph shows fluorescence intensity. All assays were carried out in three replications. E, ectopic expression of FLAG-MMP-13 was examined by immunoblotting with an anti-FLAG antibody. β-Actin expression was used as a loading control. Expression of MMP-13 in condensed conditioned medium was detected by immunoblotting with an anti-MMP-13 antibody. F, MMP-13 knockdown in HSC4 and Ho-1-N-1 cells. MMP-13 siRNA was transfected into HSC4 and Ho-1-N-1 cells. A scrambled sequence that does not show significant homology to rat, mouse, or human gene sequences was used as a control. After 48 h, cells were collected, and MMP-13 expression was examined by Western blot (WB) analysis. β-Actin expression was used as a loading control.
MMP-13 has recently been shown to play a critical role in the process of angiogenesis during the healing of fracture (15). Here, we examined the role of MMP-13 in angiogenesis. MMP-13 secretion was detected in conditioned medium from MMP-13-overexpressing-HSC3 cells by Western blot analysis (Fig. 1E). Expression level of ectopic MMP-13 in MMP-13-overexpressing HSC3 cells was similar to that of endogenous MMP-13 in Ho-1-N-1 or HSC4 cells (Fig. 1F). By using conditioned medium from MMP-13-overexpressing cells, we examined the migration of immortalized HUVECs. The HuhT1 cell line was previously established from HUVECs by transfection with human telomerase reverse transcriptase (20). Conditioned medium from MMP-13-overexpressing cells promoted migration of HuhT1 cells (Fig. 2A). Interestingly, conditioned medium from MMP-13-overexpressing cells significantly promoted capillary tube formation, in comparison with that from empty vector-transfected HSC3 cells (Fig. 2B). Moreover, we examined MMP-13 knockdown in Ho-1-N-1 or HSC4 cells with MMP-13 expression. MMP-13 siRNA reduced MMP-13 expression and protease activity (Fig. 1, D and F). Conditioned medium from MMP-13-depleted cells suppressed migration and capillary tube formation (Fig. 2, C and D).
FIGURE 2.

Involvement of MMP-13 in migration and capillary tube formation of HUVECs. A, migration activity by conditioned medium from MMP-13-overexpressing cells. Migration activity was measured by the use of a 24-well cell culture insert with 8-μm pores. The lower compartment contained 0.5 ml of conditioned medium from empty vector-transfected HSC3 cells (control CM) or MMP-13-overexpressing HSC3 cells (MMP-13 CM). After trypsinization, 5 × 104 of immortalized HUVECs (HuhT1) were resuspended in 100 μl of serum-free medium and placed in the upper compartment of the cell culture insert for 4 h. To examine the activity of migration, the cells that had penetrated onto the lower side of the filter were fixed with formalin and stained with hematoxylin. The upper panel shows the representative area of penetrated cells. The lower graph shows the average number of penetrated cells. The bars show the average values and S.D. of three independent experiments. *, significantly different from control at p < 0.05. B, upper panel shows the representative area of capillary tube formation by conditioned medium from empty vector-transfected HSC3 cells (control CM) or MMP-13-overexpressing HSC3 cells (MMP-13 CM) (×40). An angiogenesis assay kit was used according to the manufacturer's instructions with minor modifications. HUVECs were treated with mixture of conditioned medium and HuMedia-EG2 in a percentage of 1:1. The mixed media were changed every 3 days. After 12 days, the cells were fixed and stained with anti-human CD31 antibody as described under “Materials and Methods.” The lower graph shows the average capillary tube score after conditioned medium treatment. Capillary tube score was estimated with the Chalkley count method under a bright-field microscope. The values represent means of capillary tube score + S.D. based on three wells/data point in a single experiment. *, significantly different from control at p < 0.05. C, migration activity of HuhT1 cells by conditioned medium from control or MMP-13 siRNA-treated cells. MMP-13 siRNA were transfected into HSC4 and Ho-1-N-1 cells. Migration activity was measured as described in A. The upper panel shows the representative area of penetrated cells. The lower graph shows the average number of penetrated cells. The bars show the average values and S.D. of three independent experiments. *, significantly different from control at p < 0.05. D, upper panel shows the representative area of capillary tube formation by conditioned medium from control or MMP-13-depleted cells (×40). Capillary tube formation was examined as described in B. The lower graph shows the average capillary tube score after conditioned medium treatment. Capillary tube score was estimated with the Chalkley count method under a bright-field microscope. The values represent means of capillary tube score + S.D. based on three wells/data point in a single experiment. *, significantly different from control at p < 0.05.
To exclude other factors in conditioned medium, we used recombinant MMP-13 protein for in vitro angiogenesis assay. We examined the effect of recombinant MMP-13 protein on cell growth and migration of HuhT1 cells. Treatment with recombinant MMP-13 protein did not significantly promote cell growth and migration of HuhT1 cells (Fig. 3, A and B). For in vitro angiogenesis assay, we used VEGF-A as a positive control and suramin as a negative control. Similarly to conditioned medium from MMP-13-overexpressing cells, treatment with MMP-13 protein significantly promoted capillary tube formation in a concentration-dependent manner (Fig. 3C). Surprisingly, capillary tube score of MMP-13 treatment was similar to that of VEGF-A (Fig. 3C). However, MMP-13 did not significantly stimulate microvessel outgrowth from aorta comparing with control (Fig. 3D).
FIGURE 3.

MMP-13 promoted angiogenesis both in vitro and in vivo. A, effect of MMP-13 on the proliferation of HuhT1 cells. Cells were plated on 24-well plates, and trypsinized cells were counted by Cell Counter at 0, 2, 4, and 6 days after adding recombinant MMP-13 protein (100 or 200 ng/ml). B, migration activity by recombinant MMP-13 protein. Migration activity was measured as described in Fig. 1E. The upper panel shows the representative area of penetrated cells. The lower graph shows the average number of penetrated cells. The bars show the average values and S.D. of three independent experiments. C, upper panel shows the representative area of capillary tube formation by treatment with recombinant MMP-13 protein (50, 100, and 200 ng/ml) (×40). VEGF-A (2 μg/ml) was used as a positive control, and suramin (1 mm) was used as a negative control. Capillary tube formation was examined as described in Fig. 1F. The lower graph shows the average capillary tube score after treatment with recombinant MMP-13 protein. The capillary tube score was estimated with the Chalkley count method under a bright-field microscope. The values represent means of capillary tube score + S.D. based on three wells/data point in a single experiment. *, p < 0.05. D, upper panel shows representative case of culturing aortic explants in three-dimensional matrix gels with or without recombinant MMP-13 protein (100 ng/ml). Excited thoracic aorta (1-mm-long cross-sections) was placed on the Matrigel-coated wells and covered with an additional 50 μl of Matrigel. Afterward, Control was treated with EBM-2 medium only or EBM-2 medium containing recombinant MMP-13 protein. Each medium was added every other day. All assays were performed by using five aortic rings per sample. Aortic rings were photographed on day 15. The area of angiogenic sprouting was calculated using Image-Pro Plus software program (Media Cybernetics). The lower graph shows microvessel densities in square pixels.
To clarify the mechanism of MMP-13-promoted angiogenesis, we examined the involvement of several intracellular signaling molecules such as FAK, Src, and ERK by Western blotting using phosphorylation specific antibodies in HuhT1 cells after adding recombinant MMP-13 protein. Increased phosphorylation of FAK and ERK was observed after adding MMP-13 protein (Fig. 4A). To demonstrate the involvement of FAK and ERK in MMP-13-promoted angiogenesis, we examined capillary tube formation after treatment with FAK inhibitor (FAK inhibitor 14) or ERK inhibitor (U0126) together with recombinant MMP-13 protein. We confirmed that treatment with FAK inhibitor 14 or U0126 suppressed FAK or ERK activity, respectively (Fig. 4B). Both inhibitors inhibited MMP-13-promoted capillary tube formation in a concentration-dependent manner (Fig. 4C). Both inhibitors also inhibited capillary tube formation without MMP-13 treatment in a concentration-dependent manner (Fig. 4C), suggesting that the FAK and ERK signaling pathway may be a conventional pathway of angiogenesis. Moreover, FAK inhibitor did not influence on ERK activity and ERK inhibitor did not influence on FAK activity in MMP-13-treated endothelial cells (data not shown), suggesting that the inhibitory effects exerted by either FAK or ERK inhibitor are separated. These findings suggest that MMP-13 may promote angiogenesis via a conventional pathway.
FIGURE 4.

MMP-13-promoted angiogenesis is mediated by FAK and ERK signaling pathway. A, levels of total and phosphorylated forms of FAK, Src, and ERK after treatment of HuhT1 cells with MMP-13 (100 ng/ml) shown by Western blotting. β-Actin expression was used as a loading control. HuhT1 cells were seeded on a culture dish. After incubation for 24 h, medium was changed to HuMedia without FBS and growth factors. After 4 h, the recombinant MMP13 protein (100 ng/ml) was added and the cells were incubated for indicated times. B, phosphorylated forms of FAK (Tyr-576/577), Src (Tyr-416) and ERK (Thr-202/Tyr-204) in the presence of MMP-13 (100 ng/ml) after treatment with 10 μm FAK inhibitor (FAK inhibitor 14) or 10 μm ERK inhibitor (U0126). Expression of total FAK or ERK was used as a loading control. C, upper panel shows the representative area of capillary tube formation by FAK inhibitor (FAK inhibitor, 14 or 10 μm) or ERK inhibitor (U0126, 10 μm) with or without MMP-13 (100 ng/ml) (×40). The lower left graph shows the average tubule score after 10 μm FAK inhibitor (FAK inhibitor 14) or 10 μm of ERK inhibitor (U0126) with or without 100 ng/ml of recombinant MMP-13 protein. The lower right graph shows the average tubule score after FAK inhibitor (0.1 and 1 μm) or ERK inhibitor (0.1 and 1 μm) with or without 100 ng/ml of recombinant MMP-13 protein. The values represent means of capillary tube score + S.D. based on three wells/data point in a single experiment. D, to examine the effect of protease inhibition on FAK and ERK phosphorylation, CL-821198, which is a selective inhibitor of MMP-13 through the binding to the S1′ pocket of MMP-13 with its morpholine ring adjacent to the catalytic zinc atom, was used. HuhT1 cells were seeded on a culture dish. After incubation for 24 h, medium was changed to HuMedia without FBS and growth factors. After 4 h, CL-821198 (10 μg/ml) and/or recombinant MMP13 protein (100 ng/ml) were added, and the cells were incubated for 1 h. Levels of total and phosphorylated forms of FAK and ERK was examined by Western blotting. E, capillary tube formation was examined by using an angiogenesis assay kit. HUVECs were treated with the recombinant MMP-13 protein with or without CL-821198 (5 and 10 μg/ml), and the medium was changed every 3 days. After 12 days, the cells were fixed and stained with anti-human CD31 antibody. The graph shows the average capillary tube score after treatment with recombinant MMP-13 protein. The capillary tube score was estimated with the Chalkley count method under a bright-field microscope. The values represent means of capillary tube score + S.D. based on three wells/data point in a single experiment. *, p < 0.05.
To know whether ERK or FAK activity induced by MMP-13 was caused by protease activity of MMP-13, we examined the effect of protease inhibition on FAK and ERK phosphorylation by using CL-821198, which is a selective inhibitor of MMP-13 through the binding to the S1′ pocket of MMP-13 with its morpholine ring adjacent to the catalytic zinc atom. CL-821198 treatment did not influence on ERK or FAK activity in HuhT1 cells (Fig. 4D). This finding indicates that ERK or FAK activity induced by MMP-13 is not caused by protease activity of MMP-13. We also examined the effect of CL-821198 on capillary tube formation. CL-821198 inhibited MMP-13-promoted tube formation. This finding suggests that MMP-13 activity may affect to capillary tube formation via an ERK- or FAK-independent manner (Fig. 4E).
MMP-13 Promotes VEGF-A Secretion in Fibroblasts and Endothelial Cells
A recent report shows that esophageal squamous cell carcinoma-derived TGF-β regulates angiogenesis through the release of VEGF from fibroblasts (27). We examined whether MMP-13 affected the release of VEGF from fibroblasts or endothelial cells as a similar function of TGF-β. Normal fibroblasts were obtained from gingival tissues using standard explant techniques (21). The level of VEGF-A secretion by fibroblasts was measured after being induced by MMP-13 with/without the presence of TGF-β. Interestingly, MMP-13 could promote the secretion of VEGF-A, especially in the presence of TGF-β (Fig. 5A). It is known that paracrine tumor-derived growth factors activate the cancer-associated fibroblasts, which undergo a myofibroblastic transdifferentiation defined by an elongated spindle shape, and the expression of contractile α-SMA and vimentin (28). Therefore, we examined the expression of α-SMA after TGF-β or MMP-13 treatment in fibroblasts. As previously reported, TGF-β induced α-SMA expression in fibroblasts (Fig. 5B). Although MMP-13 itself did not induce α-SMA expression in fibroblasts, both TGF-β and MMP-13 induced higher expression of α-SMA in comparison with the expression level of α-SMA induced by TGF-β (Fig. 5B).
FIGURE 5.

VEGF-A secretion by MMP-13 treatment in fibroblasts. A, fibroblasts were seeded on a culture dish. After incubation for 24 h, medium was changed to DMEM without FBS. After 24 h, MMP-13 (0, 10, and 50 ng/ml) and TGF-β (1 ng/ml) with or without MMP-13 (50 ng/ml) were treated for 24 h. The concentration of VEGF-A in the culture medium was quantified with commercial ELISA kits according to the manufacturer's instructions. B, after treatment with MMP-13 (0, 10, and 50 ng/ml) or TGF-β (1 ng/ml) with or without MMP-13 (50 ng/ml) for 24 h, fibroblasts were collected. Expressions of VEGF-A, α-SMA, and β-actin were examined by immunoblotting. The densitometric analysis of VEGF-A expression was performed. VGEF-A/β-actin ratio is shown. C, HuhT1 cells were seeded on a culture dish. After incubation for 24 h, medium was changed to HuMedia without FBS and growth factors. After 4 h, the recombinant MMP13 protein (100 ng/ml) with or without 10 μm of FAK inhibitor (FAK inhibitor 14), 10 μm of ERK inhibitor (U0126) or 10 μg/ml of CL-821198 were added and the cells were incubated for 1 h. Expression of VEGF-A and β-actin were examined by immunoblotting. The concentration of VEGF-A in the culture medium was quantified with commercial ELISA kits according to the manufacturer's instructions. D, fibroblasts were seeded on a culture dish. After incubation for 24 h, medium was changed to DMEM without FBS. After 24 h, MMP-13 (0, 50, and 100 ng/ml) and TGF-β (1 ng/ml) with or without MMP-13 (100 ng/ml) were treated for 24 h. Moreover, we treated CL-821198 (10 μg/ml). The concentration of VEGF-A in the culture medium was quantified with commercial ELISA kits according to the manufacturer's instructions.
Moreover, we examined whether MMP-13-promoted VEGF-A secretion from the endothelial cell line HuhT1 or not. Interestingly, the expression and secretion levels of VEGF-A were increased by MMP-13 treatment (Fig. 5, C and D). Induction of VEGF-A in HuhT1 cells was partially dependent on ERK activity but not on FAK activity and MMP-13 protease activity (Fig. 5, C and D).
MMP-13 Is Highly Expressed in Human Cancer Tissues
To demonstrate in vitro and in vivo evidence of MMP-13-mediated angiogenesis, we examined the expression of MMP-13 and its relationship with tumor angiogenesis in clinical cancer cases. We examined the immunohistochemical expression of MMP-13 in 20 normal oral epithelium and 67 HNSCC tissues. Positive expression of MMP-13 was observed in 0 of 20 (0%) normal oral epithelium and 54 of 67 (81%) HNSCC cases (Fig. 5, A and B, and Table 1). We compared MMP13 expression with metastasis in HNSCC cases. MMP-13 expression was well correlated with metastasis (Table 1). Then, we compared MMP-13 expression with the number of blood vessels in HNSCC cases. The number of blood vessels was examined by staining using anti-CD34 antibody. CD34 is an antigen present in hematopoietic progenitor cells and endothelial cells. Anti-CD34 antibody is a highly sensitive marker for endothelial cell differentiation and has also been studied as a marker for vascular tumors. We observed an increased number of blood vessels at the invasive front of the MMP-13 positive tumor cases, compared with the MMP-13 negative cases (p < 0.05) (Fig. 6, A and B). The average number of blood vessel density assessed by histo-morphometric analysis was 41.4 ± 13.0 and 76.2 ± 26.6 in MMP-13 negative and positive cases, respectively (p < 0.05) (Fig. 6C and Table 1).
TABLE 1.
Correlation between MMP-13 expression and clinicopathologic findings in HNSCC
| MMP-13 expression |
||||
|---|---|---|---|---|
| No. of cases | Low | High | p value | |
| Non-neoplastic epithelium | 30 | 30 (100%) | 0 (0%) | p < 0.001 |
| HNSCC | 67 | 13 (19.4%) | 54 (80.6%) | |
| Metastasis | ||||
| − | 30 | 7 (23.3%) | 23 (76.7%) | |
| + | 37 | 6 (16.2%) | 31 (83.8%) | |
FIGURE 6.

MMP-13 expression is well correlated with the number of blood vessels in human cancer cases. A, immunohistochemical staining of MMP-13 and CD34 in normal oral epithelium and HNSCC. Representative cases of MMP-13 expression in normal oral epithelium and HNSCC are shown. Representative cases of CD34 expression in HNSCC cases with or without MMP-13 expression are also shown. B, graph shows the number of cases with or without MMP-13 expression in 67 HNSCC cases. *, p < 0.05. C, graph shows the average number of blood vessels in HNSCC cases with or without MMP-13 expression. *, p < 0.05.
DISCUSSION
Angiogenesis, the formation of new blood vessels from preexisting ones, is a crucial step in tumor growth, progression, and metastasis. Regulation of angiogenesis in vivo is complex and is controlled by a variety of factors. Among them, VEGF is considered to play a dominant role. It has been well established that VEGF promotes a cancer progression by up-regulating microvessel density (3). MMPs are zinc metalloenzymes with the ability to degrade the components of the ECM. Their action is crucial during the progression of cancer because they allow the remodeling of the surrounding healthy tissues and enable local invasion (8). MMP-13 is known as collagenase-3, which has the ability to degrade fibrillar collagen (29). However, it may also act as a potent gelatinase by degrading a wide variety of extracellular matrix components (30, 31). MMP-13 is overexpressed in a variety of tumors from such as head and neck, laryngeal, breast, chondrosarcoma, gastric, colorectal, vulvar carcinomas and cutaneous malignant lymphoma (17, 32–39). In most malignancies, MMP-13 has been correlated with tumor invasion, metastasis, and poor prognosis in patients (34, 35, 37, 39, 41). MMP-13 is predominantly expressed by tumor cells at the tumor invasive front and to some extent by stromal fibroblasts surrounding tumor cells (33, 41). Our immunohistochemical finding that MMP-13 expression is frequently observed, but no statistical correlation was observed in MMP-13 expression and metastasis in HNSCC (Table 1). As we used biopsy cases in this study, HNSCC cases with high expression of MMP-13 may have a potential to metastasize later. Indeed, MMP-13 expression was well correlated with number of blood vessels. Thus, it is well accepted that MMP-13 is involved in tumor progression.
In the present study, we demonstrate the novel role of MMP-13 in tumor angiogenesis. Although there is a study suggesting a role of MMP-13 in keratinocyte migration and angiogenesis during the healing of fracture (43), the role of MMP-13 in tumor angiogenesis has not been fully elucidated. It is known that some MMPs such as MMP-1, -2, -3, -7, -9, -14 and -16 are involved in tumor angiogenesis via the regulation of bioavailability of VEGF-A (11). Distinct from the function of other MMPs in angiogenesis, MMP-13 promoted angiogenesis through an increased number of blood vessels at the invasive front of the tumor and up-regulation of VEGF-A secretion from fibroblasts and endothelial cells (Fig. 7). We also found that MMP-13 promoted capillary tube formation was mediated by activation of FAK and ERK. FAK is a cytoplasmic tyrosine kinase that plays critical roles in integrin-mediated signal transductions and also participates in signaling by other cell surface receptors (44). Extensive studies in FAK knock-out mouse models indicated a critical role of FAK in angiogenesis during embryonic development (45). Moreover, the increased expression of FAK in cancer cells has been suggested to play a role in the tumor angiogenic switch to promote aggressive tumor progression and metastasis (46). However, Baek et al. (47) found that the MEK/ERK pathway is involved in endothelial cell proliferation through up-regulation of positive cell cycle proteins and down-regulation of negative cell cycle proteins. Thus, activation of FAK and ERK is critically involved in angiogenesis. Indeed, treatment with FAK inhibitor or ERK inhibitor strongly inhibited the capillary tube formation of endothelial cells, suggesting that FAK and ERK signaling are essential in angiogenesis. Although MMP-13 has central roles in modulating extracellular matrix degradation through its direct matrix degrading capability as well as having a key involvement in the activation of other MMPs (7), FAK or ERK activity induced by MMP-13 was not mediated by matrix degrading capability (Fig. 4D). Our findings suggest that MMP-13-driven angiogenesis may be mediated by a conventional pathway via activation of FAK and ERK. However, the mechanism of ERK or FAK activation by MMP-13 is still unclear.
FIGURE 7.
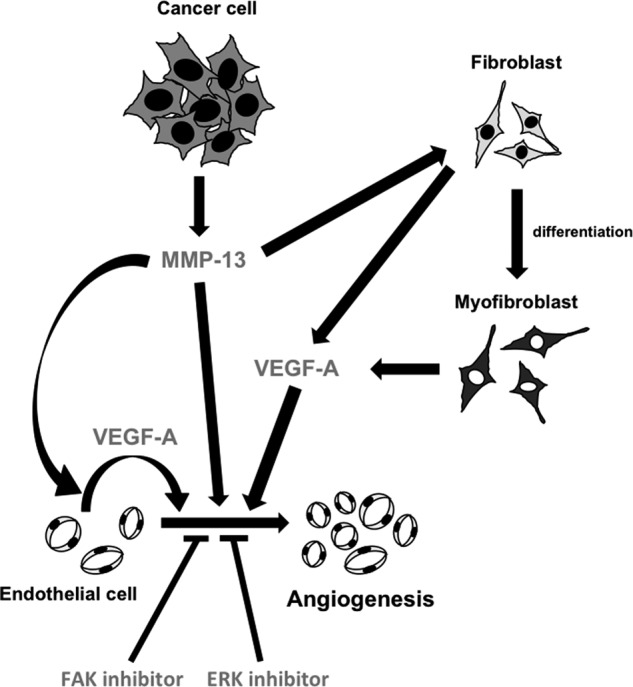
A model of MMP-13-promoted angiogenesis. MMP-13 is secreted from cancer cells. MMP-13 promotes angiogenesis through FAK and ERK signaling pathway. Moreover, MMP-13 enhances the secretion of VEGF-A from endothelial cells, fibroblasts or myofibroblasts. Secreted VEGF-A promotes angiogenesis.
The tumor tissue consists of a dynamic mixture of tumor cells, fibroblasts, endothelial cells, and immune cells that all work together to drive tumor progression (48). Activated fibroblasts, also known as cancer-associated fibroblasts within the tumor microenvironment, is preceded by the chemoattraction and migration of precursor cells, which can either arise from the surrounding host fibroblasts or from circulating mesenchymal precursor cells (40, 42, 49). Cancer-associated fibroblasts are activated by paracrine tumor-derived growth factors, which undergo a myofibroblastic transdifferentiation (28). Noma et al. (27) showed that paracrine TGF-β from the esophageal cancer cells lead to activation of the fibroblasts and vascular network formation through the release of VEGF-A. Interestingly, MMP-13 induced the secretion of VEGF-A from fibroblasts in similar to TGF-β. Moreover, TGF-β induced α-SMA expression in fibroblasts, indicating that the phenotypic switch from fibroblast to myofibroblast may be caused by TGF-β. MMP-13 itself could not induce α-SMA expression, but it enhanced α-SMA expression induced by TGF-β. Although TGF-β induced a myofibroblastic transdifferentiation of fibroblasts and VEGF-A secretion from myofibroblasts, TGF-β itself inhibited capillary tube formation of endothelial cells (data not shown). Previous report shows MMP-13 increases the expression of VEGF and its receptor, VEGFR-2 (19). In this study, we could not detect VEGFR-2 expression in fibroblasts after treatment with MMP-13 and/or TGF-β by real-time PCR analysis (data not shown). In addition, VEGF-A induction by MMP-13 in fibroblasts and endothelial cells was not dependent on MMP-13 protease activity (Fig. 5, C and D). Although it is unclear how MMP-13 promotes VEGF-A secretion from fibroblasts and endothelial cells, it is interesting to examine the detailed role of MMP-13 in the tumor microenvironment.
In summary, our findings suggest that MMP-13 may directly and indirectly promote tumor angiogenesis. In various tumors, MMP-13 is correlated with tumor invasion, metastasis, and poor prognosis. Therefore, we believe that MMP-13 can be a potential target for therapeutic intervention to additionally obstruct tumor angiogenesis in cancer patients.
Acknowledgments
The authors thank Dr. Michael Byrne (Harvard Medical School) for providing materials and Dr. Kyoko Hida (Hokkaido University) for discussion. We thank Satoko Katada (Tokushima University), Minori Manabe (Hiroshima University), and Mayumi Ohira (Hiroshima University) for technical assistance.
This work was supported by grants-in-aid from the Ministry of Education, Science, and Culture of Japan (to Y. K. and T. Ta.), a research fellowship for Young Scientists and the Excellent Young Researchers Overseas Visit Program from the Japan Society for the Promotion of Science (to S. I. and T. Ts.), and a Kurozumi Memorial Foundation grant (to Y. K.).
- ECM
- extracellular matrix
- MMP
- matrix metalloproteinase
- VEGF
- vascular endothelial growth factor
- FAK
- focal adhesion kinase
- HUVEC
- human umbilical vein endothelial cell
- HNSCC
- head and neck squamous cell carcinoma
- TGF-β
- transforming growth factor-β: α-SMA, α-smooth muscle actin.
REFERENCES
- 1. Fidler I. J. (1990) Critical factors in the biology of human cancer metastasis: twenty-eighth G.H.A. Clowes memorial award lecture. Cancer Res. 50, 6130–6138 [PubMed] [Google Scholar]
- 2. Howell G. M., Grandis J. R. (2005) Molecular mediators of metastasis in head and neck squamous cell carcinoma. Head Neck 27, 710–717 [DOI] [PubMed] [Google Scholar]
- 3. Folkman J. (2000) Tumor angiogenesis in Cancer Medicine (Holland J. F., ed) 5th Ed., pp. 132–152, B. C. Decker, Inc., Ontario, Canada [Google Scholar]
- 4. Hanahan D., Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- 5. Bouck N., Stellmach V., Hsu S. C. (1996) How tumors become angiogenic. Adv. Cancer Res. 69, 135–174 [DOI] [PubMed] [Google Scholar]
- 6. Fukumura D., Xavier R., Sugiura T., Chen Y., Park E. C., Lu N., Selig M., Nielsen G., Taksir T., Jain R. K., Seed B. (1998) Tumor induction of VEGF promoter activity in stromal cells. Cell 94, 715–725 [DOI] [PubMed] [Google Scholar]
- 7. Egeblad M., Werb Z. (2002) New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 [DOI] [PubMed] [Google Scholar]
- 8. Milner J. M., Cawston T. E. (2005) Matrix metalloproteinase knockout studies and the potential use of matrix metalloproteinase inhibitors in the rheumatic diseases. Curr. Drug Targets Inflamm. Allergy 4, 363–375 [DOI] [PubMed] [Google Scholar]
- 9. Werner J. A., Rathcke I. O., Mandic R. (2002) The role of matrix metalloproteinases in squamous cell carcinomas of the head and neck. Clin. Exp. Metastasis 19, 275–282 [DOI] [PubMed] [Google Scholar]
- 10. Coussens L. M., Fingleton B., Matrisian L. M. (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392 [DOI] [PubMed] [Google Scholar]
- 11. Littlepage L. E., Sternlicht M. D., Rougier N., Phillips J., Gallo E., Yu Y., Williams K., Brenot A., Gordon J. I., Werb Z. (2010) Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Res. 70, 2224–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bergers G., Brekken R., McMahon G., Vu T. H., Itoh T., Tamaki K., Tanzawa K., Thorpe P., Itohara S., Werb Z., Hanahan D. (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2, 737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee S., Jilani S. M., Nikolova G. V., Carpizo D., Iruela-Arispe M. L. (2005) Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J. Cell Biol. 169, 681–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ribatti D. (2009) Endogenous inhibitors of angiogenesis: a historical review. Leuk. Res. 33, 638–644 [DOI] [PubMed] [Google Scholar]
- 15. Kosaki N., Takaishi H., Kamekura S., Kimura T., Okada Y., Minqi L., Amizuka N., Chung U. I., Nakamura K., Kawaguchi H., Toyama Y., D'Armiento J. (2007) Impaired bone fracture healing in matrix metalloproteinase-13 deficient mice. Biochem. Biophys. Res. Commun. 354, 846–851 [DOI] [PubMed] [Google Scholar]
- 16. Zijlstra A., Aimes R. T., Zhu D., Regazzoni K., Kupriyanova T., Seandel M., Deryugina E. I., Quigley J. P. (2004) Collagenolysis-dependent angiogenesis mediated by matrix metalloproteinase-13 (collagenase-3). J. Biol. Chem. 279, 27633–27645 [DOI] [PubMed] [Google Scholar]
- 17. Freije J. M., Díez-Itza I., Balbín M., Sánchez L. M., Blasco R., Tolivia J., López-Otín C. (1994) Molecular cloning and expression of collagenase-3, a novel human matrix metalloproteinase produced by breast carcinomas. J. Biol. Chem. 269, 16766–16773 [PubMed] [Google Scholar]
- 18. Nielsen B. S., Rank F., López J. M., Balbin M., Vizoso F., Lund L. R., Danø K., López-Otín C. (2001) Collagenase-3 expression in breast myofibroblasts as a molecular marker of transition of ductal carcinoma in situ lesions to invasive ductal carcinomas. Cancer Res. 61, 7091–7100 [PubMed] [Google Scholar]
- 19. Lederle W., Hartenstein B., Meides A., Kunzelmann H., Werb Z., Angel P., Mueller M. M. (2010) MMP13 as a stromal mediator in controlling persistent angiogenesis in skin carcinoma. Carcinogenesis 31, 1175–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anno K., Hayashi A., Takahashi T., Mitsui Y., Ide T., Tahara H. (2007) Telomerase activation induces elongation of the telomeric single-stranded overhang, but does not prevent chromosome aberrations in human vascular endothelial cells. Biochem. Biophys. Res. Commun. 353, 926–932 [DOI] [PubMed] [Google Scholar]
- 21. Schor S. L., Schor A. M., Rushton G., Smith L. (1985) Adult, foetal and transformed fibroblasts display different migratory phenotypes on collagen gels: evidence for an isoformic transition during foetal development. J. Cell Sci. 73, 221–234 [DOI] [PubMed] [Google Scholar]
- 22. Kudo Y., Ogawa I., Kitajima S., Kitagawa M., Kawai H., Gaffney P. M., Miyauchi M., Takata T. (2006) Periostin promotes invasion and anchorage-independent growth in the metastatic process of head and neck cancer. Cancer Res. 66, 6928–6935 [DOI] [PubMed] [Google Scholar]
- 23. Bishop E. T., Bell G. T., Bloor S., Broom I. J., Hendry N. F., Wheatley D. N. (1999) An in vitro model of angiogenesis: basic features. Angiogenesis 3, 335–344 [DOI] [PubMed] [Google Scholar]
- 24. Fox S. B., Leek R. D., Weekes M. P., Whitehouse R. M., Gatter K. C., Harris A. L. (1995) Quantitation and prognostic value of breast cancer angiogenesis: comparison of microvessel density, Chalkley count, and computer image analysis. J. Pathol. 177, 275–283 [DOI] [PubMed] [Google Scholar]
- 25. Bauer K. S., Cude K. J., Dixon S. C., Kruger E. A., Figg W. D. (2000) Carboxyamido-triazole inhibits angiogenesis by blocking the calcium-mediated nitric-oxide synthase-vascular endothelial growth factor pathway. J. Pharmacol. Exp. Ther. 292, 31–37 [PubMed] [Google Scholar]
- 26. Deraz E. M., Kudo Y., Yoshida M., Obayashi M., Tsunematsu T., Tani H., Siriwardena S. B., Keikhaee M. R., Kiekhaee M. R., Qi G., Iizuka S., Ogawa I., Campisi G., Lo Muzio L., Abiko Y., Kikuchi A., Takata T. (2011) MMP-10/stromelysin-2 promotes invasion of head and neck cancer. PLoS One 6, e25438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Noma K., Smalley K. S., Lioni M., Naomoto Y., Tanaka N., El-Deiry W., King A. J., Nakagawa H., Herlyn M. (2008) The essential role of fibroblasts in esophageal squamous cell carcinoma-induced angiogenesis. Gastroenterology 134, 1981–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sappino A. P., Skalli O., Jackson B., Schürch W., Gabbiani G. (1988) Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int. J. Cancer 41, 707–712 [DOI] [PubMed] [Google Scholar]
- 29. Uitto V. J., Airola K., Vaalamo M., Johansson N., Putnins E. E., Firth J. D., Salonen J., López-Otín C., Saarialho-Kere U., Kähäri V. M. (1998) Collagenase-3 (matrix metalloproteinase-13) expression is induced in oral mucosal epithelium during chronic inflammation. Am. J. Pathol. 152, 1489–1499 [PMC free article] [PubMed] [Google Scholar]
- 30. Tardif G., Reboul P., Pelletier J. P., Martel-Pelletier J. (2004) Ten years in the life of an enzyme: the story of the human MMP-13) collagenase-3. Mod. Rheumatol. 14, 1489–1499 [DOI] [PubMed] [Google Scholar]
- 31. Ashworth J. L., Murphy G., Rock M. J., Sherratt M. J., Shapiro S. D., Shuttleworth C. A., Kielty C. M. (1999) Fibrillin degradation by matrix metalloproteinases: implications for connective tissue remodeling. Biochem. J. 340, 171–181 [PMC free article] [PubMed] [Google Scholar]
- 32. Culhaci N., Metin K., Copcu E., Dikicioglu E. (2004) Elevated expression in MMP-13 and TIMP-1 in head and neck squamous cell carcinomas may reflect increased tumor invasiveness. BMC Cancer 4, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johansson N., Vaalamo M., Grénman S., Hietanen S., Klemi P., Saarialho-Kere U., Kähäri V. M. (1999) Collagenase-3 (MMP-13) is expressed by tumor cells in invasive vulvar squamous cell carcinomas. Am. J. Pathol. 154, 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pendás A. M., Uría J. A., Jiménez M. G., Balbín M., Freije J. P., López-Otín C. (2000) An overview of collagenase-3 expression in malignant tumors and analysis of its potential value as a target in antitumor therapies. Clin. Chim. Acta 291, 137–155 [DOI] [PubMed] [Google Scholar]
- 35. Krecicki T., Fraczek M., Jelen M., Podhorska M., Szkudlarek T., Zatonski T. (2003) Expression of collagenase-1 (MMP-1), collagenase-3 (MMP-13) and tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) in laryngeal squamous cell carcinomas. Eur. Arch Otorhinolaryngol. 260, 494–497 [DOI] [PubMed] [Google Scholar]
- 36. del Casar Lizcano J. M., Vizoso Piñeiro F., González Sánchez L. O., Martin Suarez A., Gava A., Cuesta Fernandez E., Diez Santisteban M. C. (2003) Expression and clinical significance of collagenase-3 (MMP-13) in gastric cancer. Gastroenterol. Hepatol. 26, 1–7 [DOI] [PubMed] [Google Scholar]
- 37. Roeb E., Arndt M., Jansen B., Schumpelick V., Matern S. (2004) Simultaneous determination of matrix metalloproteinase (MMP)-7, MMP-1, -3, and -13 gene expression by multiplex PCR in colorectal carcinomas. Int. J. Colorectal. Dis. 19, 518–524 [DOI] [PubMed] [Google Scholar]
- 38. Corte M. D., Gonzalez L. O., Corte M. G., Quintela I., Pidal I., Bongera M., Vizoso F. (2005) Collagenase-3 (MMP-13) expression in cutaneous malignant melanoma. Int. J. Biol. Markers 20, 242–248 [DOI] [PubMed] [Google Scholar]
- 39. Luukkaa M., Vihinen P., Kronqvist P., Vahlberg T., Pyrhönen S., Kähäri V. M., Grénman R. (2006) Association between high collagenase-3 expression levels and poor prognosis in patients with head and neck cancer. Head Neck 28, 225–234 [DOI] [PubMed] [Google Scholar]
- 40. De Wever O., Mareel M. (2003) Role of tissue stroma in cancer cell invasion. J. Pathol. 200, 429–447 [DOI] [PubMed] [Google Scholar]
- 41. Johansson N., Airola K., Grénman R., Kariniemi A. L., Saarialho-Kere U., Kähäri V. M. (1997) Expression of collagenase-3 (matrix metalloproteinase-13) in squamous cell carcinomas of the head and neck. Am. J. Pathol. 151, 499–508 [PMC free article] [PubMed] [Google Scholar]
- 42. Direkze N. C., Hodivala-Dilke K., Jeffery R., Hunt T., Poulsom R., Oukrif D., Alison M. R., Wright N. A. (2004) Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 64, 8492–8495 [DOI] [PubMed] [Google Scholar]
- 43. Hattori N., Mochizuki S., Kishi K., Nakajima T., Takaishi H., D'Armiento J., Okada Y. (2009) MMP-13 plays a role in keratinocyte migration, angiogenesis, and contraction in mouse skin wound healing. Am. J. Pathol. 175, 533–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao X., Guan J. L. (2011) Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 63, 610–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ilić D., Furuta Y., Kanazawa S., Takeda N., Sobue K., Nakatsuji N., Nomura S., Fujimoto J., Okada M., Yamamoto T. (1995) Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377, 539–544 [DOI] [PubMed] [Google Scholar]
- 46. Gabarra-Niecko V., Schaller M. D., Dunty J. M. (2003) FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 22, 359–374 [DOI] [PubMed] [Google Scholar]
- 47. Baek Y. Y., Cho D. H., Choe J., Lee H., Jeoung D., Ha K. S., Won M. H., Kwon Y. G., Kim Y. M. (2011) Extracellular taurine induces angiogenesis by activating ERK-, Akt-, and FAK-dependent signal pathways. Eur. J. Pharmacol. 674, 629–640 [DOI] [PubMed] [Google Scholar]
- 48. Bissell M. J., Radisky D. (2001) Putting tumors in context. Nat. Rev. Cancer 1, 46–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dvorak H. F. (1986) Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659 [DOI] [PubMed] [Google Scholar]