

Background: Oxidative methylation and successive methylation are two possible enzymatic mechanisms of arsenite methylation.
Results: Rapid equilibrium kinetic analysis established that hAS3MT-catalyzed arsenite methylation is a completely ordered reaction.
Conclusion: The methyl transfer step occurs on hAS3MT. Reductant reduces a disulfide bond and exposes the active site cysteine residues.
Significance: This work clearly elucidates the completely ordered mechanism of arsenite methylation by a rapid equilibrium kinetic model.
Keywords: Cysteine-mediated Cross-linking, Enzyme Catalysis, Fluorescence, Kinetics, Metabolism, AdoMet, Arsenic, Fluorescence, Mechanism, Methyltransferase
Abstract
In the human body, arsenic is metabolized by methylation. Understanding this process is important and provides insight into the relationship between arsenic and its related diseases. We used the rapid equilibrium kinetic model to study the reaction sequence of arsenite methylation. The results suggest that the mechanism for arsenite methylation is a completely ordered mechanism that is also of general interest in reaction systems with different reductants, such as tris(2-carboxyethyl)phosphine, cysteine, and glutathione. In the reaction, cysteine residues of recombinant human arsenic (+3 oxidation state) methyltransferase (hAS3MT) coordinate with arsenicals and involve the methyl transfer step. S-Adenosyl-l-methionine (AdoMet) is the first-order reactant, which modulates the conformation of hAS3MT to a best matched state by hydrophobic interaction. As the second-order reactant, reductant reduces the disulfide bond, most likely between Cys-250 and another cysteine residue of hAS3MT, and exposes the active site cysteine residues for binding trivalent inorganic arsenic (iAs3+) to give monomethylarsonic dicysteine (MADC3+). In addition, the reaction can be extended to further methylate MADC3+ to dimethylarsinic cysteine (DAMC3+). In the methylation reaction, the β-pleated sheet content of hAS3MT is increased, and the hydrophobicity of the microenvironment around the active sites is decreased. Similarly, we confirm that both the high β-pleated sheet content of hAS3MT and the high dissociation ability of the enzyme-AdoMet-reductant improve the yield of dimethylated arsenicals.
Introduction
Arsenic is one of the most significant hazards in the environment, affecting millions of people around the world. Exposure to arsenic is associated with cancers of the skin, lung, urinary bladder, kidney, and liver as well as several non-cancer diseases, such as diabetes mellitus, hypertension, and cerebrovascular and cardiovascular diseases (1–4). The relationship between arsenic and its related diseases is complicated by many aspects, such as dose-response relationships, oxidative stress, cellular signaling, cell cycle control, gene amplification, and chromosomal abnormalities (5–7). In many species, including humans, methylation is a major metabolic transformation of arsenic, producing mainly monomethylated arsenicals (MMAs)2 and dimethylated arsenicals (DMAs) (8–11), both of which have been detected in human urine (12). In recent years, it has become apparent that methylation is not necessarily a detoxification process of inorganic arsenic (iAs) (13). The methylated products and intermediates may be more reactive and toxic than inorganic arsenic. For example, Kligerman et al. (14) suggested that methylation of the trivalent forms of arsenic increased their genotoxicity and cytotoxicity. Compared with iAs3+, methylated trivalent arsenicals, such as MMA3+ and DMA3+, were found to be more potent in causing DNA strand breaks in human lymphocytes and to induce a greater extent of cytotoxic and genotoxic effects, such as micronucleus formation, chromosome aberrations, and sister chromatid exchange (15, 16). A better understanding of the biotransformation metabolism of arsenic should shed light on the relationship between arsenic and its related diseases (17, 18).
Arsenic (+3 oxidation state) methyltransferase (AS3MT) has been proposed as the authentic enzyme that catalyzes the methylation of arsenite, and it is used for characterizing the methylation reaction in vitro (8–10, 18–23). Two different models have been proposed to describe the mechanism of arsenite methylation catalyzed by AS3MT. The first model arose from the work of Challenger (24, 25). Cullen et al. (22) summarized this general scheme and reported that the metabolic pathway of arsenic is oxidative methylation where the valence state of arsenic is changed in the methyl transfer step: As(V)O43− + 2e →As(III)O33− + CH3+ → MAs(V)O32− + 2e → MAs(III)O22− + CH3+ → DAs(V)O21− + 2e → DAs(III)O1−. However, Hayakawa et al. (23) proposed that the metabolic pathway of arsenic is successive methylation where the valence state of arsenic does not change in the methyl transfer step. In the reaction, the formation of the As-glutathione (GSH) complex was considered to be essential for each of the methylation steps, and the intermediates were arsenic triglutathione (ATG3+), monomethylarsonic dicysteine (MADG3+), and dimethylarsinic cysteine (DAMG3+) before DMA3+ was finally produced: iAs5+ + 2e → iAs3+ + GSH → ATG3+ + CH3+ → MADG3+ → MMA3+→ MMA5+ and MADG3+ + CH3+ → DAMG3+ → DMA3+ → DMA5+. Both models start from the reduction of iAs5+ to iAs3+ and do not further describe the function of the enzyme in the reaction. To elucidate the detailed mechanism, we studied the involvement of the cysteine residue of human AS3MT (hAS3MT) in the methylation reaction by site-directed mutagenesis and found that the conserved Cys-156 and Cys-206 were essential for catalytic activity and that Cys-72 and Cys-250 also played a key role in the reaction (20, 26). Similar to GSH, cysteine residues of hAS3MT also have a sulfhydryl group and can be involved in oxidation-reduction reactions. Therefore, the As-GSH complex might not be the required substrate for arsenite methylation. A thiol-arsenical complex might also be formed between arsenicals and the active site cysteine residues of hAS3MT. To confirm this, we studied the relationship between the reactants and hAS3MT, and we performed kinetic characterization of the methylation reaction.
In this report, three different reductants, GSH, cysteine, and tris(2-carboxyethyl)phosphine (TCEP), were used to study the kinetics of arsenite methylation catalyzed by hAS3MT. GSH is a predominant endogenous reductant in vivo and is a typical thiol reductant used to study the mechanism of methylation of arsenicals catalyzed by AS3MT. Cysteine was used as an alternative thiol reductant to GSH to show that a thiol-arsenical complex, not restricted to As-GSH, is the substrate for arsenite methylation. TCEP is a strong non-thiol reductant. Our results suggest that the valence state of arsenic is unchanged when the methyl transfer step occurs on hAS3MT. Reductant reduces the disulfide bond between the cysteine residues of hAS3MT, thereby exposing the active sites for binding to iAs3+. The highly dissociative ability of the enzyme-AdoMet-reductant and the increased content of β-pleated sheet help enable the binding of iAs3+ to the active sites.
EXPERIMENTAL PROCEDURES
Caution
Inorganic arsenic (11) is recognized as a human carcinogen. Appropriate safety measures should be taken when handling arsenic compounds.
Preparation of hAS3MT
The cloning, heterologous expression, and purification of recombinant hAS3MT were carried out as described previously (20). Details are shown in the supplemental Materials.
The Initial Velocity Assay of hAS3MT
The enzymatic methylation of arsenite was designed according to Myllylä et al. (27) and tested in a standard system (100 μl) containing hAS3MT (2.0 μm), AdoMet (1.0 mm), iAs3+ (1.0 μm), phosphate buffer (25 mm, pH 7.0), and each of the reductants (GSH, cysteine, and TCEP at 7, 10, and 0.7 mm, respectively). The initial velocity curve was obtained by varying the concentration of one of the three reactants (reductant, AdoMet, and arsenite) while the other two were fixed. All of the reactions were carried out in capped tubes at 37 °C for 30 min and then stopped by adding H2O2 to a final concentration of 3% (23). Arsenicals were analyzed by HPLC-inductively coupled plasma-MS (28). The methylation rates were calculated as mole equivalents of methyl groups transferred from AdoMet to arsenic (i.e. 1.0 pmol of CH3/1.0 pmol of MMA or 2.0 pmol of CH3/1.0 pmol of DMA) (29).
Effect of Reductant on the Conformation of hAS3MT in the Methylation Reaction
The effect of the reductant on the hAS3MT disulfide bonds was determined by the improved Ellman's test (28, 30). After incubation of hAS3MT (15 μm) with the different reductants (20 mm cysteine, 15 mm GSH, and 1.5 mm TCEP, respectively) in phosphate buffer at 37 °C for 30 min, the mixture was thoroughly dialyzed against phosphate buffer at 4 °C. The reduced hAS3MT was incubated with 0.1 mm 5,5′-dithiobis(2-nitrobenzonic acid) in Tris·HCl (20 mm, pH 7.0) at 25 °C for 90 min. The absorbance at 412 nm was then monitored to estimate the number of cysteine residues in hAS3MT. The effect of the reductant on the secondary structure was measured by CD spectroscopy after the reduced enzyme was thoroughly dialyzed against phosphate buffer at 4 °C. After the methylation reaction was catalyzed at 37 °C for 120 min, the secondary structure of hAS3MT was also determined by CD spectroscopy after being thoroughly dialyzed against phosphate buffer at 4 °C. The methylation reaction system was the same as that used in the velocity assay.
Mass Spectrometry
The methylation reaction system contained hAS3MT (4.0 μm), reductant (TCEP (0.7 mm), cysteine (10 mm), or GSH (7 mm)), AdoMet (1.0 mm), iAs3+ (1.0 μm), and phosphate buffer. After incubation at 37 °C for 60 min, the reaction mixture was immediately lyophilized and then separated by 12% non-reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The band corresponding to hAS3MT was excised and treated with 20 mm iodoacetamide in the dark at room temperature for 1.5 h followed by incubation with 5% trypsin overnight at 37 °C. Subsequently, 200 μl of 60% acetonitrile containing 0.1% (v/v) TFA was added to stop the reaction. Mass spectra were obtained on an AUTOFLEX II MALDI-TOF mass spectrometer (Bruker).
RESULTS
The Differences between hAS3MT-catalyzed Arsenite Methylation in Different Reductive Systems
Both MMA and DMA were detected in each of the reaction systems with different reductants. The differential distribution of the methylated products is shown in Fig. 1. The characteristics of the reaction in the GSH and cysteine systems were similar. MMA was generated at the beginning of the reaction, DMA increased as the reaction proceeded, and the methylation efficiency was not high. Interestingly, the yields of MMA and DMA were nearly the same in the GSH and cysteine reductive systems when the reactions were incubated for 2 h or longer. This was probably due to saturation of the reductant. However, in the TCEP system, the methylation efficiency was very high, and the methyl transfer from arsenite to MMA and then to DMA was fast. DMA was the major product and was generated rapidly at the beginning of the reaction.
FIGURE 1.

Product distribution of arsenite methylation catalyzed by hAS3MT in different reductive systems. The reaction system containing hAS3MT (2.0 μm), reductant, AdoMet (1.0 mm), iAs3+ (1.0 μm), and phosphate buffer was incubated at 37 °C for different times. The concentrations of GSH, cysteine, and TCEP were 7, 10, and 0.7 mm, respectively. Values are presented as the means ± S.D. of three separate experiments. Error bars represent S.D. from the mean of three independent experiments.
Rapid Equilibrium Kinetic Analysis of the Methylation Reaction Sequence
In this work, we used the rapid equilibrium kinetic model to study the reaction sequence of arsenite methylation (31, 32). Five distinct mechanistic steps from the completely ordered mechanism to the completely random mechanism may be involved in the enzyme-catalyzed reaction: A + B + C → products. The completely ordered mechanism can be expressed as E + A ↔ EA + B ↔ EAB + C ↔ EABC → products where E is the enzyme and EA, EAB, and EABC are the intermediate reactants (32). The deduced equations of rapid equilibrium velocity for the completely ordered mechanism are as follows.
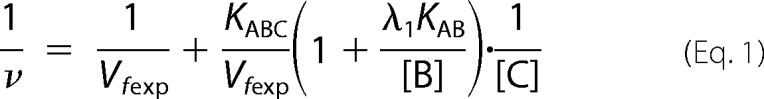 |
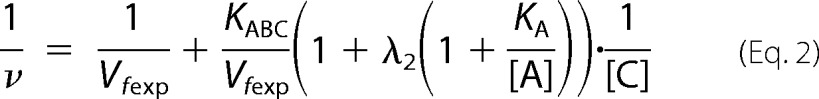 |
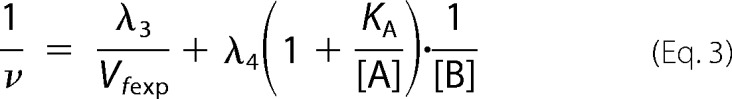 |
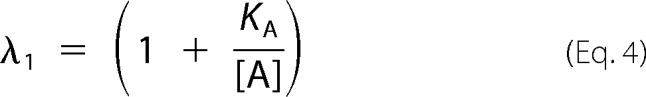 |
 |
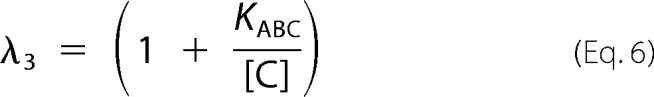 |
 |
where A, B, and C are the first-, second-, and third-order reactant, respectively; KA, KAB, and KABC are the dissociation constants of each step; Vfexp is the limiting velocity of the reaction; and λ1, λ2, λ3, and λ4 are the constants when the concentrations of the corresponding reactants are fixed in the reaction. As shown in Equations 1 and 2, when the data were plotted as velocity−1 versus C−1 at different fixed concentrations of B or A, the double reciprocal plots crossed at point (0, 1/Vfexp). In Equations 3–6, when the data were plotted as velocity−1 versus B−1 at different fixed concentrations of A, the lines intersected at point (0, λ3/Vfexp). When the arsenite concentration at different fixed concentrations of either the reductant (Fig. 2, A–C) or AdoMet (Fig. 2, D–F) was varied, the double reciprocal plots crossed the y axis at 5.40, 21.98, and 10.00 (μmol of CH3/h/mg)−1 in accordance with the value of 1/Vfexp. When the data were plotted as velocity−1 versus reductant−1 at different fixed concentrations of AdoMet (Fig. 2, G–I), the lines also intersected on the ordinate at 30.12, 44.23, and 24.98 (μmol of CH3/h/mg)−1, respectively, in accordance with the value of λ3/Vfexp.
FIGURE 2.

A–C, effect of iAs3+ at different concentrations on the rate of arsenite methylation catalyzed by hAS3MT at different fixed concentrations of TCEP (A), GSH (B), and cysteine (C). D–F, effect of iAs3+ at different concentrations on the rate of arsenite methylation catalyzed by hAS3MT at different fixed concentrations of AdoMet in the TCEP reaction system (D), GSH reaction system (E), and cysteine reaction system (F). G–I, effect of different concentrations of TCEP (G), GSH (H), and cysteine (I) on the rate of arsenite methylation catalyzed by hAS3MT at different fixed concentrations of AdoMet. Error bars represent S.D. from the mean of three independent experiments.
From the characterization of the initial velocity of arsenite methylation, a completely ordered mechanism seemed reasonable (Fig. 2). Distinctly, the reductant is the second-order reactant, and arsenite is the third-order reactant. The other characteristics of the initial velocity of arsenite methylation also supported a completely ordered mechanism (data not shown). The kinetic parameters, which were determined by the rapid equilibrium model, showed that KA and KABC were only slightly different in the three reductive systems, whereas KAB was significantly different (Table 1). The values of KA in the cysteine, GSH, and TCEP reaction systems were 62.50, 90.91, and 98.95 μmol/liter, respectively. This finding suggests that AdoMet binds to hAS3MT more easily in the cysteine system than in the GSH or TCEP system. Additionally, the values of KABC were 0.91 μmol/liter in the GSH system, 1.30 μmol/liter in the cysteine system, and 4.55 μmol/liter in the TCEP system, suggesting that the enzyme-AdoMet-TCEP-iAs3+ complex dissociates most readily among the three reductant systems. In the TCEP system, the value of KAB was 1233.74 mmol/liter, which was much higher than in either the cysteine (36.92 mmol/liter) or GSH system (5.50 mmol/liter). With the increase of KAB from GSH to cysteine and then to TCEP, the Vfexp of the reactions also increased. Thus, the second step, which forms the enzyme-AdoMet-reductant complex, is the most likely rate-limiting step for arsenite methylation. The dissociation of the enzyme-AdoMet-reductant may be essential for iAs3+ binding to hAS3MT in the third step.
TABLE 1.
Values of the kinetic parameters KA, KAB, KABC, and Vfexp for the methylation reaction
| Parameter | GSH | Cysteine | TCEP |
|---|---|---|---|
| KA (μmol/liter) | 90.91 ± 3.23 | 62.50 ± 2.12 | 98.85 ± 3.46 |
| KAB (mmol/liter) | 5.50 ± 0.67 | 36.92 ± 1.23 | 1233.74 ± 22.11 |
| KABC (μmol/liter) | 0.91 ± 0.03 | 1.30 ± 0.02 | 4.55 ± 0.06 |
| Vfexp ((pmol CH3/(h·mg)) × 10−4) | 4.55 ± 0.21 | 10.00 ± 0.62 | 18.52 ± 0.63 |
Effect of Reductant on Disulfide Bonds and the Conformation of hAS3MT in the Reaction
Trivalent arsenicals easily bind to the sulfhydryl sites of proteins (33) and have been reported to coordinate to cysteine residues of hAS3MT to catalyze the methylation reaction (8, 11, 34). Our previous work suggested that GSH reduced the disulfide bond between the cysteine residues of hAS3MT that was formed after the release of the methylated products from the enzyme (21). From our current results, reductants are involved in the second step of the reaction. Thus, we propose that the reductant might reduce the disulfide bonds of hAS3MT and change the conformation of hAS3MT to expose the active site cysteine residues for iAs3+ binding. To confirm this hypothesis, we analyzed the number of sulfhydryl groups of the reductant-treated hAS3MT using the improved Ellman's test. The numbers of cysteine residues was 6.31, 6.56, and 8.75 in cysteine-, GSH-, and TCEP-reduced hAS3MT, respectively, whereas 4.66 cysteine residues were detected on the surface of the control hAS3MT. This result agrees well with our earlier report that 4.42 thiol groups were detected on hAS3MT (28). We further analyzed the number of cysteine residues of hAS3MT by MALDI-TOF mass spectrometry. Cysteine residues of the samples were labeled by iodoacetamide before trypsin digestion and mass spectrometry analysis. As shown in Fig. 3, seven iodoacetamide-modified cysteine-corresponding peptides were detected in native hAS3MT. In the hAS3MT that underwent reaction, we detected additional peptides corresponding to two additional cysteine residues at sites 32 and 250. These results suggest that Cys-32 and Cys-250 are exposed in the reaction, although a direct disulfide bond between these two residues has yet to be confirmed.
FIGURE 3.

MALDI-TOF spectra of trypsin-digested hAS3MT. The hAS3MT was separated by 12% non-reducing SDS-PAGE and alkylated by 20 mm iodoacetamide after having catalyzed the arsenite methylation at 37 °C for 60 min. The reaction system contained hAS3MT (4.0 μm), AdoMet (1.0 mm), iAs3+ (1.0 μm), phosphate buffer (25 mm, pH 7.0), and different reductant. The reductants were TCEP (B), cysteine (C), and GSH (D) at 0.7, 10, and 7 mm, respectively. The protein under natural condition was used as a control (A). Results are the average of three determinations.
CD spectroscopy was used to evaluate the effect of reductant on the conformation of hAS3MT. Compared with native hAS3MT, the α-helix content decreased, but the β-pleated sheet content increased in the presence of reductant (Fig. 4, A and B). The secondary structure of hAS3MT changed similarly in the methylation reaction. In particular, the β-pleated sheet content increased significantly in the TCEP reaction system (Fig. 4, C and D). Regardless of whether the structural alteration of hAS3MT occurred in the reductive systems or in the methylation reaction systems, the β-pleated sheet content of hAS3MT increased. This outcome proved that the increase in β-pleated sheet content of hAS3MT was induced by the reductant.
FIGURE 4.

A, CD spectra showed the effect of reductant on the secondary structure of hAS3MT. B, the secondary structure of the reduced enzyme compared with the natural enzyme. The reaction system containing hAS3MT (15.0 μm), reductant, and phosphate buffer was incubated at 37 °C for 30 min. The concentrations of TCEP, cysteine, GSH were 1.5, 20, and 15 mm, respectively. C, CD spectra showed differences of the secondary structure of hAS3MT in different methylation reaction systems. D, the secondary structure of the enzyme that underwent reaction compared with the natural enzyme. The reaction system containing hAS3MT (2.0 μm), reductant, AdoMet (1.0 mm), iAs3+ (1.0 μm), and phosphate buffer, was incubated at 37 °C for 120 min. The concentrations of TCEP, cysteine, and GSH were 0.7, 10, and 7.0 mm, respectively. Values are presented as the means ± S.D. of three separate experiments. Error bars represent S.D. from the mean of three independent experiments. deg, degrees.
Effect of AdoMet and iAs3+ on the Conformation of hAS3MT
Tryptophan, tyrosine, and phenylalanine residues all contribute to protein fluorescence. However, phenylalanine has a very low quantum yield, and the fluorescence of tyrosine is easily quenched when it is ionized or close to an amino group, a carboxyl group, or a tryptophan (35). In hAS3MT, there are three tryptophan residues (Trp-73, Trp-203, and Trp-213) close to the cysteine residues (Cys-156, Cys-206, Cys-72, and Cys-250) that are important to the enzymatic activity (20, 26). Changes in intrinsic fluorescence intensity of hAS3MT reflect the perturbation of the active site.
The effect of AdoMet on hAS3MT fluorescence was static quenching. This effect was proved by the Stern-Volmer curves and the Lineweaver-Burk curves (36) (Fig. 5, A and B) and could be further supported by Förster's energy transfer theory because the distance we calculated between AdoMet and the tryptophan residue in hAS3MT was less than 7 nm (37). The thermodynamic parameters were calculated according to the equation proposed by Bi et al. (38) (Fig. 6). The three parameters, ΔG < 0, ΔH > 0, and ΔS > 0, suggested a hydrophobic interaction between AdoMet and hAS3MT (36). The value of ΔG showed that when hAS3MT was in the active state at 37 °C it bound AdoMet more readily in the cysteine system than in the GSH system, and hAS3MT more readily bound AdoMet in the GSH system than in the TCEP system (Fig. 7A). The values of ΔH and ΔS showed the same trend, which confirmed that the AdoMet binding reaction was an entropy-driven reaction (Fig. 7B). These findings led to the conclusion, as also suggested by KA, that AdoMet bound to hAS3MT more readily in the cysteine system than in GSH or TCEP system.
FIGURE 5.

A, the Stern-Volmer curves for the quenching of hAS3MT with AdoMet at 350 nm in different reductive systems (pH 7. 0) at 302.15 K (black line) and 310.15 K (gray line). B, the Lineweaver-Burk curves for the quenching of hAS3MT with AdoMet at the same conditions. The concentration of hAS3MT was 2.0 μm, and the concentrations of AdoMet were 0, 25, 50, 75, 100, 125, 150 and 175 μm, respectively. Each experiment was carried out three times.
FIGURE 6.

The double logarithm curves for quenching of hAS3MT with AdoMet at 302.15 K (black line) and 310.15 K (gray line). The conditions were the same as for Fig. 5. Each experiment was carried out three times.
FIGURE 7.

A, the free energy of binding reactions of AdoMet to hAS3MT in different reductive systems. B, enthalpy and entropy values of binding reactions. Each experiment was carried out three times. Error bars represent S.D. from the mean of three independent experiments.
The synchronous fluorescence spectra showed that the maximum emission wavelength of hAS3MT was slightly red-shifted in each reductive system (Table 2). Interestingly, red shifts were also observed with the titrated concentrations of AdoMet, demonstrating that the microenvironment around the active site was disturbed and that the hydrophobicity decreased in the presence of AdoMet (37). The fluorescence quenching efficiency of AdoMet on the enzyme calculated using (F0 − F)/F0 (%) was 28.91% in the cysteine system, 26.52% in the GSH system, and 19.46% in the TCEP system (Table 2), confirming that AdoMet bound to hAS3MT more readily in the cysteine reductive condition than in the GSH or TCEP reductive condition.
TABLE 2.
Synchronous fluorescence spectra of hAS3MT (Δλ = 60 nm) in different systems
| Parameter | Effect of AdoMet on rhAS3MT |
|||
|---|---|---|---|---|
| Phosphate buffer (25.00 mm) | GSH (7.00 mm) | Cysteine (10.00 mm) | TCEP (0.70 mm) | |
| F0 | 797.98 | 680.60 | 680.54 | 869.86 |
| F | 624.85 | 500.08 | 483.80 | 700.56 |
| λ0 (nm) | 277.0 | 279.0 | 279.5 | 278.0 |
| λ (nm) | 281.5 | 282.0 | 282.5 | 281.5 |
| (F0 − F)/F0 (%) | 21.70 | 26.52 | 28.91 | 19.46 |
| Δλ (nm) | 4.5 | 3.0 | 3.0 | 3.5 |
The three-dimensional fluorescence spectra of hAS3MT were notably different among the different reductive systems (data not shown). In our test range, peak 1 was mainly dominated by the microenvironments of the tryptophan and tyrosine residues, and peak 2 mainly exhibited the fluorescence character of polypeptide backbone structures (37). Peak 2 of hAS3MT disappeared in both the GSH system and the cysteine system (Table 3). After AdoMet titration, the maximum emission wavelength of peak 1 showed a slight red shift, and the fluorescence intensity decreased by 22.77 and 17.33% in the cysteine system and the GSH system, respectively. After AdoMet titration in the TCEP system, the maximum emission wavelengths of peak 1 and peak 2 had red shifts, and the fluorescence intensity decreased by 16.50 and 49.58%, respectively. The effect of AdoMet on peak 2 was more significant than that on peak 1, demonstrating a stronger impact of AdoMet on the polypeptide backbone structure of the enzyme.
TABLE 3.
Three-dimensional spectra of hAS3MT in different systems
| Peaks | hAS3MT |
hAS3MT + AdoMet |
(FAdoMet − F0)/F0 | ||
|---|---|---|---|---|---|
| Peak position λex/λem | Intensity, F0 | Peak position λex/λem | Intensity, FAdoMet | ||
| nm | nm | % | |||
| Peak 1/cysteine | 277.0/349.5 | 547.78 | 277.0/350.0 | 423.04 | −22.77 |
| Peak 1/GSH | 277.0/350.0 | 600.67 | 277.0/350.5 | 496.58 | −17.32 |
| Peak 1/TCEP | 277.0/350.0 | 718.44 | 277.0/351.0 | 599.88 | −16.50 |
| Peak 2/TCEP | 228.0/350.5 | 453.41 | 228.0/353.0 | 228.61 | −49.58 |
The effect of iAs3+ on the secondary structure of hAS3MT is very limited and irregular. A further analysis showed that the values of (α + β)% and (β/(α + β))% increase slightly with increasing iAs3+ at 37 °C, whereas they were nearly unchanged at 29 °C (data not shown) (39).
DISCUSSION
It has been reported previously that the nonenzymatic methylation of arsenite by methylcobalamin proceeds via nucleophilic attack of the As-GSH complex on cobalt (40) and that the methylation of arsenite catalyzed by arsenic methyltransferase also proceeds via the formation of As-GSH complexes (23). These reports suggested that arsenicals are capable of forming a thiol-arsenical complex with cysteine in the reaction system. However, TCEP is a non-thiol reductant, which cannot form thiol-arsenical complexes with arsenicals in the reaction. Therefore, our results suggested that the thiol-arsenical complex formed by arsenicals and exogenous reductant might only exist in a thiol reductive reaction system and is not necessarily a substrate for the hAS3MT-catalyzed methylation reaction.
Based on our results, we propose that the methyl group is transferred from AdoMet to iAs3+ on the enzyme. In the reaction, AdoMet first modulates the peptide backbone of hAS3MT to a best matched state, and then the reductant reduces the disulfide bond of hAS3MT to expose cysteine residues in the active site for binding iAs3+ to form arsenic tricysteine (ATC3+). An ion pair of ATC3+ attacks the cationic sulfur of AdoMet, and a methyl group of AdoMet is transferred to ATC3+, resulting in monomethylarsonic dicysteine (MADC3+) and S-adenosylhomocysteine. The extra reductant may function in cleaving S-adenosylhomocysteine from the enzyme. A portion of MADC3+ also dissociates from the enzyme to form MMA3+ in the presence of reductant (11, 28). The remaining MADC3+ is further methylated into dimethylarsinic cysteine (DAMC3+) on hAS3MT in the presence of AdoMet and reductant and then dissociated into DMA3+. MMA3+ and DMA3+ can be further oxidized by environmental oxygen into MMA5+ and DMA5+, respectively (21) (Fig. 8, A and B).
FIGURE 8.

A, a putative mechanism of arsenite methylation catalyzed by hAS3MT. B, the conformational transition of hAS3MT in the sequential process.
Early studies showed that, in the GSH reaction system, arsenite and GSH form ATG3+, which easily binds to hAS3MT (11, 23), and further form ATC3+ by exchanging the GSH with cysteine residues of the enzyme. GSH can also competitively coordinate with the arsenic of MADC3+. This coordination leads to the dissociation of MADC3+ from the enzyme into MADG3+, which can then be transformed into MMA3+. Therefore, the transformation from iAs3+ to MMA3+ is the major methylation step in the initiation of the reaction, and MMA is the major methylated product at the beginning of the GSH reaction system. However, arsenite did not exist in thiol-arsenical form in the TCEP reaction system. The high reductive potential of TCEP leads to rapid dissociation of S-adenosylhomocysteine from hAS3MT, which favors the further methylation of MADC3+ into DAMC3+ and accelerates the velocity of the methyl transfer reaction. A large amount of DMA is thus generated at the beginning of the reaction with TCEP.
The completely ordered mechanism of AdoMet + reductant + iAs3+ → products and the role of each reactant in the reaction are detailed in Fig. 8B. As the first-order reactant, AdoMet affects the peptide backbone of hAS3MT and decreases the hydrophobicity of the microenvironment around the active site. The conformational change of the enzyme increases the exposure of the originally buried hydrophobic regions (41). Fomenko et al. (42) analyzed the structure model of mouse AS3MT and confirmed that the cysteine residues in the active site are surface-exposed on the β-pleated sheet of the enzyme. In our research, the β-pleated sheet content increased, and the hydrophobicity of the microenvironment around the active site decreased when reductant was added into the enzymatic system. Therefore, the reductant alters the conformation of hAS3MT to an active state before the methylation reaction. The effect of iAs3+ on the conformation of hAS3MT is insignificant.
The enzyme was thoroughly inactive in the GSH reaction system when any of the cysteine residues at positions 72, 156, 206, and 250 were mutated into serine residues (20, 26). Cys-156 and Cys-206 of hAS3MT were proved to be the cysteine residues in the active site that bind to iAs3+ (20). The results of our MALDI-TOF mass spectroscopy experiment suggest that the disulfide bond reduced by the reductant is associated with Cys-250. Therefore, we concluded that the third active-site cysteine residue, which bound iAs3+ in the reaction, might be Cys-250, although a possible involvement of Cys-72 cannot be ruled out. The enzyme had no catalytic activity until the disulfide bond between Cys-250 and another cysteine residue was cleaved by reductant.
In conclusion, the methylation of arsenite by hAS3MT is a completely ordered mechanism that is of general interest in different reductive systems. The methyl transfer process occurs on hAS3MT, which agrees well with the views of Marapakala et al. (11) and Naranmandura et al. (34). In this process, arsenicals form thiol-arsenical complexes with the cysteine residues of hAS3MT, and the valence state of arsenic does not change. Although this mechanism is a successive methylation, arsenicals do not necessarily coordinate with GSH in the form of As-GSH complexes to facilitate the methylation reaction (23). The reductant cleaves the disulfide bonds of hAS3MT and exposes the active site cysteine residues for iAs3+ binding. Similarly, we further confirmed that the high β-pleated content increases the exposure of the active site on the surface of hAS3MT. The high dissociation ability of the enzyme-AdoMet-reductant intermediate might be beneficial to the binding of iAs3+ to the active site. This work used a rapid equilibrium kinetic model to analyze the reaction sequence of a real enzyme-catalyzed reaction and provided new insight into the mechanism of arsenite methylation catalyzed by hAS3MT. A deep understanding of the mechanism in vitro will guide further studies on the metabolism of arsenic in vivo in which GSH is a predominant endogenous reductant and assist in the endeavor to seek the antidotal pathway of arsenic poisoning.
Acknowledgment
We thank Dr. S. Lu for critical reading of the manuscript.
This work was supported by National Natural Science Foundation of China Grants 21075064, 21027013, 21021062, 21275072, and 21201101.

This article contains supplemental Materials.
- MMA
- monomethylated arsenical
- iAs
- inorganic arsenic
- iAs3+
- trivalent inorganic arsenic
- DMA
- dimethylated arsenical
- ATG
- arsenic triglutathione
- AS3MT
- arsenic (+3 oxidation state) methyltransferase
- hAS3MT
- human arsenic (+3 oxidation state) methyltransferase
- MADG
- monomethylarsonic diglutathione
- DAMG
- dimethylarsinic glutathione
- ATC
- arsenic tricysteine
- MADC
- monomethylarsonic dicysteine
- DAMC
- dimethylarsinic cysteine
- AdoMet
- S-adenosyl-l-methionine
- GSH
- glutathione
- TCEP
- tris(2-carboxyethyl)phosphine hydrochloride.
REFERENCES
- 1. Chen C. J., Chen C. W., Wu M. M., Kuo T. L. (1992) Cancer potential in liver, lung, bladder and kidney due to ingested inorganic arsenic in drinking water. Br. J. Cancer 66, 888–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tsai S. M., Wang T. N., Ko Y. C. (1999) Mortality for certain diseases in areas with high levels of arsenic in drinking water. Arch. Environ. Health 54, 186–193 [DOI] [PubMed] [Google Scholar]
- 3. Chen C. J., Hsueh Y. M., Lai M. S., Shyu M. P., Chen S. Y., Wu M. M., Kuo T. L., Tai T. Y. (1995) Increased prevalence of hypertension and long-term arsenic exposure. Hypertension 25, 53–60 [PubMed] [Google Scholar]
- 4. Tseng C. H., Chong C. K., Tseng C. P., Hsueh Y. M., Chiou H. Y., Tseng C. C., Chen C. J. (2003) Long-term arsenic exposure and ischemic heart disease in arseniasis-hyperendemic villages in Taiwan. Toxicol. Lett. 137, 15–21 [DOI] [PubMed] [Google Scholar]
- 5. Tapio S., Grosche B. (2006) Arsenic in the aetiology of cancer. Mutat. Res. 612, 215–246 [DOI] [PubMed] [Google Scholar]
- 6. Muñiz Ortiz J. G., Shang J., Catron B., Landero J., Caruso J. A., Cartwright I. L. (2011) A transgenic Drosophila model for arsenic methylation suggests a metabolic rationale for differential dose-dependent toxicity endpoints. Toxicol. Sci. 121, 303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thomas D. J., Styblo M., Lin S. (2001) The cellular metabolism and systemic toxicity of arsenic. Toxicol. Appl. Pharmacol. 176, 127–144 [DOI] [PubMed] [Google Scholar]
- 8. Thomas D. J., Waters S. B., Styblo M. (2004) Elucidating the pathway for arsenic methylation. Toxicol. Appl. Pharmacol. 198, 319–326 [DOI] [PubMed] [Google Scholar]
- 9. Thomas D. J., Nava G. M., Cai S. Y., Boyer J. L., Hernández-Zavala A., Gaskins H. R. (2010) Arsenic (+3 oxidation state) methyltransferase and the methylation of arsenicals in the invertebrate chordate Ciona intestinalis. Toxicol. Sci. 113, 70–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki K. T., Iwata K., Naranmandura H., Suzuki N. (2007) Metabolic differences between two dimethylthioarsenicals in rats. Toxicol. Appl. Pharmacol. 218, 166–173 [DOI] [PubMed] [Google Scholar]
- 11. Marapakala K., Qin J., Rosen B. P. (2012) Identification of catalytic residues in the As(III) S-adenosylmethionine methyltransferase. Biochemistry 51, 944–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Concha G., Vogler G., Nermell B., Vahter M. (2002) Intra-individual variation in the metabolism of inorganic arsenic. Int. Arch. Occup. Environ. Health 75, 576–580 [DOI] [PubMed] [Google Scholar]
- 13. Thomas D. J., Li J., Waters S. B., Xing W., Adair B. M., Drobna Z., Devesa V., Styblo M. (2007) Arsenic (+3 oxidation state) methyltransferase and the methylation of arsenicals. Exp. Biol. Med. 232, 3–13 [PMC free article] [PubMed] [Google Scholar]
- 14. Kligerman A. D., Doerr C. L., Tennant A. H., Harrington-Brock K., Allen J. W., Winkfield E., Poorman-Allen P., Kundu B., Funasaka K., Roop B. C., Mass M. J., DeMarini D. M. (2003) Methylated trivalent arsenicals as candidate ultimate genotoxic forms of arsenic: induction of chromosomal mutations but not gene mutations. Environ. Mol. Mutagen. 42, 192–205 [DOI] [PubMed] [Google Scholar]
- 15. Mass M. J., Tennant A., Roop B. C., Cullen W. R., Styblo M., Thomas D. J., Kligerman A. D. (2001) Methylated trivalent arsenic species are genotoxic. Chem. Res. Toxicol. 14, 355–361 [DOI] [PubMed] [Google Scholar]
- 16. Dopp E., Hartmann L. M., Florea A. M., von Recklinghausen U., Pieper R., Shokouhi B., Rettenmeier A. W., Hirner A. V., Obe G. (2004) Uptake of inorganic and organic derivatives of arsenic associated with induced cytotoxic and genotoxic effects in Chinese hamster ovary (CHO) cells. Toxicol. Appl. Pharmacol. 201, 156–165 [DOI] [PubMed] [Google Scholar]
- 17. Aposhian H. V., Aposhian M. M. (2006) Arsenic toxicology: five questions. Chem. Res. Toxicol. 19, 1–15 [DOI] [PubMed] [Google Scholar]
- 18. Thomas D. J. (2007) Molecular processes in cellular arsenic metabolism. Toxicol. Appl. Pharmacol. 222, 365–373 [DOI] [PubMed] [Google Scholar]
- 19. Thomas D. J. (2009) Unraveling arsenic-glutathione connections. Toxicol. Sci. 107, 309–311 [DOI] [PubMed] [Google Scholar]
- 20. Geng Z., Song X., Xing Z., Geng J., Zhang S., Zhang X., Wang Z. (2009) Effects of selenium on the structure and function of recombinant human S-adenosyl-l-methionine dependent arsenic (+3 oxidation state) methyltransferase in E. coli. J. Biol. Inorg. Chem. 14, 485–496 [DOI] [PubMed] [Google Scholar]
- 21. Song X., Geng Z., Li X., Hu X., Bian N., Zhang X., Wang Z. (2010) New insights into the mechanism of arsenite methylation with the recombinant human arsenic (+3) methyltransferase (hAS3MT). Biochimie 92, 1397–1406 [DOI] [PubMed] [Google Scholar]
- 22. Cullen W. R., McBride B. C., Reglinski J. (1984) The reaction of methyl arsenicals with thiols: some biological implications. J. Inorg. Biochem. 21, 179–193 [Google Scholar]
- 23. Hayakawa T., Kobayashi Y., Cui X., Hirano S. (2005) A new metabolic pathway of arsenite: arsenic-glutathione complexes are substrates for human arsenic methyltransferase Cyt19. Arch. Toxicol. 79, 183–191 [DOI] [PubMed] [Google Scholar]
- 24. Challenger F. (1945) Biological methylation. Chem. Rev. 36, 315–361 [Google Scholar]
- 25. Challenger F. (1951) Biological methylation. Adv. Enzymol. 12, 429–491 [DOI] [PubMed] [Google Scholar]
- 26. Song X., Geng Z., Li X., Zhao Q., Hu X., Zhang X., Wang Z. (2011) Functional and structural evaluation of cysteine residues in the human arsenic (+3 oxidation state) methyltransferase (hAS3MT). Biochimie 93, 369–375 [DOI] [PubMed] [Google Scholar]
- 27. Myllylä R., Tuderman L., Kivirikko K. I. (1977) Mechanism of the prolyl hydroxylase reaction. 2. Kinetic analysis of the reaction sequence. Eur. J. Biochem. 80, 349–357 [DOI] [PubMed] [Google Scholar]
- 28. Song X., Geng Z., Zhu J., Li C., Hu X., Bian N., Zhang X., Wang Z. (2009) Structure-function roles of four cysteine residues in the human arsenic (+3 oxidation state) methyltransferase (hAS3MT) by site-directed mutagenesis. Chem. Biol. Interact. 179, 321–328 [DOI] [PubMed] [Google Scholar]
- 29. Walton F. S., Waters S. B., Jolley S. L., LeCluyse E. L., Thomas D. J., Styblo M. (2003) Selenium compounds modulate the activity of recombinant rat AsIII-methyltransferase and the methylation of arsenite by rat and human hepatocytes. Chem. Res. Toxicol. 16, 261–265 [DOI] [PubMed] [Google Scholar]
- 30. Zhang L. Y., Xu X. L., Luo Z. F., Wu H., Shen D. K., Peng L. L., Liu Y. Z. (2010) Small-molecule reductants inhibit multicatalytic activity of AA-NADase from Agkistrodon acutus venom by reducing the disulfide-bonds and Cu(II) of enzyme. Biopolymers 93, 141–149 [DOI] [PubMed] [Google Scholar]
- 31. Alberty R. A. (2009) Determination of rapid-equilibrium kinetic parameters of ordered and random enzyme-catalyzed reaction A+B=P+Q. J. Phys. Chem. B 113, 10043–10048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alberty R. A. (2009) Determination of kinetic parameters of enzyme-catalyzed reaction a + B + C → products with the minimum number of velocity measurements. J. Phys. Chem. B 113, 1225–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kitchin K. T., Wallace K. (2005) Arsenite binding to synthetic peptides based on the Zn finger region and the estrogen binding region of the human estrogen receptor-α. Toxicol. Appl. Pharmacol. 206, 66–72 [DOI] [PubMed] [Google Scholar]
- 34. Naranmandura H., Suzuki N., Suzuki K. T. (2006) Trivalent arsenicals are bound to proteins during reductive methylation. Chem. Res. Toxicol. 19, 1010–1018 [DOI] [PubMed] [Google Scholar]
- 35. Sułkowska A., Bojko B., Równicka J., Sułkowski W. W. (2006) Paracetamol and cytarabine binding competition in high affinity binding sites of transporting protein. J. Mol. Struct. 792, 249–256 [Google Scholar]
- 36. Cui F., Zhang Q., Yao X., Luo H., Yang Y., Qin L., Qu G., Lu Y. (2008) The investigation of the interaction between 5-iodouracil and human serum albumin by spectroscopic and modeling methods and determination of protein by synchronous fluorescence technique. Pestic. Biochem. Physiol. 90, 126–134 [Google Scholar]
- 37. Zhang Y. Z., Zhou B., Liu Y. X., Zhou C. X., Ding X. L., Liu Y. (2008) Fluorescence study on the interaction of bovine serum albumin with p-aminoazobenzene. J. Fluoresc. 18, 109–118 [DOI] [PubMed] [Google Scholar]
- 38. Bi S., Song D., Tian Y., Zhou X., Liu Z., Zhang H. (2005) Molecular spectroscopic study on the interaction of tetracyclines with serum albumins. Spectrochim. Acta A Mol. Biomol. Spectrosc. 61, 629–636 [DOI] [PubMed] [Google Scholar]
- 39. Yang J. T., Wu C. S., Martinez H. M. (1986) Calculation of protein conformation from circular dichroism. Methods Enzymol. 130, 208–269 [DOI] [PubMed] [Google Scholar]
- 40. Zakharyan R. A., Aposhian H. V. (1999) Arsenite methylation by methylvitamin B12 and glutathione does not require an enzyme. Toxicol. Appl. Pharmacol. 154, 287–291 [DOI] [PubMed] [Google Scholar]
- 41. Tian J., Liu J., Hu Z., Chen X. (2005) Interaction of wogonin with bovine serum albumin. Bioorg. Med. Chem. 13, 4124–4129 [DOI] [PubMed] [Google Scholar]
- 42. Fomenko D. E., Xing W., Adair B. M., Thomas D. J., Gladyshev V. N. (2007) High-throughput identification of catalytic redox-active cysteine residues. Science 315, 387–389 [DOI] [PubMed] [Google Scholar]