

Background: 1,N6-(2-Hydroxy-3-hydroxymethylpropan-1,3-diyl)-2′-deoxyadenosine (1,N6-γ-HMHP-dA) adducts are formed in DNA by 1,2,3,4-diepoxybutane (metabolite of human carcinogen 1,3-butadiene).
Results: hpols η and κ carry out translesion synthesis, incorporating T, G, or A opposite the 1,N6-γ-HMHP-dA adduct.
Conclusion: Translesion bypass of 1,N6-γ-HMHP-dA adducts by TLS polymerases is error-prone.
Significance: This study identifies 1,N6-γ-HMHP-dA as the DNA adduct potentially responsible for A→T and A→C transversions and deletions induced by 1,3-butadiene.
Keywords: DNA Damage, DNA Polymerase, DNA Replication, DNA Sequencing, Mass Spectrometry (MS), Adduct, Bypass, Lesion, Mutations
Abstract
The 1,N6-(2-Hydroxy-3-hydroxymethylpropan-1,3-diyl)-2′-deoxyadenosine (1,N6-γ-HMHP-dA) adducts are formed upon bifunctional alkylation of adenine nucleobases in DNA by 1,2,3,4-diepoxybutane, the putative ultimate carcinogenic metabolite of 1,3-butadiene. The presence of a substituted 1,N6-propano group on 1,N6-γ-HMHP-dA is expected to block the Watson-Crick base pairing of the adducted adenine with thymine, potentially contributing to mutagenesis. In this study, the enzymology of replication past site-specific 1,N6-γ-HMHP-dA lesions in the presence of human DNA polymerases (hpols) β, η, κ, and ι and archebacterial polymerase Dpo4 was investigated. Run-on gel analysis with all four dNTPs revealed that hpol η, κ, and Dpo4 were able to copy the modified template. In contrast, hpol ι inserted a single base opposite 1,N6-γ-HMHP-dA but was unable to extend beyond the damaged site, and a complete replication block was observed with hpol β. Single nucleotide incorporation experiments indicated that although hpol η, κ, and Dpo4 incorporated the correct nucleotide (dTMP) opposite the lesion, dGMP and dAMP were inserted with a comparable frequency. HPLC-ESI-MS/MS analysis of primer extension products confirmed the ability of bypass polymerases to insert dTMP, dAMP, or dGMP opposite 1,N6-γ-HMHP-dA and detected large amounts of −1 and −2 deletion products. Taken together, these results indicate that hpol η and κ enzymes bypass 1,N6-γ-HMHP-dA lesions in an error-prone fashion, potentially contributing to A→T and A→C transversions and frameshift mutations observed in cells following treatment with 1,2,3,4-diepoxybutane.
Introduction
Cellular DNA is constantly damaged by physical and chemical factors, including reactive oxygen species, lipid peroxidation products, UV light, environmental pollutants, and dietary carcinogens (1). If not repaired, the resulting DNA adducts can block the progression of replicative DNA polymerases along the damaged strand (2, 3). In this case, a specialized group of polymerases, translesion synthesis polymerases (TLS),6 are recruited to the damaged site and can carry out DNA polymerization across the lesion (4, 5). Five TLS DNA polymerases are primarily involved in translesion synthesis in humans as follows: hpol η, hpol ι, hpol κ, and Rev1 from the Y family and hpol ζ from the B family of polymerases (6–8). The archebacterial translesion DNA polymerase from Sulfolobus solfataricus P2 (Dpo4) has been widely used as a model TLS polymerase for kinetic and structural studies of DNA lesions due its ready availability and its functional similarity to human hpol η (9, 10). This damage tolerance mechanism allows the cell to overcome replication blocks induced by bulky lesions such as UV-induced thymine dimers, facilitating cell survival. However, TLS polymerases are less catalytically efficient than replicative DNA polymerases and have a relatively low fidelity due to an increased size and flexibility of their active sites (11–15).
An important DNA lesion that occurs upon exposure to the human carcinogen 1,3-butadiene (BD) is 1,N6-(2-hydroxy-3-hydroxymethylpropan-1,3-diyl)-2′-deoxyadenosine (1,N6-γ-HMHP-dA) and its isomer 1,N6-(1-hydroxymethyl-2-hydroxypropan-1,3-diyl)-2′-deoxyadenosine (1,N6-α-HMHP-dA) (Scheme 1) (16). BD is an important industrial chemical widely used in the production of synthetic rubber, resins, and polymers, as well as an environmental contaminant present in cigarette smoke, automobile exhaust, and urban air (17). BD is metabolically activated to 1,2,3,4-diepoxybutane (DEB), which can alkylate adenine residues of DNA to give 1,N6-HMHP-dA (16). HPLC-ESI+-MS/MS analysis of DEB-treated calf thymus DNA revealed a concentration-dependent formation of 1,N6-α-HMHP-dA and 1,N6-γ-HMHP-dA adducts (16). Under physiological conditions, the two lesions interconvert via a Dimroth rearrangement-like mechanism (16). Both isomers of 1,N6-HMHP-dA have been detected in liver, lung, and kidney tissues of laboratory mice exposed to 6.25–625 ppm BD (18). Although these lesions are less abundant than guanine monoadducts and guanine-guanine cross-links of DEB, they are persistent in mouse tissues for at least 10 days, suggesting that they may contribute to the biological effects of BD (19).
SCHEME 1.

Structures of 1,N6-γ-HMHP-dA and 1,N6-α-HMHP-dA adducts formed by 1,2,3,4-diepoxybutane.
Because both the N6 and the N-1 positions of adenine in 1,N6-HMHP-dA adducts are blocked by the HMHP exocycle, they can no longer be used to form complementary hydrogen bonds with dT. Structurally related 1,N6-ethenodeoxyadenosine (1,N6-ϵdA) adducts are known to adopt the syn conformation about the glycosidic bond, allowing them to mispair with protonated dC in the active site of human DNA polymerase ι (20). We therefore hypothesized that 1,N6-HMHP-dA adducts can similarly form Hoogsteen base pairs with dG and protonated dA, potentially leading to A→C and A→T transversions, respectively. Such mispairing could explain the mutational spectra observed upon treatment of cells and laboratory animals with BD and DEB (21–23).
The main aim of this study was to investigate the ability of human TLS polymerases to carry out DNA synthesis past site-specific (R,S)-1,N6-γ-HMHP-dA lesions. We have determined steady-state kinetic parameters for nucleotide insertion opposite the lesion and identified primer extension products following in vitro replication in the presence of polymerases κ, η, ι, and Dpo4. Our results indicate that hpol κ, hpol η, and Dpo4 are able to bypass (R,S)-1,N6-γ-HMHP-dA lesions and to extend the primer beyond the damaged site by incorporating either correct (T) or incorrect base (A or G) opposite the adduct, along with introducing −1 and −2 frameshift mutations. In contrast, replicative hpol β is completely blocked by the lesion, and hpol ι inserts the correct nucleotide but is unable to extend past the lesion site.
EXPERIMENTAL PROCEDURES
Materials
Full-length recombinant hpol η, hpol κ, and hpol ι enzymes used for steady-state kinetics experiments were obtained from Enzymax (Lexington, KY). Recombinant human DNA pol κ (amino acids 19–526), recombinant human DNA pol ι (amino acids 1–420), and recombinant human DNA pol η (amino acids 1–437) employed in full-length primer extension and mass spectrometry experiments were expressed and purified as described previously (24–26). Dpo4 polymerase was expressed and purified by Professor Michael Stone (Vanderbilt University) as per the previously published methodology (10). hpol β was purchased from Trevigen (Gaithersburg, MD). T4 polynucleotide kinase and Escherichia coli uracil DNA glycosylase (UDG) were obtained from New England Biolabs (Beverly, MA). [γ-32P]ATP was purchased from PerkinElmer Life Sciences. The unlabeled dNTPs were obtained from Omega Bio-Tek (Norcross, GA). 40% 19:1 acrylamide/bis solution was purchased from Bio-Rad. All the other chemicals and reagents were obtained from Sigma and Fisher. Urea, Sigmacote, Tris, boric acid, ammonium acetate, formamide, BSA, DTT, magnesium chloride, and N,N,N′,N′-tetramethylethylenediamine were purchased from Sigma. Ammonium persulfate, acetonitrile, and EDTA were obtained from Fisher.
Oligonucleotide Synthesis
The 18-mer oligodeoxynucleotide templates containing site- and stereospecific (R,S)-1,N6-γ-HMHP-dA lesions at the 5th position (5′-TCATXGAATCCTTCCCCC-3′) were synthesized by the post-oligomerization methodology developed in our laboratory (27). Briefly, 18-mer oligodeoxynucleotides containing site-specific 6-chloropurine at position X were coupled with (R,R)-N-Fmoc-1-amino-2-hydroxy-3,4-epoxybutane to generate the corresponding oligomers containing site- and stereospecific (R,R)-N6-(2-hydroxy-3,4-epoxybut-1-yl) adenine (N6-HEB-dA) adducts. N6-HEB-dA containing oligomers were isolated by HPLC and incubated in water at room temperature to allow the cyclization of (R,R)-N6-HEB-dA to (R,S) 1,N6-γ-HMHP-dA (27). The corresponding unmodified 18-mer (5′-TCATAGAATCCTTCCCCC-3′) and 13-mer primers (5′-GGGGGAAGGATTC-3′ and 5′-GGGGGAAGGAUTC-3′) were purchased from Integrated DNA Technologies (Coralville, IA). All DNA strands were purified by HPLC, characterized by HPLC-ESI−-MS/MS, and quantified by UV spectrophotometry.
Generation of Primer-Template DNA Substrate for in Vitro Assays
The 13-mer oligodeoxynucleotide primer (5′-GGGGGAAGGATTC-3′) was 5′-end-labeled in the presence of [γ-32P]ATP (>6000 Ci/mmol) and T4 polynucleotide kinase in 50 mm Na-MOPS buffer (pH 7.5), 10 mm MgCl2, and 5 mm DTT for 1 h at 37 °C. Excess [γ-32P]ATP was removed by gel filtration through Bio-Spin 6 filters (Bio-Rad). The 32P-end-labeled primer was annealed to an equimolar amount of 18-mer oligodeoxynucleotide templates containing either dA or 1,N6-γ-HMHP-dA (1:1 molar ratio) by heating at 95 °C for 3 min in the presence of 40 mm NaCl, followed by slow cooling overnight.
Reaction Conditions for Enzyme Assays
Primer extension assays and steady-state kinetics experiments were conducted for each of the five DNA polymerases (hpol β, hpol η, hpol κ, hpol ι, and Dpo4). The assays were conducted at 37 °C in buffered solutions containing 50 mm Na-MOPS (pH 7.5), 50 mm NaCl, 5 mm DTT, 100 μg/ml BSA, 10% glycerol (v/v) with 50 nm radiolabeled primer-template complexes, and 5 nm polymerase enzymes (except for Dpo4, which was used at 25 nm and hpol β, 2 units). The reactions were initiated by the addition of individual nucleotides or a mixture of all four dNTPs and MgCl2 (final concentration, 5 mm). The reactions were stopped after pre-selected time intervals by the addition of 36 μl of stop solution (95% formamide (w/v), 10 mm EDTA, 0.03% bromphenol blue (w/v), 0.03% xylene cyanol (w/v)) to a 4-μl aliquot of the sample.
Primer Extension Assays
32P-End-labeled primer-template complexes containing either unmodified dA or 1,N6-γ-HMHP-dA at position X of the template strand were incubated with DNA polymerases as described above in the presence of all four dNTPs (500 μm) for 0–60 min. The reaction products were separated using a 20% (w/v) denaturing polyacrylamide gel containing 7 m urea at a constant voltage (2500 V) for 3 h. The radioactive products were visualized using a phosphorimager (Bio-Rad).
Steady-state Kinetics Analyses
32P-End-labeled primer-template complexes were incubated with TLS polymerases in the presence of increasing concentrations of individual dNTPs (0–800 μm) for 0–60 min. The resulting oligodeoxynucleotide products were separated by gel electrophoresis as described above and visualized with a phosphorimager. DNA amounts in each band were quantified using Quantity One image software (Bio-Rad), and the steady-state kinetic values were determined by plotting product formation versus dNTP concentration using nonlinear regression analysis (one-site hyperbolic fits in GraphPad Prism).
HPLC-ESI-MS/MS Analysis of Primer Extension Products from DNA Polymerase Reactions
Oligodeoxynucleotide 18-mers (5′-TCATXGAATCCTTCCCCC-3′, where X = dA or (R,S)-1,N6-γ-HMHP-dA) were annealed to the 13-mer primer (5′-GGGGGAAGGAUTC-3′) (100 pmol each) in 20 μl of 50 mm NaCl to form primer-template complexes. The resulting partial duplexes were incubated with individual DNA polymerases (hpol κ, 40 pmol; hpol η, 40 pmol; or Dpo4, 100 pmol) in 50 mm Tris-HCl (pH 7.8) buffer containing 5% glycerol, 5 mm DTT, 5 mm MgCl2, 100 μg/ml BSA at 37 °C for 4–6 h. Primer extension reactions were initiated by the addition of all four dNTPs (1 mm each) in a final reaction volume of 75 μl. The reactions were terminated by removing excess dNTPs by size exclusion using a Bio-Spin 6 column (Bio-Rad). Tris-HCl, DTT, and EDTA were added to the filtrate to restore their concentrations to 50, 5, and 1 mm, respectively.
To facilitate MS/MS analyses, oligodeoxynucleotide products were cleaved into smaller fragments by incubation with UDG (6 units, 37 °C for 6 h), followed by heating with 0.25 m piperidine at 95 °C for 1 h (28). The final solution was dried under vacuum, and the residue was reconstituted in 25 μl of water. The 14-mer oligonucleotide (5′p-CTTCACGAGCCCCC-3′; 40 pmol) was added as an internal standard for mass spectrometry. The injection volume for mass spectrometric analysis was 8 μl. Primer extension products following reactions catalyzed by hpol β (10 units) and hpol ι (80 pmol) were not cleaved with UDG/piperidine because only short extension products were observed that could be directly sequenced by MS/MS.
HPLC-ESI−-MS/MS analysis was conducted on an Eksigent HPLC system (Eksigent, Dublin, CA) interfaced to a Thermo LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Waltham, MA). The instrument was operated in the negative ion ESI MS/MS mode. Polymerase extension products were separated with an Agilent Zorbax SB 300 C18 (0.5 × 150 mm, 5 μm) column using a gradient of 15 mm ammonium acetate (Buffer A) and acetonitrile (Buffer B). The column was eluted at a flow rate of 15 μl/min. The solvent composition was linearly changed from 1 to 10% Buffer B in 18 min, further to 75% B in 2 min, held at 75% B for 3 min, and brought back to 1% B in 2 min and held at 1% B for 10 min. Under these conditions, all DNA oligodeoxynucleotides, including extension products, eluted between 10 and 13 min.
Electrospray ionization conditions were as follows: ESI source voltage, 3.5 kV; source current, 6.7 μA; auxiliary gas flow rate setting, 0; sweep gas flow rate setting, 0; sheath gas flow setting, 30; capillary temperature, 275 °C; and S-lens RF level, 50%. The most abundant ions from the ESI−-FTMS spectra were selected and subjected to collision-induced dissociation (CID) analysis using the linear ion trap. The MS/MS conditions were as follows: normalized collision energy, 35%; activation Q, 0.250; activation time, 10 ms; product ion scan range, m/z 300–2000. The amount of each extension product was calculated by comparing peak areas corresponding to each product in extracted ion chromatograms with the peak area of the internal standard. Expected CID fragmentation patterns of oligonucleotides were obtained using the Mongo Oligo mass calculator version 2.06 available from the Mass Spectrometry Group of Medicinal Chemistry, University of Utah.
RESULTS
Primer Extension Studies in the Presence of All Four dNTPs
Our initial studies investigated the ability of DNA polymerases to bypass 1,N6-γ-HMHP-dA lesions in the presence of all four dNTPs. 1,N6-γ-HMHP-dA was site-specifically incorporated into an 18-mer template (5′-TCATXGAATCCTTCCCCC-3′, where X = 1,N6-γ-HMHP-dA), which was annealed to a 13-mer primer (5′-GGGGGAAGGATTC-3′). In the resulting duplex (Scheme 2), the primer terminus is positioned immediately prior to the lesion site. The primer-template complexes were subjected to in vitro replication in the presence of hpol β, η, κ, ι, and Dpo4. Control experiments with template containing only native nucleotides (X = dA) confirmed a complete primer extension by hpol β, hpol κ, hpol η, and Dpo4, to form an 18-mer product (left panels in Figs. 1A and 2). In addition, some 19-mer products have also been observed with Dpo4 (Fig. 2C, left panel), which could be attributed to template-independent nucleotide incorporation (blunt end addition) as observed previously (29, 30). pol ι formed primarily a 16-mer product (Fig. 1B) due to its low processivity as compared with other Y family polymerases (31–34).
SCHEME 2.

Sequences of DNA substrates employed in primer extension assays.
FIGURE 1.
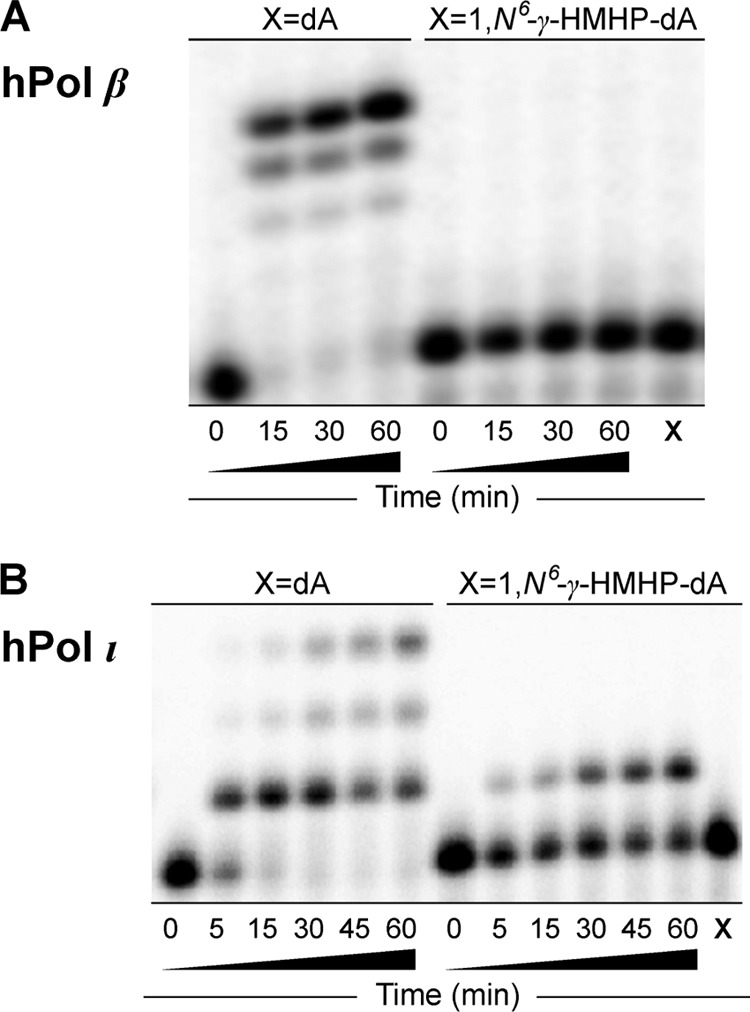
Extension of 32P-labeled primer opposite templates containing unmodified dA or 1,N6-γ-HMHP-dA adduct by hpol β (A) and hpol ι (B). The 13-mer primers (Scheme 2) were annealed with 18-mer template containing either unmodified dA or 1,N6-γ-HMHP-dA adduct. The resulting primer-template complexes (50 nm) were incubated at 37 °C in the presence of hpol β (2 units) or hpol ι (5 nm). Reactions were started by the addition of all four dNTPs (500 μm) and quenched at the indicated time points. The extension products were separated by 20% (w/v) denaturing PAGE and visualized by phosphorimaging analysis. X represents the migration of the starting primer.
FIGURE 2.
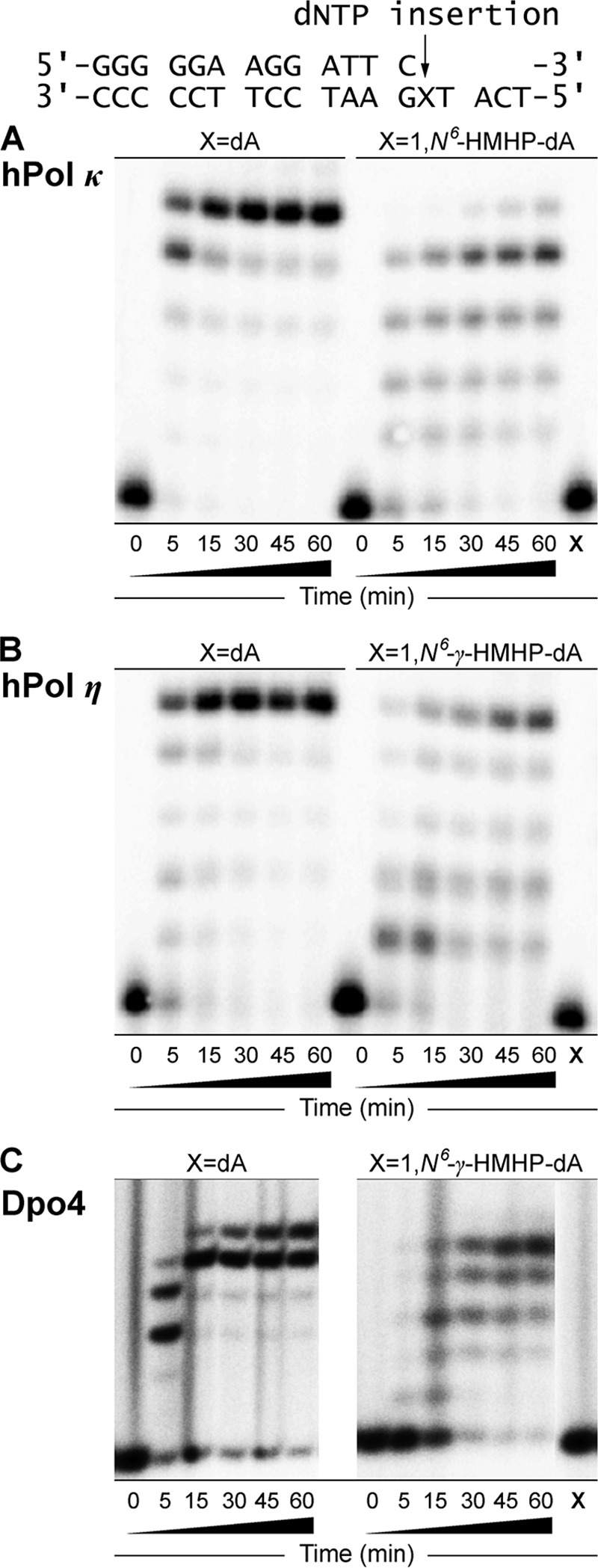
Extension of 32P-labeled primers opposite templates containing unmodified dA or 1,N6-γ-HMHP-dA adduct by hpol κ (A), hpol η (B), and Dpo4 (C). The 13-mer primers (Scheme 2) were annealed with 18-mer template containing either unmodified dA or 1,N6-γ-HMHP-dA adduct. The resulting primer-template complexes (50 nm) were incubated in the presence of hpol κ, hpol η (5 nm), or Dpo4 (25 nm). The polymerase reactions were started by the addition of the four dNTPs (500 μm) and quenched at the indicated time points. The quenched samples were separated by 20% (w/v) denaturing PAGE and visualized by phosphorimaging analysis. X represents the migration of the starting primer.
The presence of 1,N6-γ-HMHP-dA at position X led to a complete blockage of primer extension by hpol β, whereas hpol ι inserted a single nucleotide but was unable to extend past the lesion site (right panels in Fig. 1). In contrast, hpol η and hpol κ were able to extend the primer to the terminus, forming the expected 18-mer products (right panels in Fig. 2). The efficiency of primer extension was markedly reduced in the presence of 1,N6-γ-HMHP-dA as compared with unmodified dA, as evident by the presence of incomplete extension products (15-, 16-, and 17-mers). No primer extension was observed in Dpo4 reactions with 1,N6-γ-HMHP-dA-containing DNA template until the enzyme/DNA ratio was increased to 1:2.
To determine whether the incomplete extension products generated by hpol ι (which is stalled after the insertion of dTMP opposite the 1,N6-γ-HMHP-dA adduct, Fig. 1B) can be extended by hpol η or κ, 32P-end-labeled primer-template complexes were first incubated in the presence of hpol ι, resulting in the extension of primer by one base to form a 14-mer product. Upon subsequent addition of hpol κ to the above reaction mixture, the 14-mer product was completely extended to form the expected 18-mer product (Fig. 3). Similar results were obtained in the presence of hpol η (data not shown). This provides preliminary evidence for possible cooperativity of TLS polymerases during the replication of 1,N6-γ-HMHP-dA-containing DNA, with hpol ι adding one nucleotide across the damaged base, polymerase switching, and hpol η or κ completing the bypass synthesis.
FIGURE 3.

Cooperativity of human TLS polymerases ι and κ during the bypass of 1,N6-γ-HMHP-dA adduct. Primer-template complex containing 1,N6-γ-HMHP-dA (50 nm) was incubated with hpol ι (10 nm) for 0–30 min. After 30 min, 5 nm of hpol κ was added to the reaction mixture to determine whether the primer extension started by hpol ι and stalled by the adduct can be completed by hpol κ. The extension products were resolved by PAGE.
Steady-state Kinetic Analysis of dNTP Incorporation Opposite 1,N6-γ-HMHP-dA Adducts
Single nucleotide insertion assays were conducted to compare the rates of incorporation of each nucleotide opposite 1,N6-γ-HMHP-dA lesion by individual TLS DNA polymerases. Primer-template complexes containing 1,N6-γ-HMHP-dA or unmodified dA (control) were incubated with polymerase enzymes in the presence of each of the four individual dNTPs. Our pilot experiments have revealed that hpol η, hpol κ, and Dpo4 incorporate dTMP, dAMP, and dGMP, but not dCMP, opposite the 1,N6-γ-HMHP-dA lesion (Fig. 4). hpol ι preferentially inserted dTMP opposite the lesion. Subsequently, the rates of incorporation of dAMP, dGMP, and dTMP opposite 1,N6-γ-HMHP-dA by hpol η, hpol κ, and Dpo4 were determined in kinetic experiments. No kinetic analysis was possible for hpol β, because it was completely blocked by 1,N6-γ-HMHP-dA (Fig. 1). In the case of hpol ι, the kinetics experiments were limited to dTMP, because no incorporation of other nucleotides was observed. The insertion rate for each nucleotide was calculated by plotting reaction velocity against dNTP concentration. The kinetic results are summarized in Table 1, where the catalytic specificity constant (kcat/Km) provides a measure of catalytic efficiency for each dNTP insertion, and the misinsertion frequency ( f) reflects the probability of insertion of an incorrect dNTP as compared with that of the correct one (dTTP).
FIGURE 4.

Single nucleotide insertion by hpol η (A) and hpol κ (B) opposite unmodified dA (control) and 1,N6-γ-HMHP-dA. Primer-template complexes (50 nm) were incubated with polymerases (5 nm) in the presence of specific dNTPs (500 μm) for 0–30 min at 37 °C. The reaction mixtures were quenched and separated by 20% PAGE.
TABLE 1.
Steady-state kinetic parameters for single nucleotide incorporation opposite dA and 1,N6-γ-HMHP-dA adduct by hpol κ, hpol η, and Dpo4
f = misinsertion frequency = (kcat/Km)incorrect dNTP/(kcat/Km)correctdNTP (dTTP).
| Polymerase | Template | Incoming nucleotide | kcat | Km | kcat/Km) | f |
|---|---|---|---|---|---|---|
| min−1 | μm | (μm−1 min−1) | ||||
| hpol κ | dA | dTTP | 0.3 ± 0.01 | 0.8 ± 0.1 | 0.38 | 1 |
| dATP | 0.06 ± 0.005 | 20 ± 4.6 | 0.003 | 0.008 | ||
| dGTP | 0.27 ± 0.01 | 73 ± 10.4 | 0.004 | 0.01 | ||
| 1,N6-γ-HMHP-dA | dTTP | 0.3 ± 0.02 | 4.0 ± 1.0 | 0.07 | 1 | |
| dATP | 0.2 ± 0.004 | 13 ± 3.5 | 0.01 | 0.2 | ||
| dGTP | 1.8 ± 0.008 | 35 ± 7.4 | 0.05 | 0.7 | ||
| hpol η | dA | dTTP | 1.6 ± 0.1 | 3.2 ± 0.9 | 0.5 | 1 |
| dATP | 0.04 ± 0.002 | 5.6 ± 2.3 | 0.007 | 0.01 | ||
| dGTP | 0.09 ± 0.02 | 35 ± 25 | 0.002 | 0.004 | ||
| 1,N6-γ-HMHP-dA | dTTP | 0.6 ± 0.03 | 4.4 ± 1.2 | 0.14 | 1 | |
| dATP | 0.7 ± 0.03 | 9.2 ± 2.2 | 0.08 | 0.57 | ||
| dGTP | 0.9 ± 0.04 | 10 ± 2.0 | 0.09 | 0.65 | ||
| Dpo4 | dA | dTTP | 0.6 ± 0.03 | 2.8 ± 0.4 | 0.21 | 1 |
| dATP | 0.05 ± 0.005 | 471 ± 112 | 0.00016 | 0.0004 | ||
| 1,N6-γ-HMHP-dA | dTTP | 0.35 ± 0.04 | 979 ± 194 | 0.00035 | 1 | |
| dATP | 0.14 ± 0.05 | 884 ± 272 | 0.00016 | 0.45 | ||
| dGTP | 0.07 ± 0.004 | 596 ± 77 | 0.00011 | 0.31 | ||
| hpol ι | dA | dTTP | 1.6 ± 0.2 | 21 ± 7 | 0.08 | 1 |
| 1,N6-γ-HMHP-dA | dTTP | 0.06 ± 0.004 | 4.6 ± 1.5 | 0.01 | 1 |
Our kinetic data indicate that hpol η, hpol κ, and hpol ι polymerization rates for primer-template complexes containing 1,N6-γ-HMHP-dA were 3–8-fold slower than for the control templates containing unmodified dA (Table 1). In the case of Dpo4, the efficiency of incorporation of correct base (dTMP) opposite 1,N6-γ-HMHP-dA was 600-fold lower than dT incorporation opposite unmodified dA. The preference order for the insertion of individual dNTPs opposite the 1,N6-γ-HMHP-dA adduct by hpol η was T > G > A, with ∼1.5-fold preference for incorporation of dTMP as compared with dGMP and dAMP (Table 1). A similar specificity (T > G > A) was observed for hpol κ, which was 1.5-fold more likely to incorporate dTMP rather than dGMP opposite the lesion and 5-fold more likely to insert dTMP rather than dAMP (Table 1). The preference order for Dpo4 was T > A > G, and this polymerase was 2-fold more likely to incorporate dTMP rather than dAMP and 3-fold more likely to incorporate dTMP rather than dGMP (Table 1).
LC-MS/MS Analysis of Primer Extension Products
A mass spectrometry-based strategy was employed to confirm the identities of nucleotides incorporated opposite 1,N6-γ-HMHP-dA adduct by various DNA polymerases and to detect any insertion/deletion events. Synthetic primer-template complexes were incubated with individual polymerases in the presence of all four dNTPs, and the primer extension products were sequenced by capillary HPLC-ESI−-MS/MS using an LTQ Orbitrap Velos instrument. Because of the high sensitivity of this method, 100 pmol of DNA was sufficient to obtain sequence information for the extension products.
Parallel reactions were conducted with unmodified and 1,N6-γ-HMHP-dA-containing primer-template complexes (Scheme 2, bottom). To facilitate MS analysis of the products, one of the thymine residues within the primer was substituted for uridine (5′-GGGGGAAGGAUTC-3′) to allow for selective cleavage of the extension products with UDG/hot piperidine. This generates oligonucleotides (5-, 6-, or 7-mers) that are short enough to be readily sequenced by HPLC-ESI-MS/MS (10). However, the extension products from hpol β- and hpol ι-catalyzed reactions were not cleaved because these polymerases generated short products by adding 0–1 nucleotides (Fig. 1).
Each primer extension reaction mixture was analyzed twice. First, samples were analyzed by full scan HPLC-ESI−-FTMS to detect the molecular ions of the major extension products (Fig. 5). The same samples were then re-analyzed in the HPLC-ESI−-MS/MS mode (Fig. 6). MS/MS spectra of the observed oligodeoxynucleotide products (Figs. 7 and 8) were compared with the predicted CID spectra of each extension product to determine their nucleobase sequence (supplemental Tables S1–S6).
FIGURE 5.

HPLC-ESI−-MS/MS analysis of hpol κ in vitro replication products on 1,N6-γ-HMHP-dA-adduct containing template. Primer-template complexes (100 pmol) were incubated at 37 °C for 4 h in the presence of hpol κ (40 pmol) and the four dNTPs. The primer extension products were subjected to UDG hydrolysis and piperidine treatment to yield shorter oligonucleotide fragments that can be sequenced by HPLC-ESI-MS/MS. A, Total Ion Chromatogram of primer extension mixtures; B, mass spectra of the primer extension products eluting between 10 and 13 min.
FIGURE 6.
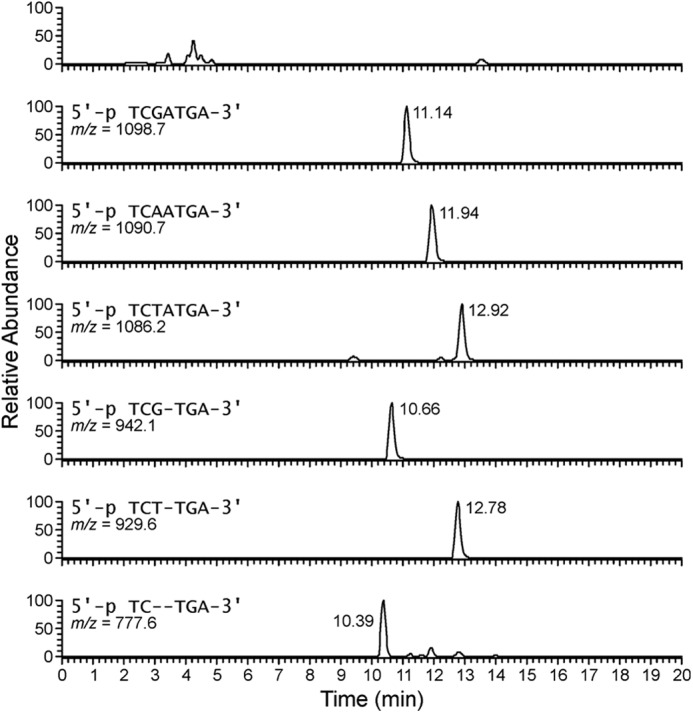
Extracted ion chromatograms of the six major polymerase extension products formed upon in vitro replication on 1,N6-γ-HMHP-dA- adduct template by hpol κ (Fig. 5).
FIGURE 7.

MS/MS spectra of the extension products observed following in vitro replication of 1,N6-γ-HMHP-dA-containing template by hpol κ. A, CID spectrum of the extension product 5′-pTCGATGA-3′. B, CID spectrum of the extension product 5′-pTCAATGA-3′. C, CID spectrum of the extension product 5′-pTCTATGA-3′.
FIGURE 8.

CID spectra of the −1 and −2 deletion products observed following in vitro replication of 1,N6-γ-HMHP-dA-containing template by hpol κ. A, CID spectrum of the product 5′-pTCG_TGA-3′. B, CID spectrum of the extension product 5′-pTCT_TGA-3′. C, CID spectrum of the extension product 5′-pTC__TGA-3′.
HPLC-ESI−-MS/MS analysis of the hpol β reactions with unmodified primer-template complex and all four dNTPs revealed the presence of the fully extended primer (5′-GGGGGAAGGAUTCTATGA-3′; m/z 1015.17; [M − 4H]4−) as the major product (>95%) (data not shown). This is consistent with our previous observation (Fig. 1A, left panel) that in the absence of the lesion the primer is completely extended by hpol β. In contrast, the corresponding reaction mixtures from hpol β extension of 1,N6-γ-HMHP-dA containing primer-template complexes contained mainly the unextended primer (5′-GGGGGAAGGAUTC-3′; m/z 1406.23; [M − 4H]4−) (results not shown). These results indicate that primer extension by human pol β is completely blocked by the 1,N6-γ-HMHP-dA lesion, in accordance with our previous data from the gel electrophoresis analysis (Fig. 1A, right panel).
HPLC-ESI−-MS/MS analysis of the primer extension products generated upon incubation of 1,N6-γ-HMHP-dA containing primer-template complexes with hpol κ has revealed six major peaks at m/z as follows: 777.6, 929.6, 942.1, 1086.2, 1090.7, and 1098.7, which correspond to the doubly charged ions of 5′-pTC__TGA-3′, 5′-pTCT_TGA-3′, 5′-pTCG_TGA-3′, 5′-pTCTATGA-3′, 5′-pTCAATGA-3′, and 5′-pTCGATGA-3′, respectively (Scheme 3 and Fig. 6). CID spectra of each extension product were obtained to determine their exact nucleobase sequence (Figs. 7 and 8). This sequence information cannot be obtained from molecular weight only. For instance, the molecular weight of the doubly charged ions at m/z 1086.2 (M = 2175.4) is consistent with an oligonucleotide product containing three Ts, two As, one C, and one G. Extension products 5′-pTCTATGA-3′ (insertion of T opposite the lesion and A opposite the next base) and 5′-pTCATTGA-3′ (insertion of A opposite the lesion and T opposite the next base) are indistinguishable by gel electrophoresis or by molecular weight. By comparing the CID spectra obtained from MS/MS analysis with the predicted CID fragments (a-B and w ions, Fig. 7C andsupplemental Table S3), the sequence of the product was determined as 5′p-TCTATGA-3′, with the correct base (T) inserted opposite the adduct. Detailed MS/MS analysis was conducted for each of the hpol κ extension products detected by HPLC-ESI−-FTMS. According to HPLC-ESI−-FTMS peak areas, 18% of the extension products were error-free, with the incorporation of correct base (T) opposite the lesion (Scheme 3). Approximately 21 and 25% of the extension products were formed by misincorporation of dAMP and dGMP opposite the lesion, respectively. In addition, three deletion products were detected. 5′-pTCG_TGA-3′ (22%) was formed as a result of misinsertion of G opposite 1,N6-γ-HMHP-dA and deletion of the next base. 5′-pTCT_TGA-3′ (13%) was formed upon incorporation of the correct base (T) opposite the lesion and skipping the next base. A minor −2 deletion product, 5′-pTC__TGA-3′ (< 1%), was also detected, in which no base was incorporated opposite the lesion or the 3′-neighboring base. These results are summarized in Scheme 3. Overall, our HPLC-ESI−-FTMS results for hpol κ are consistent with the gel electrophoresis data, but they revealed several additional products generated upon deletion of one or two bases.
SCHEME 3.

Summary of primer extension products formed by hpol κ as identified by capillary HPLC-ESI-MS/MS.
HPLC-ESI−-MS/MS analysis of the primer extension products from hpol η-catalyzed reactions revealed the same six major products as observed with hpol κ (5′-pTC__TGA-3′, 5′-pTCT_TGA-3′, 5′-pTCG_TGA-3′, 5′-pTCTATGA-3′, 5′-pTCAATGA-3′, and 5′-pTCGATGA-3′), but their relative contributions were entirely different (Scheme 4). Error-free replication past the adduct (5′-pTCTATGA-3′) accounted for over 80% of the total products. The relative yields of 5′-pTCGATGA-3′ and 5′-pTCAATGA-3′ corresponding to the incorporation of incorrect bases, G and A, opposite the lesion, accounted for <10%, whereas single deletion products, 5′-pTCG_TGA-3′ and 5′-pTCT_TGA-3′, accounted for <5%. The double deletion product (5′-pTC__TGA-3′) was also detected, with the relative yield of <1%. These results are summarized in Scheme 4. Taken together, these results indicate that hpol η makes fewer errors than does hpol κ upon replicating 1,N6-γ-HMHP-dA containing DNA.
SCHEME 4.

Summary of primer extension products formed by hpol η as identified by capillary HPLC-ESI-MS/MS.
HPLC-ESI-FTMS analysis of hpol ι extension products of 1,N6-γ-HMHP-dA containing 18-mer oligonucleotide primer-template complexes revealed the formation of a single product with an m/z of 1091.20 (data not shown). CID fragmentation of m/z 1091.20 confirmed that the product was 5′-GGGGGAAGGAUTCT-3′, which is formed upon incorporation of the correct base (T) opposite the lesion, but no further extension by the enzyme (results not shown). As discussed above, UDG/piperidine cleavage was not employed for hpol ι-generated products due to their relatively short length, enabling their direct sequencing by MS/MS. These results confirm that human hpol ι is able to insert the correct base (T) opposite 1,N6-γ-HMHP-dA, but it is unable to extend the primer beyond the damaged site. This result is consistent with our gel electrophoresis results (see Fig. 1B above).
HPLC-ESI−-MS/MS analysis of the Dpo4 primer extension mixtures has revealed the presence of five major DNA peaks at m/z 929.6, 934.2, 1086.2, 1090.7, and 1098.7 (Scheme 5). The nucleobase sequences of these products were determined by MS/MS as described above. We found that the major products were 5′-pTCTATGA-3′ (59%) and 5′-pTCAATGA-3′ (30%), corresponding to error-free replication and the misinsertion of A opposite the lesion, respectively. A low abundance product, 5′-pTCGATGA-3′ (4%) formed by the misinsertion of G, was also observed. Analogous to our results for hpol κ, a single nucleotide deletion product 5′-pTCT_TGA-3′ (5%) was found. A novel deletion product, 5′-pTCA_TGA-3′ (2%), was formed by misinsertion of A opposite the adduct, followed by skipping of the neighboring 3′ base. These results are summarized in Scheme 5.
SCHEME 5.

Summary of primer extension products formed by Dpo4 as identified by capillary HPLC-ESI-MS/MS.
DISCUSSION
DEB is a key carcinogenic metabolite of BD, an important industrial and environmental chemical present in urban air and in cigarette smoke. DEB is considered the ultimate carcinogenic metabolite of BD due to its potent genotoxicity (17). Studies in the HPRT gene have revealed that DEB induces a large number of A→T mutations and deletions, along with smaller numbers of A→G and A→C base substitutions (21, 35). Furthermore, exposure of Rat2 laci transgenic cells and human TK6 lymphoblasts to DEB induced an increased frequency of A→T transversions and partial deletions (23).
Despite significant efforts, specific DNA adducts responsible for DEB-induced genetic changes have not been previously identified. DEB-mediated DNA alkylation gives rise to a complex mixture of nucleobase adducts, including 2,3,4-trihydroxybut-1-yl (THB) monoadducts, 2,3-dihydroxybutane-1,4-yl DNA-DNA cross-links, DNA-protein lesions, and exocyclic dG and dA adducts (22, 36–39). Polymerase bypass studies of synthetic DNA templates containing N6-THB-adenine monoadducts revealed essentially error-free replication, producing very low levels of A→G and A→C base substitutions (<0.3%) (22). N2-Trihydroxybutylguanine lesions completely blocked DNA replication (37). The N1-trihydroxybutane-deoxyinosine lesions originating from deamination of the corresponding N1-dA adducts induced high numbers of A→G transitions (38). Similar studies conducted with putative N2-guanine and N6-adenine intrastrand DNA-DNA cross-links have revealed that bis-N2G-BD cross-links are not bypassed by DNA polymerases, whereas bis-N6A-BD lesions induce A→G base substitutions at the 3′-base (36, 38). It should be noted that N1-trihydroxybutane-deoxyinosine, bis-N6A-BD, and bis-N2G-BD have not yet been detected in tissues of BD-treated animals and may not be relevant in vivo. In summary, studies to date have not uncovered the origins of A→T transversions that predominate in DEB-treated cells and tissues.
Previous studies in our laboratory have revealed that DEB can sequentially react with the N1 and the N6 positions of adenine in DNA to form 1,N6-α-HMHP-dA adducts (Scheme 1) (16). 1,N6-α-HMHP-dA can undergo a slow, reversible Dimroth-like rearrangement in water at room temperature to 1,N6-γ-HMHP-dA adducts (16). The concentrations of both exocyclic 1,N6-HMHP-dA adducts increased linearly when calf thymus DNA was treated in vitro with increasing concentrations of DEB (50–1000 μm) (16). Furthermore, 1,N6-HMHP-dA adducts were formed in a dose-dependent manner in liver, kidney, and lung DNA of B6C3F1 mice exposed to BD by inhalation (18). Importantly, these lesions persisted in vivo, with an estimated half-life of >40 days (19). Based on their stability and their chemical structure that resembles known promutagenic lesions such as 1,N6-etheno-dA and 1,N6-ethano-dA, 1,N6-HMHP-dA adducts have been proposed to contribute to the mutagenicity of BD (19).
In this study, in vitro polymerase bypass experiments were conducted employing synthetic DNA templates containing site-specific 1,N6-γ-HMHP-dA adducts. Human lesion bypass polymerases hpol β, hpol η, hpol κ, hpol ι, and archebacterial DNA polymerase Dpo4 were investigated, because these polymerases are known to conduct DNA replication past a variety of DNA lesions. For example, hpol η is capable of bypassing 8-oxoguanine, O6-methylguanine, and cis-syn-TT dimer (15), whereas hpol κ efficiently bypasses bulky N2-guanine adducts (8, 40). Additionally, hpol ι has been proven to conduct translesion synthesis across several DNA lesions, including N2-ethylguanine (31). 1,N2-Ethenoguanine and M1dG lesions are bypassed by archebacterial polymerase Dpo4 (10, 41).
Primer extension experiments combined with gel electrophoresis and HPLC-MS/MS analyses of the products revealed that hpol η, hpol κ, and Dpo4 were able to bypass the lesion and to extend the primer completely to the terminus (Fig. 6). In contrast, hpol β was completely blocked by the lesion, whereas hpol ι incorporated the correct nucleotide (dTMP) opposite the lesion but was unable to extend the primer further (Fig. 1B). However, incomplete primer extension products generated by hpol ι can be completed either by hpol η or hpol κ (Fig. 3). Similar cooperativity experiments conducted for N2-ethylguanine adducts revealed that hpol η can successfully complete the polymerization started by hpol α (42).
Steady-state kinetic analysis of the incorporation of single nucleotide opposite the lesion was completed for human pol η, pol κ, pol ι, and Dpo4 to determine the specificity constants (kcat/Km) and to obtain the misinsertion frequencies (f). The kcat/Km values for the incorporation of correct nucleotide (dTMP) opposite 1,N6-γ-HMHP-dA by hpol η and hpol κ were 0.14 and 0.07 μm−1 min−1, respectively; in comparison, the corresponding values for dTMP insertion opposite structurally analogous 1,N6-ϵdA lesions were 0.004 and 0.001 μm−1 min−1, respectively (43). The efficiency of incorporation of dTMP opposite the lesion by hpol η and hpol κ was 3–8-fold lower than those for the control template containing unmodified dA (Table 1). An even greater decrease (600-fold) was observed for archebacterial DNA polymerase Dpo4. Similar decreases in the efficiency of Dpo4 upon replication of damaged templates have been previously reported for the incorporation of dCTP opposite guanine adducts 7,8-dihydro-8-oxodeoxyguanosine (44), O6-methylguanine (45), and 1,N2-ethenoguanine (10).
In addition to the correct base (dTMP), hpol η, κ, and Dpo4 also inserted incorrect bases (dGMP and dAMP) opposite 1,N6-γ-HMHP-dA lesions. The misinsertion frequency was between 0.2 and 0.7, depending on specific polymerase (Table 1). For example, the frequency of dAMP incorporation opposite 1,N6-γ-HMHP-dA by hpol κ was 0.7 (Table 1). If observed in vivo, such mis-incorporation is expected to cause A→T transversion mutations.
An important limitation of gel electrophoresis experiments is that they cannot determine the nucleotide sequence of the primer extension products, potentially yielding misleading or incomplete results. Zang et al. (10) have developed a robust methodology for the analysis of primer extension products by HPLC-ESI-MS/MS. In their approach, a uracil residue is introduced into the primer, and the extension products are cleaved with UDG/hot piperidine to facilitate their sequencing by tandem mass spectrometry. This methodology has been previously applied in polymerase bypass studies of several DNA lesions (28, 30, 40, 44, 45). We adopted a similar methodology to sequence the in vitro replication products of 1,N6-γ-HMHP-dA containing templates by HPLC-ESI-MS/MS (Figs. 5–7 and Schemes 3–5). To enhance HPLC-ESI−-MS/MS detection sensitivity, a capillary HPLC column was employed instead of the conventional 1.0–2.0-mm inner diameter columns used previously (28, 30, 40, 44, 45). Using this modified HPLC-ESI−-MS/MS methodology, it was possible to characterize primer extension products using only 100 pmol of modified oligonucleotide. In comparison, the previous analytical methods required 1–4 nmol of modified oligonucleotide and large amounts of recombinant polymerases (28, 30, 40, 44, 45). The observed CID spectra of the primer extension products were in good agreement with the predicted CID spectra (Figs. 7 and 8 and supplemental Tables S1–S6).
HPLC-MS/MS sequencing has revealed that among the three polymerases that are able to conduct DNA synthesis past 1,N6-γ-HMHP-dA (hpol η, hpol κ, and Dpo4), primer extension by hpol κ was the most error-prone. hpol κ displayed a greater preference for the incorporation of incorrect nucleotide dGMP opposite the lesion (Scheme 3) as compared with the correct nucleotide dTMP. Only 18% of the total extension products formed corresponded to error-free replication products. In addition, −1 frameshift products accounted for ∼35% of the total hpol κ extension products (Scheme 3 and Fig. 8). In these cases, dGMP or dTMP was incorporated opposite the lesion, and the next base was skipped, followed by correct primer extension all the way to the terminus (Scheme 3). However, in vitro replication past 1,N6-γ-HMHP-dA by hpol η and Dpo4 was less error-prone, with a higher production of error-free extension products that accounted for more than 60% of the products and a lower percentage of single nucleotide deletion products (<10%) (Schemes 4 and 5). For both hpol η and Dpo4, misincorporation of dGMP opposite the lesion accounted for less than 10% of total products. However, Dpo4 reaction mixtures also contained significant amounts of the extension product corresponding to the incorporation of dAMP opposite the lesion (30%). In addition, small amounts of −2 frameshift deletion products were observed in primer extensions catalyzed by pol κ and η but not Dpo4.
Taken together, our gel electrophoresis and tandem mass spectrometry results indicate that human TLS polymerases η, κ, and Dpo4 are able to bypass DEB-induced 1,N6-γ-HMHP-dA lesions and to extend the primer to the terminus, but they are potentially error-prone. In addition to correct nucleotide (dTMP), these bypass polymerases incorporate dAMP and dGMP opposite the lesion and produce −1 and −2 deletion products (Schemes 3–5). In contrast, hpol ι is unable to extend beyond the 1,N6-γ-HMHP-dA lesion, and hpol β is completely blocked at the modification site.
A possible model for the insertion of dA, dT, dG opposite 1,N6-γ-HMHP-dA is shown in Scheme 6. 1,N6-γ-HMHP-dA adduct can exist in two tautomeric forms. The N1-C6 immonium ion tautomer can be envisioned to adopt a syn confirmation and form a stable Hoogsteen base pair with dT or dG. The C6-N6 imino tautomer could form a Hoogsteen base pair with protonated dA. The ability of 1,N6-γ-HMHP-dA adduct to adopt a syn confirmation and mispair with dG and protonated dA is not unprecedented. For example, 1,N6-etheno-dA adducts have been shown to adopt the syn confirmation in the active site of hpol ι, forming a Hoogsteen base pair with dT or protonated dC (20). Taken together, our results provide a possible mechanism for the induction of A→T and A→C transversions and frameshift mutations by DEB and its metabolic precursor 1,3-butadiene.
SCHEME 6.

Models for the correct insertion of dT and misinsertion of dG and dA opposite 1,N6-γ-HMHP-dA adduct by translesion DNA polymerases.
Acknowledgments
We thank Dr. Michael Stone (Vanderbilt University) for providing recombinant Dpo4 polymerase for these experiments. We are also thankful to Dr. Sarah Shuck (Vanderbilt University) for help with experimental protocols and PAGE experimental setup. HPLC-MS/MS analyses were performed in the Analytical Biochemistry Facility of the Masonic Cancer Center, which is supported in part by National Institutes of Health Grant CA-77598 from the NCI.
This work was supported, in whole or in part, by National Institutes of Health Grants CA-100670 and ES-010375.

This article contains supplemental Tables 1–6.
- TLS
- translesion synthesis
- 1,N6-HMHP-dA
- 1,N6-(2-hydroxy-3-hydroxymethylpropan-1,3-diyl)-2′-deoxyadenosine
- 1,N6-γ-HMHP-dA
- 1,N6-(2-hydroxy-3-hydroxymethylpropan-1,3-diyl)-2′-deoxyadenosine
- 1,N6-α-HMHP-dA
- 1,N6-(1-hydroxymethyl-2-hydroxypropan-1,3-diyl)-2′-deoxyadenosine
- N6-HEB-dA
- N6-(2-hydroxy-3,4-epoxybut-1-yl) adenine
- BD
- 1,3-butadiene
- DEB
- 1,2,3,4-diepoxybutane
- bis-N6A-BD
- 1,4-bis-(adenine-6-yl)-2,3-butanediol
- bis-N2G-BD
- 1,4-bis-(guan-2-yl)-2,3-butanediol
- hpol
- human polymerase
- Dpo4
- S. solfataricus S2 DNA polymerase IV
- UDG
- uracil DNA gylcosylase
- FTMS
- Fourier transform mass spectrometry
- CID
- collision induced dissociation.
REFERENCES
- 1. De Bont R., van Larebeke N. (2004) Endogenous DNA damage in humans. A review of quantitative data. Mutagenesis 19, 169–185 [DOI] [PubMed] [Google Scholar]
- 2. Lehmann A. R., Niimi A., Ogi T., Brown S., Sabbioneda S., Wing J. F., Kannouche P. L., Green C. M. (2007) Translesion synthesis. Y-family polymerases and the polymerase switch. DNA Repair 6, 891–899 [DOI] [PubMed] [Google Scholar]
- 3. Prakash S., Prakash L. (2002) Translesion DNA synthesis in eukaryotes. A one- or two-polymerase affair. Genes Dev. 16, 1872–1883 [DOI] [PubMed] [Google Scholar]
- 4. Friedberg E. C., Lehmann A. R., Fuchs R. P. (2005) Trading places. How do DNA polymerases switch during translesion DNA synthesis? Mol. Cell 18, 499–505 [DOI] [PubMed] [Google Scholar]
- 5. Woodgate R. (1999) A plethora of lesion-replicating DNA polymerases. Genes Dev. 13, 2191–2195 [DOI] [PubMed] [Google Scholar]
- 6. Ohmori H., Friedberg E. C., Fuchs R. P., Goodman M. F., Hanaoka F., Hinkle D., Kunkel T. A., Lawrence C. W., Livneh Z., Nohmi T., Prakash L., Prakash S., Todo T., Walker G. C., Wang Z., Woodgate R. (2001) The Y-family of DNA polymerases. Mol. Cell 8, 7–8 [DOI] [PubMed] [Google Scholar]
- 7. Burgers P. M., Koonin E. V., Bruford E., Blanco L., Burtis K. C., Christman M. F., Copeland W. C., Friedberg E. C., Hanaoka F., Hinkle D. C., Lawrence C. W., Nakanishi M., Ohmori H., Prakash L., Prakash S., Reynaud C. A., Sugino A., Todo T., Wang Z., Weill J. C., Woodgate R. (2001) Eukaryotic DNA polymerases. Proposal for a revised nomenclature. J. Biol. Chem. 276, 43487–43490 [DOI] [PubMed] [Google Scholar]
- 8. Waters L. S., Minesinger B. K., Wiltrout M. E., D'Souza S., Woodruff R. V., Walker G. C. (2009) Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 73, 134–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boudsocq F., Iwai S., Hanaoka F., Woodgate R. (2001) Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4). An archaeal DinB-like DNA polymerase with lesion-bypass properties akin to eukaryotic poleta. Nucleic Acids Res. 29, 4607–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zang H., Goodenough A. K., Choi J. Y., Irimia A., Loukachevitch L. V., Kozekov I. D., Angel K. C., Rizzo C. J., Egli M., Guengerich F. P. (2005) DNA adduct bypass polymerization by Sulfolobus solfataricus DNA polymerase Dpo4. Analysis and crystal structures of multiple base pair substitution and frameshift products with the adduct 1,N2-ethenoguanine. J. Biol. Chem. 280, 29750–29764 [DOI] [PubMed] [Google Scholar]
- 11. Plosky B. S., Woodgate R. (2004) Switching from high fidelity replicases to low fidelity lesion-bypass polymerases. Curr. Opin. Genet. Dev. 14, 113–119 [DOI] [PubMed] [Google Scholar]
- 12. Goodman M. F. (2002) Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 71, 17–50 [DOI] [PubMed] [Google Scholar]
- 13. Yang W., Woodgate R. (2007) What a difference a decade makes. Insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 15591–15598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang W. (2005) Portraits of a Y-family DNA polymerase. FEBS Lett. 579, 868–872 [DOI] [PubMed] [Google Scholar]
- 15. Prakash S., Johnson R. E., Prakash L. (2005) Eukaryotic translesion synthesis DNA polymerases. Specificity of structure and function. Annu. Rev. Biochem. 74, 317–353 [DOI] [PubMed] [Google Scholar]
- 16. Seneviratne U., Antsypovich S., Goggin M., Dorr D. Q., Guza R., Moser A., Thompson C., York D. M., Tretyakova N. (2010) Exocyclic deoxyadenosine adducts of 1,2,3,4-diepoxybutane. Synthesis, structural elucidation, and mechanistic studies. Chem. Res. Toxicol. 23, 118–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Swenberg J. A., Bordeerat N. K., Boysen G., Carro S., Georgieva N. I., Nakamura J., Troutman J. M., Upton P. B., Albertini R. J., Vacek P. M., Walker V. E., Sram R. J., Goggin M., Tretyakova N. (2011) 1,3-Butadiene. Biomarkers and application to risk assessment. Chem. Biol. Interact. 192, 150–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goggin M., Seneviratne U., Swenberg J. A., Walker V. E., Tretyakova N. (2010) Column switching HPLC-ESI+-MS/MS methods for quantitative analysis of exocyclic dA adducts in the DNA of laboratory animals exposed to 1,3-butadiene. Chem. Res. Toxicol. 23, 808–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goggin M., Sangaraju D., Walker V. E., Wickliffe J., Swenberg J. A., Tretyakova N. (2011) Persistence and repair of bifunctional DNA adducts in tissues of laboratory animals exposed to 1,3-butadiene by inhalation. Chem. Res. Toxicol. 24, 809–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nair D. T., Johnson R. E., Prakash L., Prakash S., Aggarwal A. K. (2006) Hoogsteen base pair formation promotes synthesis opposite the 1,N6-ethenodeoxyadenosine lesion by human DNA polymerase ι. Nat. Struct. Mol. Biol. 13, 619–625 [DOI] [PubMed] [Google Scholar]
- 21. Cochrane J. E., Skopek T. R. (1994) Mutagenicity of butadiene and its epoxide metabolites. II. Mutational spectra of butadiene, 1,2-epoxybutene, and diepoxybutane at the hprt locus in splenic T cells from exposed B6C3F1 mice. Carcinogenesis 15, 719–723 [DOI] [PubMed] [Google Scholar]
- 22. Carmical J. R., Nechev L. V., Harris C. M., Harris T. M., Lloyd R. S. (2000) Mutagenic potential of adenine N6 adducts of monoepoxide and diolepoxide derivatives of butadiene. Environ. Mol. Mutagen. 35, 48–56 [PubMed] [Google Scholar]
- 23. Recio L., Steen A. M., Pluta L. J., Meyer K. G., Saranko C. J. (2001) Mutational spectrum of 1,3-butadiene and metabolites 1,2-epoxybutene and 1,2,3,4-diepoxybutane to assess mutagenic mechanisms. Chem. Biol. Interact. 135, 325–341 [DOI] [PubMed] [Google Scholar]
- 24. Pence M. G., Blans P., Zink C. N., Fishbein J. C., Perrino F. W. (2011) Bypass of N2-ethylguanine by human DNA polymerase κ. DNA Repair 10, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pence M. G., Choi J. Y., Egli M., Guengerich F. P. (2010) Structural basis for proficient incorporation of dTTP opposite O6-methylguanine by human DNA polymerase ι. J. Biol. Chem. 285, 40666–40672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Choi J. Y., Guengerich F. P. (2005) Adduct size limits efficient and error-free bypass across bulky N2-guanine DNA lesions by human DNA polymerase η. J. Mol. Biol. 352, 72–90 [DOI] [PubMed] [Google Scholar]
- 27. Seneviratne U., Antsypovich S., Dorr D. Q., Dissanayake T., Kotapati S., Tretyakova N. (2010) DNA oligomers containing site-specific and stereospecific exocyclic deoxyadenosine adducts of 1,2,3,4-diepoxybutane. Synthesis, characterization, and effects on DNA structure. Chem. Res. Toxicol. 23, 1556–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Christov P. P., Angel K. C., Guengerich F. P., Rizzo C. J. (2009) Replication past the N5-methyl-formamidopyrimidine lesion of deoxyguanosine by DNA polymerases and an improved procedure for sequence analysis of in vitro bypass products by mass spectrometry. Chem. Res. Toxicol. 22, 1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fiala K. A., Brown J. A., Ling H., Kshetry A. K., Zhang J., Taylor J. S., Yang W., Suo Z. (2007) Mechanism of template-independent nucleotide incorporation catalyzed by a template-dependent DNA polymerase. J. Mol. Biol. 365, 590–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choi J. Y., Zang H., Angel K. C., Kozekov I. D., Goodenough A. K., Rizzo C. J., Guengerich F. P. (2006) Translesion synthesis across 1,N2-ethenoguanine by human DNA polymerases. Chem. Res. Toxicol. 19, 879–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pence M. G., Blans P., Zink C. N., Hollis T., Fishbein J. C., Perrino F. W. (2009) Lesion bypass of N2-ethylguanine by human DNA polymerase ι. J. Biol. Chem. 284, 1732–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson R. E., Washington M. T., Haracska L., Prakash S., Prakash L. (2000) Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 406, 1015–1019 [DOI] [PubMed] [Google Scholar]
- 33. Tissier A., McDonald J. P., Frank E. G., Woodgate R. (2000) polι, a remarkably error-prone human DNA polymerase. Genes Dev. 14, 1642–1650 [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y., Yuan F., Wu X., Wang Z. (2000) Preferential incorporation of G opposite template T by the low fidelity human DNA polymerase ι. Mol. Cell. Biol. 20, 7099–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Steen A. M., Meyer K. G., Recio L. (1997) Analysis of hprt mutations occurring in human TK6 lymphoblastoid cells following exposure to 1,2,3,4-diepoxybutane. Mutagenesis 12, 61–67 [DOI] [PubMed] [Google Scholar]
- 36. Carmical J. R., Kowalczyk A., Zou Y., Van Houten B., Nechev L. V., Harris C. M., Harris T. M., Lloyd R. S. (2000) Butadiene-induced intrastrand DNA cross-links. A possible role in deletion mutagenesis. J. Biol. Chem. 275, 19482–19489 [DOI] [PubMed] [Google Scholar]
- 37. Carmical J. R., Zhang M., Nechev L., Harris C. M., Harris T. M., Lloyd R. S. (2000) Mutagenic potential of guanine N2 adducts of butadiene mono- and diolepoxide. Chem. Res. Toxicol. 13, 18–25 [DOI] [PubMed] [Google Scholar]
- 38. Kanuri M., Nechev L. V., Tamura P. J., Harris C. M., Harris T. M., Lloyd R. S. (2002) Mutagenic spectrum of butadiene-derived N1-deoxyinosine adducts and N6,N6-deoxyadenosine intrastrand cross-links in mammalian cells. Chem. Res. Toxicol. 15, 1572–1580 [DOI] [PubMed] [Google Scholar]
- 39. Michaelson-Richie E. D., Loeber R. L., Codreanu S. G., Ming X., Liebler D. C., Campbell C., Tretyakova N. Y. (2010) DNA-protein cross-linking by 1,2,3,4-diepoxybutane. J. Proteome. Res. 9, 4356–4367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maddukuri L., Eoff R. L., Choi J. Y., Rizzo C. J., Guengerich F. P., Marnett L. J. (2010) In vitro bypass of the major malondialdehyde- and base propenal-derived DNA adduct by human Y-family DNA polymerases κ, ι, and Rev1. Biochemistry 49, 8415–8424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eoff R. L., Stafford J. B., Szekely J., Rizzo C. J., Egli M., Guengerich F. P., Marnett L. J. (2009) Structural and functional analysis of Sulfolobus solfataricus Y-family DNA polymerase Dpo4-catalyzed bypass of the malondialdehyde-deoxyguanosine adduct. Biochemistry 48, 7079–7088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perrino F. W., Blans P., Harvey S., Gelhaus S. L., McGrath C., Akman S. A., Jenkins G. S., LaCourse W. R., Fishbein J. C. (2003) The N2-ethylguanine and the O6-ethyl- and O6-methylguanine lesions in DNA. Contrasting responses from the “bypass” DNA polymerase η and the replicative DNA polymerase α. Chem. Res. Toxicol. 16, 1616–1623 [DOI] [PubMed] [Google Scholar]
- 43. Levine R. L., Miller H., Grollman A., Ohashi E., Ohmori H., Masutani C., Hanaoka F., Moriya M. (2001) Translesion DNA synthesis catalyzed by human pol η and pol κ across 1,N6-ethenodeoxyadenosine. J. Biol. Chem. 276, 18717–18721 [DOI] [PubMed] [Google Scholar]
- 44. Zang H., Irimia A., Choi J. Y., Angel K. C., Loukachevitch L. V., Egli M., Guengerich F. P. (2006) Efficient and high fidelity incorporation of dCTP opposite 7,8-dihydro-8-oxodeoxyguanosine by Sulfolobus solfataricus DNA polymerase Dpo4. J. Biol. Chem. 281, 2358–2372 [DOI] [PubMed] [Google Scholar]
- 45. Eoff R. L., Irimia A., Egli M., Guengerich F. P. (2007) Sulfolobus solfataricus DNA polymerase Dpo4 is partially inhibited by “wobble” pairing between O6-methylguanine and cytosine, but accurate bypass is preferred. J. Biol. Chem. 282, 1456–1467 [DOI] [PubMed] [Google Scholar]