

Background: Increase in tripeptidyl-peptidase 1 (TPP1) is a possible therapeutic approach for late infantile Batten disease or neuronal ceroid lipofuscinosis (LINCL).
Results: Gemfibrozil and fenofibrate, FDA-approved drugs for hyperlipidemia, stimulate TPP1 in brain cells via the PPARα/RXRα pathway.
Conclusion: These results delineate a novel TPP1 up-regulating property of gemfibrozil and fenofibrate.
Significance: Gemfibrozil and fenofibrate may be of therapeutic benefit in Batten disease.
Keywords: Astrocytes, Gene Regulation, Lysosomes, Neurons, Peroxisome Proliferator-activated Receptor (PPAR)
Abstract
The classical late infantile neuronal ceroid lipofuscinosis (LINCLs) is an autosomal recessive disease, where the defective gene is Cln2, encoding tripeptidyl-peptidase I (TPP1). At the molecular level, LINCL is caused by accumulation of autofluorescent storage materials in neurons and other cell types. Currently, there is no established treatment for this fatal disease. This study reveals a novel use of gemfibrozil and fenofibrate, Food and Drug Administration-approved lipid-lowering drugs, in up-regulating TPP1 in brain cells. Both gemfibrozil and fenofibrate up-regulated mRNA, protein, and enzymatic activity of TPP1 in primary mouse neurons and astrocytes as well as human astrocytes and neuronal cells. Because gemfibrozil and fenofibrate are known to activate peroxisome proliferator-activated receptor-α (PPARα), the role of PPARα in gemfibrozil- and fenofibrate-mediated up-regulation of TPP1 was investigated revealing that both drugs up-regulated TPP1 mRNA, protein, and enzymatic activity both in vitro and in vivo in wild type (WT) and PPARβ−/−, but not PPARα−/−, mice. In an attempt to delineate the mechanism of TPP1 up-regulation, it was found that the effects of the fibrate drugs were abrogated in the absence of retinoid X receptor-α (RXRα), a molecule known to form a heterodimer with PPARα. Accordingly, all-trans-retinoic acid, alone or together with gemfibrozil, up-regulated TPP1. Co-immunoprecipitation and ChIP studies revealed the formation of a PPARα/RXRα heterodimer and binding of the heterodimer to an RXR-binding site on the Cln2 promoter. Together, this study demonstrates a unique mechanism for the up-regulation of TPP1 by fibrate drugs via PPARα/RXRα pathway.
Introduction
Neuronal ceroid lipofuscinosis (NCL)2 is a group of neurodegenerative diseases mainly composed of typical autosomal recessive lysosomal storage disorders. The NCLs can be characterized by clinical manifestations like progressive mental deterioration, cognitive impairment, visual failures, seizures, and deteriorating motor function accompanied by histological findings such as the accumulation of autofluorescent storage material in neurons or other cell types (1). The NCLs have been subdivided into several groups (types 1–10) based on the age of onset, ultrastructural variations in accumulated storage materials, and genetic alterations unique to each specific disease type (2, 3).
Late infantile neuronal ceroid lipofuscinosis (Jansky-Bielschowsky disease, LINCL, type 2) typically produces symptoms at the age of 2–4 years, progresses rapidly, and ends in death between ages 8 and 15 as a result of a dramatic decrease in the number of neurons and other cells (2, 4). LINCL is associated with mutations in the Cln2 gene, a 13 exon and 12 intron gene of a total length of 6.65 kb mapped to chromosome 11p15.5. The Cln2 gene encodes lysosomal tripeptidyl-tripeptidase I (TPP-I or pepstatin-insensitive protease), a 46-kDa protein that functions in the acidic environment of the lysosomal compartment to remove tripeptides from the amino terminus of proteins (5, 6). This mutation in the Cln2 gene results in a deficiency and/or loss of function of the TPP1 protein that leads to intralysosomal accumulation of autofluorescent lipopigments known as ceroid-lipofuscin (5). Currently, there is no established treatment or drugs available for this disease; all approaches are merely supportive or symptomatic, indicating a need for novel therapeutic approaches (7). However, there are different variants of Cln2 mutations, and there have been reports that residual TPP-I activity can be found in patients with LINCL, indicating that there must be a few copies of the normal Cln2 gene remaining in patients affected with LINCL (8, 9). Thus, one approach for treatment may be to find ways to enhance the levels and residual activity of the TPP1 protein to ameliorate the disease.
Gemfibrozil and fenofibrate, two of the most significant FDA-approved lipid-lowering drugs, reduce the level of triglycerides in the blood circulation and decrease the risk of hyperlipidemia (10–12). However, a number of recent studies reveal that apart from its lipid-lowering effects, these drugs, especially gemfibrozil, can also regulate many other signaling pathways responsible for inflammation, such as switching of T-helper cells, cell-to-cell contact, migration, and oxidative stress (13–16). Here, we describe that both gemfibrozil and fenofibrate are capable of enhancing TPP1 in cultured neurons and glial cells and in vivo in the brain. We also demonstrate that PPARα, but not PPARβ and PPARγ, is involved in gemfibrozil- and fenofibrate-mediated up-regulation of TPP1. Furthermore, we also demonstrate that fibrate drugs up-regulate TPP1 via activation of the PPARα/RXRα heterodimer. Collectively, this study suggests that gemfibrozil and fenofibrate, FDA-approved drugs for hyperlipidemia, may be of therapeutic value in the treatment of LINCL.
MATERIALS AND METHODS
Reagents
DMEM/F-12, 50:50, 1×, Hanks' balanced salt solution, and 0.05% trypsin were purchased from Mediatech (Washington, D. C.). Fetal bovine serum (FBS) was obtained from Atlas Biologicals (Fort Collins, CO). Antibiotic-antimycotic, gemfibrozil, and Akt-inhibitor (Akt-i) were obtained from Sigma. Wortmannin and LY294002 were purchased from Calbiochem.
Isolation of Mouse Primary Astroglia
Astroglia were isolated from mixed glial cultures as described (17, 18) and according to the procedure of Giulian and Baker (19). Briefly, on day 9, the mixed glial cultures were washed three times with Dulbecco's modified Eagle's medium/F-12 and subjected to shaking at 240 rpm for 2 h at 37 °C on a rotary shaker to remove microglia. After 2 days, the shaking was repeated for 24 h for the removal of oligodendroglia and to ensure the complete removal of all non-astroglial cells. The attached cells were seeded onto new plates for further studies.
Isolation of Primary Human Astroglia
Primary human astroglia were prepared as described (20, 21). All experimental protocols were reviewed and approved by the Institutional Review Board of the Rush University Medical Center. Briefly, 11–17-week-old fetal brains obtained from the Human Embryology Laboratory (University of Washington, Seattle) were dissociated by trituration and trypsinization. On the 9th day, these mixed glial cultures were placed on a rotary shaker at 240 rpm at 37 °C for 2 h to remove loosely attached microglia. On the 11th day, the flasks were shaken again at 190 rpm at 37 °C for 18 h to remove oligodendroglia. The attached cells remaining were primarily astrocytes. These cells were trypsinized and subcultured in complete media at 37 °C with 5% CO2 in air to yield more viable and healthy cells. By immunofluorescence assay, these cultures homogeneously expressed GFAP, a marker for astrocytes (22).
Isolation of Neurons from Different Brain Regions
Fetal (E18–E16) mouse neurons were prepared as described previously (23) with modifications. Whole brains were removed, and cortical, hippocampal, striatal, and cerebellar fractions were dissected in serum-free Neurobasal media. The cells were washed by centrifugation three times at 1200 rpm for 10 min; the pellet was dissociated, and the cells were plated at 10% confluence in 8-well chamber slides pretreated for >2 h with poly-d-lysine (Sigma). After 4 min, the nonadherent cell suspension was aspirated, and 500 ml of complete Neurobasal media (Invitrogen) supplemented with 2% B27 was added to each well. The cells were incubated for 4 days prior to experimentation. Double-label immunofluorescence with β-tubulin and either GFAP or CD11b revealed that neurons were more than 98% pure (data not shown). The cells were stimulated with gemfibrozil in Neurobasal media supplemented with 2% B27 minus antioxidants (Invitrogen) for 24 h prior to methanol fixation and immunostaining.
Semi-quantitative Reverse Transcriptase-coupled PCR (RT-PCR)
Total RNA was isolated from mouse primary astrocytes and human primary astrocytes using RNA-Easy Qiagen (Valencia, CA) kit and following the manufacturer's protocol. Semi-quantitative RT-PCR was carried out as described earlier (24) using oligo(dT)12–18 as primer and Moloney murine leukemia virus reverse transcriptase (Invitrogen) in a 20-μl reaction mixture. The resulting cDNA was appropriately amplified using Promega Master Mix (Madison, WI) and the following primers (Invitrogen) for murine genes: mouse Cln1, sense 5′-ACACAGAGGACCGCCTGGGG-3′ and antisense 5′-TCATGCACGGCCCACACAGC-3′; mouse Cln2, sense 5′-CACCATCCAGTTACTTCAATGC-3′ and antisense 5′-CTGACCCTCCACTTCTTCATTC-3′; mouse Cln3, sense 5′-TGCTGCCCTGCCATCGAGTG-3′ and antisense 5′-GGCAGCGCTCAGCATCACCA-3′; and mouse Gapdh, sense 5′- GCACAGTCAAGGCCGAGAAT-3′ and antisense 5′-GCCTTCTCCATGGTGGTGAA-3′.
Amplified products were electrophoresed on 2% agarose (Invitrogen) gels and visualized by ethidium bromide (Invitrogen) staining. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) mRNA was used as a loading control to ascertain that an equivalent amount of cDNA was synthesized from each sample.
Quantitative Real Time PCR
The mRNA quantification was performed using the ABI-Prism7700 sequence detection system (Applied Biosystems, Foster City, CA) using iTaqTM Fast Supermix with ROX (Bio-Rad) and the following 6-FAM/ZEN/IBFQ-labeled primers for murine genes Cln2 and Gapdh (Integrated DNA Technologies, Coralville, IA). The mRNA expression of the targeted genes was normalized to the level of Gapdh mRNA, and data were processed by the ABI Sequence Detection System 1.6 software.
Immunostaining of Cells
Immunocytochemistry was performed as described earlier (25). Briefly, 8-well chamber slides containing mouse primary astrocytes, mouse neurons, human primary astrocytes, or SH-SY5Y cells cultured to 70–80% confluence were fixed with chilled methanol (Fisher) overnight, followed by two brief rinses with filtered PBS. Samples were blocked with 2% BSA (Fisher) in PBS containing Tween 20 (Sigma) and Triton X-100 (Sigma) for 30 min and incubated at room temperature under shaking conditions for 2 h in PBS containing the following anti-mouse primary antibodies: TPP1 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA); GFAP (1:100; Santa Cruz Biotechnology), and β-tubulin (1:5000; Millipore). After four 15-min washes in filtered PBS, the slides were further incubated with Cy2- or Cy5-labeled secondary antibodies (all 1:200; Jackson ImmunoResearch, West Grove, PA) for 1 h under similar shaking conditions. Following four 15-min washes with filtered PBS, cells were incubated for 4–5 min with 4′,6-diamidino-2-phenylindole (DAPI, 1:10,000; Sigma). The samples were run in an EtOH and xylene (Fisher) gradient, mounted, and observed under Olympus BX41 fluorescence microscope.
Immunostaining of Tissue Sections
After 21 days of treatment, mice were sacrificed, and their brains were fixed, embedded, and processed. Sections were made from different brain regions and for immunofluorescence staining on fresh frozen sections, and anti-mouse TPP1 (1:200), goat anti-mouse GFAP (1:100) were used. The samples were mounted and observed under Olympus BX41 fluorescence microscope (26).
Immunoblotting
Western blotting was conducted as described earlier (27, 28) with modifications. Briefly, cells were scraped in double-distilled H2O and SDS and electrophoresed on NuPAGE® Novex® 4–12% BisTris gels (Invitrogen), and proteins were transferred onto a nitrocellulose membrane (Bio-Rad) using the Thermo-Pierce Fast Semi-Dry Blotter. The membrane was then washed for 15 min in TBS plus Tween 20 (TBST) and blocked for 1 h in TBST containing BSA. Next, membranes were incubated overnight at 4 °C under shaking conditions with the following 1° antibodies: TPP1 (1:250, Santa Cruz Biotechnology) and β-actin (1:800; Abcam, Cambridge, MA). The next day, membranes were washed in TBST for 1 h, incubated in 2° antibodies against 1° antibody hosts (all 1:10,000; Jackson ImmunoResearch) for 1 h at room temperature, washed for 1 more h, and visualized under the Odyssey® Infrared Imaging System (Li-COR, Lincoln, NE).
TPP1 Activity Assay
TPP-I activity was assayed in 96-well format plates using the following modification of the method described by Vines and Warburton (6). Briefly, samples and substrate (40 μl) were mixed in individual wells of a polystyrene 96-well plate (Nalge Nunc International). The substrate solution consisted of 250 μmol/liter Ala-Ala-Phe 7-amido-4-methylcoumarin (catalog no. A3401; Sigma; diluted freshly from a 25 mmol/liter stock solution in dimethyl sulfoxide stored at −20 °C) in 0.15 mol/liter NaCl, 1 g/liter Triton X-100, 0.1 mol/liter sodium acetate, adjusted to pH 4.0 at 20 °C. Plates were centrifuged briefly to dispel bubbles and placed in a 37 °C. Plates were mixed for 10 s before each reading. The plates were read from the bottom using 360/20 nm excitation and 460/25 nm emission filters. Prior to the assay, the optimum substrate concentration and total protein in cell extract that can used to get the best results were determined in the same manner described above, using different substrate concentrations and protein concentrations in the cell extract.
Immunoprecipitation from Nuclear Extract
After treatment, cells were washed with PBS, scraped into 1.5-ml tubes, and centrifuged in 4 °C for 5 min at 500 rpm. The supernatant was aspirated, and the pellet was resuspended in a membrane lysis buffer consisting of HEPES (pH 8.0), MgCl2, KCl, dithiothreitol (DTT), and protease/phosphatase inhibitors, vortexed, and centrifuged at 4 °C at 15,000 rpm for 3 min. Again, the supernatant was aspirated, and the pellet was resuspended in a high salt nuclear envelope lysis buffer consisting of HEPES (pH 8.0), MgCl2, glycerol, NaCl, EDTA, DTT, and protease/phosphatase inhibitors, rotated vigorously at 4 °C for 30 min, and centrifuged at 4 °C at 15,000 rpm for 15 min. The resultant nuclear pellet was resuspended in IP buffer, and a fraction was kept separately as lysate. The remaining nuclear extract was then precleared with 25 μl of protein A-agarose (50%, v/v). The supernatants were immunoprecipitated with 5 μg of anti-RXRα or anti-PPARα or normal IgG (Santa Cruz Biotechnology) overnight at 4 °C, followed by incubation with protein A-agarose for 4 h at 4 °C. Protein A-agarose-antigen-antibody complexes were collected by centrifugation at 12,000 rpm for 60 s at 4 °C. The pellets were washed five times with 1 ml of IP buffer (20 mm Tris-HCl (pH 8.0), 137 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, 10% glycerol, 0.1 mm phenylmethylsulfonyl fluoride) for 20 min each time at 4 °C. Bound proteins were resolved by SDS-PAGE, followed by Western blotting with the anti-RXRα (1:2000, Santa Cruz Biotechnology) and/or anti-PPARα (1:250, Santa Cruz Biotechnology). The lysate was resolved by SDS-PAGE followed by immunoblot for PPARα, RXRα, and H3.
Chromatin Immunoprecipitation Assay
ChIP assays were performed using the method described by Nelson et al. (29), with certain modifications. Briefly, mouse primary astrocytes were stimulated by 10 μm gemfibrozil and 0.5 μm RA together for 6 h followed by fixing with formaldehyde (1.42% final volume) and quenching with 125 mm glycine. The cells were pelleted and lysed in IP buffer containing 150 mm NaCl, 50 mm Tris-HCl (pH 7.5), 5 mm EDTA, Nonidet P-40 (0.5% v/v), Triton X-100 (1.0% v/v). For 500 ml, add 4.383 g of NaCl, 25 ml of 100 mm EDTA (pH 8.0), 25 ml of 1 m Tris-HCl (pH 7.5), 25 ml of 10% (v/v) Nonidet P-40, and 50 ml of 10% (v/v) Triton X-100 containing the following inhibitors: 10 μg/ml leupeptin, 0.5 mm phenylmethylsulfonyl fluoride (PMSF), 30 mm p-nitrophenyl phosphate, 10 mm NaF, 0.1 mm Na3VO4, 0.1 mm Na2MoO4, and 10 mm β-glycerophosphate. After one wash with 1.0 ml of IP buffer, the pellet was resuspended in 1 ml of IP buffer (containing all inhibitors), and sonicated and sheared chromatin was split into two fractions (one to be used as Input). The remaining fraction was incubated overnight under rotation at 4 °C with 5–7 μg of anti-PPARα or anti-RXRα Abs or normal IgG (Santa Cruz Biotechnology) followed by incubation with protein G-agarose (Santa Cruz Biotechnology) for 2 h at 4 °C under rotation. Beads were then washed five times with cold IP buffer, and a total of 100 μl of 10% Chelex (10 g/100 ml H2O) was added directly to the washed protein G beads and vortexed. After 10 min boiling, the Chelex/protein G bead suspension was allowed to cool to room temperature. Proteinase K (100 μg/ml) was then added, and the beads were incubated for 30 min at 55 °C while shaking, followed by another round of boiling for 10 min. The suspension was centrifuged, and the supernatant was collected. The Chelex/protein G beads fraction was vortexed with another 100 μl of water, centrifuged again, and the first and the second supernatants combined. Eluate was used directly as a template in PCR. The following primers were used to amplify fragments flanking the RXR-binding element in the mouse Cln2 promoter: Set1, sense 5′-CAG CTG CCA TGT CCC CCA GC-3′ and antisense 5′-TGC GCA GCT CTG TGT CAT CCG-3′; Set2, sense 5′-GCT CCC TCT CCT CAG CTG CCA-3′ and antisense 5′-CAT CCG GAG GCT CCA GGC CA-3′. The PCRs were repeated by using varying cycle numbers and different amounts of templates to ensure that results were in the linear range of PCR.
Densitometric Analysis
Protein blots were analyzed using ImageJ (National Institutes of Health, Bethesda), and bands were normalized to their respective β-actin loading controls. Data are representative of the average fold change with respect to control for three independent experiments.
Statistics
Values are expressed as means ± S.E. of at least three independent experiments. Statistical analyses for differences were performed via Student's t test. This criterion for statistical significance was p < 0.05.
RESULTS
Fibrate Drugs Up-regulate TPP1 mRNA and Protein in Mouse Primary Astrocytes
There have been reports that residual TPP-I activity can be found in patients indicating that a few copies of normal Cln2 gene are left in patients affected with LINCL (8, 9, 30, 31). We examined if FDA-approved lipid-lowering drugs like gemfibrozil and fenofibrate were capable of up-regulating the expression of TPP1 in brain cells. Mouse primary astrocytes were treated in serum-free media with gemfibrozil with different doses and for different time points. Both RT-PCR and real time quantitative PCR (qPCR) analyses clearly indicated that gemfibrozil up-regulated Cln2 mRNA levels in mouse primary astrocytes in a time- and dose-dependent manner with a maximum increase at 24 h of 25 μm gemfibrozil treatment (Fig. 1, A–D). Other lysosomal genes like Cln1 and Cln3, which are responsible for infantile NCL and juvenile NCL, respectively, were also found to increase within 12–24 h (Fig. 1A). The mRNA data were validated by Western blot where a 3–4-fold increase in TPP1 protein level was found with 25 μm gemfibrozil and 10 μm fenofibrate treatment for 24 h (Fig. 1, E and F). Immunofluorescence of primary mouse astrocytes stimulated with gemfibrozil and fenofibrate also revealed a dose-dependent increase in TPP1 protein (Fig. 1G).
FIGURE 1.

Gemfibrozil and fenofibrate up-regulate TPP1 mRNA and functionally active protein in mouse brain cells. A and B, mouse primary astrocytes were treated with 25 μm gemfibrozil (Gem) in serum-free DMEM/F-12 for 2, 6, 12, and 24 h followed by monitoring the mRNA expression of Cln1, Cln2, and Cln3 by semi-quantitative RT-PCR (A) and qPCR (B) (for Cln2). C and D, mouse astrocytes were treated with different concentrations of gemfibrozil for 24 h under the same culture conditions followed by monitoring the mRNA expression of Cln2 by semi-quantitative RT-PCR (C) and real time PCR (D). E, mouse primary astrocytes were treated with 25 μm gemfibrozil and 10 μm fenofibrate (Feno) for 24 h under the same culture conditions followed by Western blot for TPP1. F, densitometric analysis of TPP1 expression (relative to β-actin) by gemfibrozil and fenofibrate treatment. G, mouse primary astrocytes were treated with different concentrations of gemfibrozil and fenofibrate under similar culture conditions and were double-labeled for TPP1 (red) and GFAP (green). UN, untreated. Scale bar, 10 μm. H, J, and L, mouse primary neurons were isolated from different parts of the brain and were treated with 25 μm gemfibrozil and 10 μm fenofibrate in neurobasal media containing B27-AO for 24 h and were double-labeled for TPP1 (red) and β-tubulin (β-TUB) (green) (H, cortical neurons; J, hippocampal neurons; L, striatal neurons.) DAPI was used to stain nuclei. Scale bar, 20 μm. I, K, and M, mouse neurons were treated with 25 μm gemfibrozil under same culture conditions for 24 h followed by Western blot for TPP1 (I, cortical neurons; K, hippocampal neurons; M, striatal neurons.) Graphs represent the densitometric analysis of TPP1 level (relative to β-actin). N, mouse primary neurons were treated with different concentrations of gemfibrozil and fenofibrate in B27-AO containing Neurobasal media for 24 h followed by the activity assay using cell extract containing 5 μg of total protein. O, mouse primary astrocytes were treated with different concentrations of gemfibrozil and fenofibrate in serum-free DMEM/F-12 medium for 24 h followed by the activity assay using cell extract containing 5 μg of total protein. All results are mean ± S.E. of at least three independent experiments. *, p < 0.05 versus control; **, p < 0.01 versus control.
Gemfibrozil and Fenofibrate Up-regulates TPP1 in Neurons from Different Parts of Mouse Brain
Lack of TPP1 enzyme causes accumulation of lipofuscins in neurons leading to loss of neurons in brain causing the disease to progress (5). Hence, we examined the effect of the fibrate drugs in neurons and determined whether the induction of TPP1 occurs throughout the brain. Mouse primary neurons were isolated from different brain regions, viz. cortex, hippocampus, and striatum, and were cultured and treated with gemfibrozil and fenofibrate. The immunofluorescence showed a significant increase in the mouse neurons from all three brain regions (Fig. 1, H, J, and L). Furthermore, the neurons from those brain regions were treated with gemfibrozil for 24 h followed by Western blotting for TPP1, which showed about 2–3-fold increase in TPP1 protein, as determined by densitometric quantification (Fig. 1, I, K, and M).
TPP1 Proteins Up-regulated by the Fibrate Drugs Are Functionally Active
Because the functional activity of the TPP1 protein is of critical importance in the clinical setting for LINCL (6), activity of the enzyme was measured. The cells were homogenized, and the cell extracts were subjected to TPP1 activity assay. Prior to that, the optimal substrate concentration and optimal amount of extract for the assay was determined by using different concentrations of substrate and sample, respectively (supplemental Fig. 1, A and B). TPP1 activity was measured (as described under “Materials and Methods”) in mouse primary neurons and mouse primary astrocytes. The product formation increased with increasing doses of treatment indicating an increase in activity of the protein in the cell extracts (Fig. 1, N and O). This can be attributed to the increase in the levels of proteins in the cells observed in the earlier experiments. Collectively, these data strongly suggest that fibrate drugs can enhance both the mRNA and protein levels resulting in an increased activity of the protein in the cell.
Fibrate Drugs Up-regulate TPP1 mRNA and Protein in Human Brain Cells
We further examined whether a similar increase in Cln2 mRNA and protein was obtained upon treatment of human cells with gemfibrozil and fenofibrate. Human astrocytes were treated in the same way as the mouse cells, and the mRNA levels were quantified. Again, both RT and qPCR data indicated an increase in Cln2 mRNA levels in human astrocytes in a dose- and time-dependent manner with maximum at a dose of 25 μm gemfibrozil (∼15-fold) and at 12 h (∼10-fold) (Fig. 2, A–D). However, fenofibrate was seen to increase the mRNA levels at a relatively lower dose (10 μm) but at same time point (12 h) as that of gemfibrozil (Fig. 2, E and F). Once again, the protein levels were assessed in human astrocytes and SH-SY5Y cell lines by immunofluorescence, and in both the cell types, a considerable increase in the level of TPP1 protein was observed. (Fig. 2, G and H).
FIGURE 2.

Gemfibrozil and fenofibrate up-regulate TPP1 mRNA and protein in human primary astrocytes and SHSY5Y neuronal cells. A and B, human primary astrocytes were treated with 25 μm gemfibrozil in serum-free DMEM/F-12 for 2, 6, 12, and 24 h followed by monitoring the mRNA expression of Cln2 by semi-quantitative RT-PCR (A) and real time PCR (B). C–F, human primary astrocytes were treated with different concentrations of gemfibrozil (Gem) and fenofibrate (Feno) for 12 h under the same culture conditions followed by monitoring the mRNA expression of Cln2 by semi-quantitative RT-PCR (C, gemfibrozil; E, fenofibrate) and real time PCR (D, gemfibrozil; F, fenofibrate). G, human primary astrocytes were treated with 25 μm gemfibrozil and 10 μm fenofibrate for 24 h under similar culture conditions and were double-labeled for TPP1 (red) and GFAP (green). H, SH-SY5Y cells were treated with 25 μm gemfibrozil and 10 μm fenofibrate in B27-AO containing Neurobasal media for 24 h and were double-labeled for TPP1 (red) and β-tubulin (β-TUB) (green). DAPI was used to stain nuclei. All results are mean ± S.E. of at least three independent experiments. *, p < 0.05 versus control. UN, untreated. Scale bar, 10 μm.
PPARα Is Involved in Fibrate Drug-mediated Up-regulation of TPP1
Because it is known that PPARs are activated by fibrate drugs, the role of these receptors in mediating up-regulation of TPP1 protein was examined (15). Astrocytes isolated from PPARα−/− and PPARβ−/− and wild type (WT) mice were treated with gemfibrozil and fenofibrate, and Cln2 mRNA levels were measured. The data from semi-quantitative RT-PCR and qRT-PCR showed that WT and PPARβ−/− cells showed similar patterns of up-regulation, whereas PPARα−/− cells showed little or no effect on the up-regulation of Cln2 mRNA expression upon gemfibrozil treatment (Fig. 3, A and B) and fenofibrate treatment (Fig. 3, C and D). When mouse primary astrocytes were treated with GW9662, a PPARγ antagonist, followed by gemfibrozil or fenofibrate treatment, there was increased expression of Cln2 mRNA, even in presence of the antagonist (Fig. 3, G and H).
FIGURE 3.

Involvement of PPARα in fibrate drug-mediated up-regulation of TPP1 mRNA and protein. A–E, mouse primary astrocytes isolated from PPARα−/− and PPARβ−/− and wild type mice were treated with different concentrations of gemfibrozil (Gem) and fenofibrate (Feno) in serum-free DMEM/F-12 for 24 h followed by monitoring the mRNA expression of Cln2 by semi-quantitative RT-PCR (A and C) and real time PCR (B and D) and protein level of TPP1 by Western blot (E). F, densitometric analysis of TPP1 levels (relative to β-actin) in PPARα−/− and PPARβ−/− and wild type astrocytes by gemfibrozil and fenofibrate treatment. a, p <0.05 versus WT control; b, p <0.05 versus PPARβ−/− control; ns, not significant with respect to PPARα−/− control. G and H, mouse primary astrocytes isolated from WT mice were pretreated with GW9662 (GW) for 30 min followed by treatment with 25 μm gemfibrozil under similar culture conditions. The mRNA expression of Cln2 was monitored by semi-quantitative RT-PCR (G) and real time PCR (H). c, p <0.05 versus control; d, p <0.05 versus only GW9662-treated. I–K, mouse primary astrocytes isolated from PPARα−/− and PPARβ−/− and WT mice were treated with 25 μm gemfibrozil and 10 μm fenofibrate in serum-free DMEM/F-12 for 24 h and double-labeled for TPP1 (red) and GFAP (green) (I, WT; J, PPARα−/−; K, PPARβ−/− astrocytes.) DAPI was used to stain nuclei. UN, no treatment. Scale bar, 10 μm. L–N, mouse primary astrocytes isolated from PPARα−/− and PPARβ−/−, and WT mice were treated with 25 μm gemfibrozil and 10 μm fenofibrate in serum-free DMEM/F-12 for 24 h. Whole cell extracts containing 5 μg of total protein was incubated at 37 °C with 250 μm 7-amido-4-methylcoumarin in 96-well plates, and readings were taken at an interval of 30, 45, 60, 90, and 120 min. Mean values were taken and plotted in graphical format (L, wild type cells; M, PPARα−/− cells; N, PPARβ−/− cells.) All results are mean ± S.E. of at least three independent experiments.
To confirm the mRNA measurements, the WT and PPARα−/− and PPARβ−/− astrocytes were processed for protein analysis. The cells were treated similarly with gemfibrozil and fenofibrate, and immunofluorescence and Western blotting were performed. The immunoblot and densitometric analysis of the blots showed no significant increase in TPP1 levels in PPARα−/− cells but about 4–5-fold increase in WT and PPARβ−/− cells (Fig. 3, E and F). The data from the Western blot was confirmed by immunofluorescence where a similar effect of the drugs on WT and KO astrocytes was observed, i.e. little or no increase in TPP1 in PPARα−/− cells, compared with WT and PPARβ−/− cells (Fig. 3, I–K).
Furthermore, the presence of active TPP1 enzyme was also confirmed by measurement of TPP1 activity in WT and KO cell types. The enzymatic activity was drastically increased in WT and PPARβ−/− cells upon treatment with gemfibrozil or fenofibrate (Fig. 3, L and N), whereas PPARα−/− cell extracts showed no significant increase in TPP1 enzymatic activity (Fig. 3M). Collectively, these data indicate that PPARα, but neither PPARβ nor PPARγ, is involved in the gemfibrozil- and fenofibrate-mediated up-regulation of TPP1.
TPP1 Is Up-regulated by Fibrate Drugs in Vivo in the CNS of WT and PPARβ−/−, but Not PPARα−/−, Mice
Once we confirmed the involvement of PPARα in the fibrate-mediated up-regulation of TPP1 protein, we further checked whether the same results could be replicated in in vivo settings. WT, PPARα−/−, and PPARβ−/− mice from same background were treated orally for 21 days with 7.5 mg/kg body weight/day gemfibrozil dissolved in 0.1% methylcellulose, which was also used as vehicle. At the end of the treatment, the mice were killed, and different regions of their brain, viz. substantia nigra pars compacta, cortex, hippocampus, and dentate gyrus were sectioned, and immunofluorescence was performed for the presence of TPP1. Gemfibrozil treatment markedly increased the level of TPP1 both in GFAP-positive cortical astrocytes (Fig. 4, A1–A4) and NeuN-positive cortical neurons (Fig. 4, B1–B4) in WT and PPARβ−/−, but not PPARα−/−, mice. Similarly, gemfibrozil treatment also increased the level of TPP1 in GFAP-positive astrocytes (Fig. 4, C1–C4) and tyrosine hydroxylase-positive neurons (Fig. 4, D1–D4) in the substantia nigra of WT and PPARβ−/−, but not PPARα−/−, mice. Gemfibrozil also increased TPP1 mostly in the non-neuronal cells in the dentate gyrus (Fig. 5, A–D) and CA1 region of the hippocampus (Fig. 5, E–H) of WT and PPARβ−/−, but not PPARα−/−, mice. These data clearly indicate that gemfibrozil increases TPP1 in vivo in the CNS via PPARα.
FIGURE 4.

Oral administration of gemfibrozil up-regulates TPP1 in vivo in cortical and nigral astrocytes and neurons of WT and PPARβ−/−, but not PPARα−/− mice. WT, PPARα−/−, and PPARβ−/− mice (n = 4 in each group) were treated with 7.5 mg/kg body weight/day gemfibrozil (dissolved in 0.1% methylcellulose) or vehicle (0.1% methylcellulose) via gavage. After 21 days of treatment, mice were killed, and cortical and nigral sections were double-labeled for TPP1 (red) along with either GFAP (green) or NeuN (green) or TH (green). DAPI was used to visualize the nucleus. A1–A3 and B1–B3, TPP1 levels were compared between the astrocytes and neurons of the cortical sections of vehicle-treated and gemfibrozil-treated (A1 and B1), WT (A2 and B2), PPARα−/− and PPARβ−/− (A3 and B3) mice. (A1–A3, GFAP and TPP1; B1–B3, NeuN and TPP1.) Scale bar, 20 μm. A4 and B4, higher magnification images showing co-localization of TPP1 and GFAP in the (A4) cortical astroglia and co-localization of NeuN and TPP1 in (B4) cortical neurons of gemfibrozil treated mice (WT and PPARβ−/−). Scale bar, 10 μm. C1–C3 and D1–D3, TPP1 levels were compared between the astrocytes and neurons of the nigral sections of vehicle treated and gemfibrozil treated (C1 and D1) WT, (C2 and D2) PPARα−/− & (C3 and D3) PPARβ−/− mice. Scale bar, 20 μm. (C1–C3, GFAP and TPP1; D1–D3, TH and TPP1). C4 and D4, higher magnification images showing co-localization of TPP1 and GFAP in the (C4) nigral astroglia and co-localization of TH and TPP1 in (D4) nigral TH neurons of gemfibrozil treated mice (WT and PPARβ−/−). Scale bar, 10 μm. All results represent analysis of each of three cortical and nigral sections of each of four different mice per group.
FIGURE 5.

Oral administration of gemfibrozil up-regulates TPP1 in vivo in the hippocampus of WT and PPARβ−/− but not PPARα−/− mice. WT, PPARα−/−, and PPARβ−/− mice (n = 4 in each group) were treated with 7.5 mg/kg body weight/day of gemfibrozil (dissolved in 0.1% methylcellulose) or vehicle (0.1% methylcellulose) via gavage. After 21 days of treatment, mice were killed, and hippocampal (CA1 and dentate gyrus) sections were double-labeled for TPP1 (red) and GFAP (green). DAPI was used to visualize the nucleus. A–C, TPP1 levels were compared between the dentate gyrus region of vehicle-treated and gemfibrozil-treated WT (A), PPARα−/− (B), and PPARβ−/− (C) mice. Scale bar, 20 μm. D, higher magnification images showing co-localization of TPP1 and GFAP in the dentate gyrus of gemfibrozil-treated mice (WT, PPARα−/−, and PPARβ−/−). Scale bar, 10 μm. E–G, TPP1 levels were compared between the CA1 region of vehicle-treated and gemfibrozil-treated WT (E), PPARα−/− (F), and PPARβ−/− (G) mice. Scale bar, 20 μm. H, higher magnification images showing co-localization of TPP1 and GFAP in the CA1 region of gemfibrozil-treated mice (WT, PPARα−/−, and PPARβ−/−). Scale bar, 10 μm. All results represent analysis of each of three hippocampal sections of each of four different mice per group.
Up-regulation of TPP1 by Fibrate Drugs Involves Both PPARα and RXRα
Next, we investigated the mechanism of this up-regulation. We observed that Cln2 gene promoter lacked the PPAR-binding site but contained an RXR-binding site instead. Because of the facts that RXRα is abundant in the brain and astrocytes (32, 33) and that PPARα and RXRα form a heterodimer, we thought the mechanism of up-regulation of TPP1 may involve cooperative action of both PPARα and RXRα and not PPARα alone. To verify our hypothesis, we performed an array of experiments. First, we checked whether activating RXR by RA, a known activator of RXRs, caused any change in the mRNA or protein levels of TPP1. Interestingly, quantitative real time PCR data showed that even RA alone enhanced the mRNA levels of Cln2 (Fig. 6A). RA at a concentration of 0.5 μm caused about a 3.5-fold increase in Cln2 mRNA levels, which is comparable with the effect of 10 μm gemfibrozil treatment (∼4-fold) (Fig. 6A). Moreover, when cells were treated with a low dose of gemfibrozil (10 μm) together with RA (at different concentrations), there was a profound increase in the Cln2 levels with an optimum concentration of the combination being at 10 μm gemfibrozil and 0.5 μm RA (about 12-fold increase) (Fig. 6A). These mRNA data were validated by Western blot performed in mouse astrocytes using similar treatments (Fig. 6B). The densitometry analysis showed a similar pattern of increase in the protein levels of TPP1 as observed from the mRNA data (Fig. 6C). In both real time PCR and immunoblot experiments, the increase of TPP1 expression with the combinatorial treatment (10 μm gemfibrozil and 0.5 μm RA) was found to be statistically significant when compared with either gemfibrozil (10 μm) or RA (0.5 μm) treatment alone. This finding clearly indicates the possible involvement of RXR in the up-regulation of the Cln2 gene. Second, to confirm the involvement of RXRα, we knocked down RXRα in astrocytes by RXRα siRNA followed by treatment with gemfibrozil and RA. The siRNA was found to specifically knock down RXRα but neither RXRβ nor RXRγ (Fig. 6D). The effect of gemfibrozil and RA was found to be abrogated in the absence of RXRα, as observed from the quantitative real time PCR data (Fig. 6E). There was an almost 8–10-fold increase in the Cln2 level in both untransfected cells as well as in cells transfected with scrambled siRNA, whereas cells with RXRα knockdown were almost unresponsive to the treatment of gemfibrozil or RA alone as well as the combination (Fig. 6E). Similar results were obtained with the protein analysis. The densitometric analysis for the TPP1 Western blot showed almost no enhancement of TPP1 levels in RXRα siRNA-transfected cells (Fig. 6, F and G). Finally, to validate our hypothesis that both PPARα and RXRα are involved in the up-regulation process, we checked whether activation of RXRα alone (in the absence of PPARα) can induce the expression of Cln2. Mouse astrocytes from wild type (WT), PPARα−/−, and PPARβ−/− mice were treated with RA (0.5 μm) and the combination of gemfibrozil (10 μm) and RA (0.5 μm) followed by immunoblot analysis for TPP1. It was observed that neither RA alone nor the combination could induce TPP1 in PPARα−/− cells, whereas WT and PPARβ−/− cells were responsive to the treatment (about 5–6-fold induction of TPP1) (Fig. 6, H and I). These data suggest that either PPARα or RXRα alone is not sufficient for the up-regulation of TPP1.
FIGURE 6.

Up-regulation of TPP1 by fibrate drugs involves both PPARα and RXRα. A–C, mouse primary astrocytes were treated with different concentrations of all-trans-retinoic acid (RA) and gemfibrozil (Gem) and the combination of the two in serum-free DMEM/F-12 medium for 24 h followed by monitoring of mRNA expression of Cln2 by quantitative RT-PCR (A) and protein expression of TPP1 by Western blot (B). C, densitometric analysis of TPP1 (relative to β-actin) with RA and gemfibrozil treatment. a, p <0.05 versus WT control; b, p <0.05 versus 0.5 μm RA-only treatment; c, p <0.05 versus 10 μm gemfibrozil-only treatment. D and E, mouse primary astrocytes were untransfected and transfected with scrambled siRNA (1.0 μg) or RXRα siRNA (1.0 μg) for 36 h followed by treatment with RA (0.5 μm) and gemfibrozil (10 μm) alone and in combination for 24 h of serum-free DMEM/F-12 medium followed by RT-PCR for RXRα, RXRβ, and RXRγ (D) and quantitative real time PCR for Cln2 (E). *, p < 0.05 versus untransfected control; **, p < 0.05 versus scrambled siRNA transfected control; d, p < 0.05 versus untransfected and gemfibrozil-treated sample; e, p < 0.05 versus untransfected and RA-treated sample; f, p <0.05 versus scrambled siRNA-transfected and gemfibrozil-treated sample; g, p < 0.05 versus scrambled siRNA-transfected and RA-treated sample; ns, not significant with respect to RXR-α siRNA transfected control. F, mouse primary astrocytes were transfected with scrambled siRNA (1.0 μg) or RXRα siRNA (1.0 μg) and treated with gemfibrozil (10 μm) and RA (0.5 μm) alone and in combination under similar culture conditions, and the protein expressions of TPP1 were estimated by Western blot. G, densitometric analysis of TPP1 (relative to β-actin) with RA and gemfibrozil treatment. **, p < 0.05 versus scrambled siRNA-transfected control; h, p <0.05 versus only gemfibrozil treatment; i, p <0.05 versus only RA treatment; ns, not significant w.r.t. RXR-α siRNA-transfected control. H, mouse primary astrocytes isolated from wild type, PPARα−/−, and PPARβ−/− mice were treated with RA (0.5 μm) alone and in combination with gemfibrozil (10 μm) under similar culture conditions and cells, were subjected to Western blot for TPP1. I, densitometric analysis of TPP1 levels (relative to β-actin) in PPARα−/− and PPARβ−/− and wild type astrocytes after RA and gemfibrozil + RA treatment. †, p <0.05 versus WT control; ††, p <0.05 versus PPARβ−/− control; j, p <0.05 versus only RA treatment in WT cells; k, p <0.05 versus only RA treatment in PPARβ−/− cells; ns, not significant w.r.t. PPARα−/− control. All results are means ± S.E. of at least three independent experiments.
Fibrate Drugs Up-regulate TPP1 via Activation of PPARα/RXRα Heterodimer
After confirming the involvement of both PPARα and RXRα, we were interested to find out the actual role of the two factors. First, we examined whether there was any actual physical interaction between PPARα and RXRα. Mouse astrocytes were treated with gemfibrozil and RA separately as well as in combination, and the nuclear extract was subjected to co-immunoprecipitation for both PPARα and RXR. Immunoprecipitation with PPARα Ab showed increased presence of RXR in the immunoblot for the treated samples compared with control (Fig. 7A (i)). Similarly, increased abundance of PPARα was also observed when the nuclear extracts were immunoprecipitated with RXR Ab (Fig. 7A (ii)). These results demonstrate the presence of the PPARα/RXRα heterodimer in the nucleus of cells stimulated with gemfibrozil and RA. These results are specific as we did not find any bands with IgG (Fig. 7A (iii)). Levels of PPARα and RXRα and histone 3 (H3) have been shown as loading controls (Fig. 7A (iv)). Next, we performed ChIP studies to show the recruitment of the PPARα and RXRα on the RXR-binding site on the Cln2 gene (Fig. 7B). Chromatin fragments from cells treated with gemfibrozil and RA were immunoprecipitated with both PPARα Ab and RXRα Ab, and the DNA obtained was amplified by PCR with primers spanning the RXR-binding site on the Cln2 gene promoter. In both cases, we were able to amplify 200-bp fragments flanking the RXR-binding site (Fig. 7C). In contrast, no amplification product was observed in any of the immunoprecipitates obtained with control IgG (Fig. 7C), suggesting the specificity of these interactions. These results suggest that gemfibrozil and RA are capable of recruiting both PPARα and RXRα to the RXR-binding site of the Cln2 gene promoter (Fig. 7C).
FIGURE 7.

Fibrate drugs up-regulate TPP1 via activation of PPARα/RXRα heterodimer. A, mouse primary astrocytes were treated with gemfibrozil (10 μm) and RA (0. 5 μm) alone and in combination in serum-free DMEM/F-12 for 6 h, and the nuclear extract was subjected to the following: (i) immunoprecipitation by PPARα Ab followed by immunoblot for both RXRα and PPARα; (ii) immunoprecipitation by RXRα Ab followed by immunoblot for both PPARα and RXRα; (iii) immunoprecipitation by control IgG followed by immunoblot for both PPARα and RXRα, and (iv) nuclear extract was subjected to immunoblot for PPARα, RXRα, and histone 3 (H3). B, schematic diagram for RXR-binding site on the Cln2 promoter with the core sequence and amplicon length. C, mouse astrocytes were treated with the combination of gemfibrozil (10 μm) and RA (0.5 μm) for 6 h, and recruitment of PPARα and RXRα on the RXR-binding site of Cln2 promoter was monitored by ChIP analysis as described under “Materials and Methods.” Normal IgG was used as control. All results are representative of at least three independent experiments.
DISCUSSION
The NCL family of disease can be considered to be one of the most important hereditary neurodegenerative lysosomal storage diseases in children (34). Mutations in the Cln2 gene result in deficiency or loss of function of the TPP1 enzyme (9, 30, 35). There have been reports of over 68 missense mutations in the Cln2 gene, including 35 single amino acid substitutions. Studies with 14 different naturally occurring disease-associated mutations showed alteration of lysosomal transport, increased half-life of the proenzyme, and improper folding, resulting to loss of function of the enzyme (9). Currently, there is no established drug-mediated therapy for LINCL, a classic subtype of the NCLs. Studies using adeno-associated virus and other viral vectors expressing recombinant TPP1 demonstrate widespread expression of TPP1, and treatment of Cln2-targeted mice with these recombinant vectors show slowing of disease-associated pathology and an increase in survival in mutant mice (36–38). However, levels of TPPI activity achievable by adeno-associated virus-mediated gene therapy can vary and depend on various critical parameters, and there is considerable doubt whether similar effects can be achieved in humans (36, 39).
Nevertheless, restoration of activity at even low levels could prove helpful for most lysosomal storage diseases, where restoration of even <10% of normal activity may have therapeutic benefits (40). Studies with hypomorphs of Cln2 mutant mice, expressing different levels of TPP1 enzyme, indicate that even 3% of normal TPP1 activity is capable of delaying the onset of the disease, and 6% of the normal activity attenuates the disease and increases the life span of mice (40). Also, two specific variants of mutated TPP1 were responsive to molecular chaperone treatment, indicating that folding improvement strategies can be used to restore the enzymatic activity (9). Recent studies suggest that some misfolded variants or misprocessed proteins may also be rescued by treatment in permissive temperatures under suitable conditions (9). There have also been reports of some mutations in TPP1 (R447H), which apparently may not have any pathogenic effect (31). Moreover, a sensitive enzyme activity assay detected residual levels of TPP1 activity in various biological samples from patients who were confirmed to have LINCL by genetic analysis (30). This study also showed the presence of enzyme activity in various animals having NCL-like neurodegenerative symptoms rendering them unsuitable for being a model for classical LINCL (30). Furthermore, using a highly sensitive capillary electrophoresis technique, Viglio et al. (8) reported that lymphocytes from patients affected with LINCL exhibited TPP1 activity, although at low levels (in a range between 0.1 and 0.8 milliunits/mg). These findings about the presence of residual enzymatic activity in LINCL patients are very interesting as they indicate the presence of at least a few copies of the functional gene in the system. Therefore, identifying specific drugs and understanding the mechanisms by which these drugs can up-regulate the endogenous normal copies of the gene may be a critical step for LINCL therapy.
Gemfibrozil, marketed as “Lopid,” and fenofibrate, known as “Tricor,” are FDA-approved drugs prescribed for hyperlipidemia (10, 12). Here, we delineate for the first time that these drugs are capable of up-regulating TPP1 in brain cells. This finding was confirmed by both mRNA and protein studies in both mouse and human cells. The increase in protein levels was throughout the brain as neurons isolated from different brain regions of mice showed increased TPP1 expression upon treatment with gemfibrozil. In the case of LINCL, the presence of the functionally active TPP1 enzyme is critical for therapy, as we have to rely on the up-regulation of residual enzyme activity in patients. The TPP1 activity assay, performed in different cell types, clearly showed that there was significant increase in the activity of the enzyme, which is a result of increased levels of the protein. Considering the possibility of treatment by up-regulation of the endogenous Cln2 gene, this finding could be of importance in the therapy of LINCL.
Over the last few years, a number of studies emphasized the role of PPARs in different regulatory and modulatory pathways. It is also well known that PPARα is activated by polyunsaturated fatty acids and oxidized derivatives and by lipid-modifying drugs of the fibrate family, including fenofibrate and gemfibrozil (41, 42). PPARα is present in the cytoplasm as an inactive complex with heat-shock protein 90 (HSP-90) and hepatitis virus B-X-associated protein-2 (XAP-2), which act as an inhibitor of PPARα. Fibrate drugs replace the HSP90 repressor complex and help to rescue the transcriptional activity of PPARα (15). Therefore, we investigated the role of the PPAR group of receptors in this phenomenon. We examined all three PPARs, viz. PPARα, PPARβ, and PPARγ, for their involvement in up-regulation of TPP1. These studies clearly indicate the involvement of PPARα, but not PPARβ and PPARγ, in this process. In astrocytes from WT and PPARα−/− and PPARβ−/− mice, both the TPP1 mRNA and protein analysis showed the involvement of only PPARα. Involvement of PPARγ was ruled out as studies using known antagonist of PPARγ revealed no effect. TPP1 enzyme activity in the cell extracts was also increased in WT and PPARβ−/−, but not PPARα−/−, cells. The in vitro studies were further validated by in vivo studies, where we used the knock-out mice for PPARα and PPARβ. Our in vivo results also supported the cell culture data.
To delineate the mechanism of fibrate drug-mediated up-regulation of TPP1, we analyzed the promoter region of the Cln2 gene. Surprisingly, no PPAR-binding site was found in the mouse Cln2 promoter, but further analysis of the promoter revealed an RXR-binding site. It is well known that to bind to DNA and activate transcription, PPAR requires the formation of s heterodimer with the RXR (43). Together, the PPAR/RXR heterodimer regulates the transcription of genes for which products are involved in lipid homeostasis, cell growth, and differentiation (44, 45). This led us to think whether the pathway of TPP1 up-regulation requires a cooperative effect of both PPAR and RXR. It was observed that the activation of RXR by low doses of RA alone (0.5 μm) was capable of up-regulating TPP1 to a comparable level of that of gemfibrozil (10 μm). Also, when cells were treated with both gemfibrozil and RA together, they cooperatively enhanced the expression of TPP1 by almost more than 3-fold compared with the levels achieved by either gemfibrozil or RA alone, which implies that a combinatorial therapy could be more useful than using the compounds separately for treatment. Furthermore, the effects of both gemfibrozil and RA were abrogated in the absence of either RXRα or PPARα. The co-immunoprecipitation studies performed with the nuclear extracts on astrocytes stimulated with gemfibrozil and RA demonstrated physical interaction between PPARα and RXR. These data clearly suggest that the treatment with gemfibrozil and RA activates both PPARα and RXRα, which forms a heterodimer in the nucleus. The ChIP data indicated the recruitment of the PPARα and RXRα on the RXR-binding site of the Cln2 promoter, hence validating our hypothesis. Collectively, these data outline a unique mechanism where gemfibrozil, a known activator of PPARα, and RA, an agonist of RXRα, together can up-regulate TPP1 in brain cells via the PPARα/RXRα heterodimer Fig. 8.
FIGURE 8.
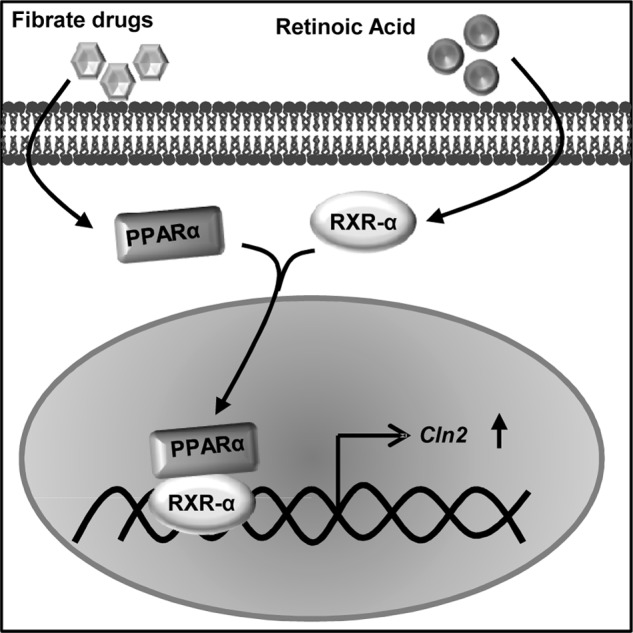
Schematic representation of the mechanism of up-regulation of TPP1 by fibrate drugs via PPARα/RXRα pathway.
Gemfibrozil and other fibrate drugs are known to reduce superoxide, lipid peroxidation products. It also strengthens the cellular defense by stimulating the activity of anti-oxidant proteins such as paraoxonase and is associated with the free radical scavenging ability as well as metal ion chelation. Therefore, apart from its lipid-lowering effects, these drugs also have anti-inflammatory, immunomodulatory, and anti-oxidative properties (14, 27, 46–48). In the NCL cases, predominantly in LINCL, different brain regions have been shown to be immunoreactive for 4-hydroxynonenal or 8-hydroxydeoxyguanosine, popular markers for evaluation of oxidative stress that may be caused due to accumulation of lipofuscins and elevated cytokine response (1). Therefore, treatment with these drugs will not only lead to the up-regulation of endogenous normal TPP1 leading to clearance of lipofuscins but also can be beneficial for the peripheral immune system by down-regulating the inflammatory pathways generated due to accumulation of lipofuscins.
In summary, we have delineated that gemfibrozil and fenofibrate, FDA-approved lipid-lowering drugs, up-regulate TPP1 in cultured mouse and human brain cells and in vivo in mouse brain via the PPARα/RXRα pathway. Although the in vitro situation of mouse and human brain cells in culture and its treatment with gemfibrozil and fenofibrate may not truly resemble the in vivo situation of the CNS of patients with LINCLs, our results clearly identify these two drugs as possible therapeutic agents for LINCL that can be immediately taken to clinical trials for testing.
This work was supported, in whole or in part, by National Institutes of Health Grants AT6681, NS64564, and NS71479. This work was also supported by a generous donation from Noah's Hope Foundation.

This article contains supplemental Fig. 1.
- NCL
- neuronal ceroid lipofuscinosis
- LINCL
- late infantile neuronal ceroid lipofuscinosis
- FDA
- Food and Drug Administration
- PPARα
- peroxisome proliferator-activated receptor-α
- RXRα
- retinoid X receptor-α
- GFAP
- glial fibrillary acidic protein
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- RA
- all-trans-retinoic acid
- Ab
- antibody
- qPCR
- quantitative PCR.
REFERENCES
- 1. Hachiya Y., Hayashi M., Kumada S., Uchiyama A., Tsuchiya K., Kurata K. (2006) Mechanisms of neurodegeneration in neuronal ceroid-lipofuscinoses. Acta Neuropathol. 111, 168–177 [DOI] [PubMed] [Google Scholar]
- 2. Lane S. C., Jolly R. D., Schmechel D. E., Alroy J., Boustany R. M. (1996) Apoptosis as the mechanism of neurodegeneration in Batten disease. J. Neurochem. 67, 677–683 [DOI] [PubMed] [Google Scholar]
- 3. Mole S. E., Williams R. E., Goebel H. H. (2005) Correlations between genotype, ultrastructural morphology, and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 6, 107–126 [DOI] [PubMed] [Google Scholar]
- 4. Sleat D. E., Donnelly R. J., Lackland H., Liu C. G., Sohar I., Pullarkat R. K., Lobel P. (1997) Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science 277, 1802–1805 [DOI] [PubMed] [Google Scholar]
- 5. Goebel H. H. (1995) The neuronal ceroid-lipofuscinoses. J. Child Neurol. 10, 424–437 [DOI] [PubMed] [Google Scholar]
- 6. Vines D. J., Warburton M. J. (1999) Classical late infantile neuronal ceroid lipofuscinosis fibroblasts are deficient in lysosomal tripeptidyl-peptidase I. FEBS Lett. 443, 131–135 [DOI] [PubMed] [Google Scholar]
- 7. Chang M., Cooper J. D., Sleat D. E., Cheng S. H., Dodge J. C., Passini M. A., Lobel P., Davidson B. L. (2008) Intraventricular enzyme replacement improves disease phenotypes in a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol. Ther. 16, 649–656 [DOI] [PubMed] [Google Scholar]
- 8. Viglio S., Marchi E., Wisniewski K., Casado B., Cetta G., Iadarola P. (2001) Diagnosis of late-infantile neuronal ceroid lipofuscinosis. A new sensitive method to assay lysosomal pepstatin-insensitive proteinase activity in human and animal specimens by capillary electrophoresis. Electrophoresis 22, 2343–2350 [DOI] [PubMed] [Google Scholar]
- 9. Walus M., Kida E., Golabek A. A. (2010) Functional consequences and rescue potential of pathogenic missense mutations in tripeptidyl-peptidase I. Hum. Mutat. 31, 710–721 [DOI] [PubMed] [Google Scholar]
- 10. Robins S. J., Collins D., Wittes J. T., Papademetriou V., Deedwania P. C., Schaefer E. J., McNamara J. R., Kashyap M. L., Hershman J. M., Wexler L. F., Rubins H. B. (2001) Relation of gemfibrozil treatment and lipid levels with major coronary events. VA-HIT. A randomized controlled trial. JAMA 285, 1585–1591 [DOI] [PubMed] [Google Scholar]
- 11. Rubins H. B., Robins S. J. (1992) Effect of reduction of plasma triglycerides with gemfibrozil on high density lipoprotein-cholesterol concentrations. J. Intern. Med. 231, 421–426 [DOI] [PubMed] [Google Scholar]
- 12. Rubins H. B., Robins S. J., Collins D., Fye C. L., Anderson J. W., Elam M. B., Faas F. H., Linares E., Schaefer E. J., Schectman G., Wilt T. J., Wittes J. (1999) Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high density lipoprotein cholesterol. Veterans Affairs High Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 341, 410–418 [DOI] [PubMed] [Google Scholar]
- 13. Dasgupta S., Roy A., Jana M., Hartley D. M., Pahan K. (2007) Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-α. Mol. Pharmacol. 72, 934–946 [DOI] [PubMed] [Google Scholar]
- 14. Ghosh A., Pahan K. (2012) Gemfibrozil, a lipid-lowering drug, induces suppressor of cytokine signaling 3 in glial cells. Implications for neurodegenerative disorders. J. Biol. Chem. 287, 27189–27203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pahan K., Jana M., Liu X., Taylor B. S., Wood C., Fischer S. M. (2002) Gemfibrozil, a lipid-lowering drug, inhibits the induction of nitric-oxide synthase in human astrocytes. J. Biol. Chem. 277, 45984–45991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roy A., Pahan K. (2009) Gemfibrozil, stretching arms beyond lipid lowering. Immunopharmacol. Immunotoxicol. 31, 339–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brahmachari S., Pahan K. (2007) Sodium benzoate, a food additive and a metabolite of cinnamon, modifies T cells at multiple steps and inhibits adoptive transfer of experimental allergic encephalomyelitis. J. Immunol. 179, 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saha R. N., Pahan K. (2007) Differential regulation of Mn-superoxide dismutase in neurons and astroglia by HIV-1 gp120. Implications for HIV-associated dementia. Free Radic. Biol. Med. 42, 1866–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giulian D., Baker T. J. (1986) Characterization of ameboid microglia isolated from developing mammalian brain. J. Neurosci. 6, 2163–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jana A., Pahan K. (2010) Fibrillar amyloid-β-activated human astroglia kill primary human neurons via neutral sphingomyelinase. Implications for Alzheimer disease. J. Neurosci. 30, 12676–12689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saha R. N., Jana M., Pahan K. (2007) MAPK p38 regulates transcriptional activity of NF-κB in primary human astrocytes via acetylation of p65. J. Immunol. 179, 7101–7109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jana M., Jana A., Pal U., Pahan K. (2007) A simplified method for isolating highly purified neurons, oligodendrocytes, astrocytes, and microglia from the same human fetal brain tissue. Neurochem. Res. 32, 2015–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jana M., Pahan K. (2005) Redox regulation of cytokine-mediated inhibition of myelin gene expression in human primary oligodendrocytes. Free Radic. Biol. Med. 39, 823–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khasnavis S., Jana A., Roy A., Mazumber M., Bhushan B., Wood T., Ghosh S., Watson R., Pahan K. (2012) Suppression of nuclear factor-κB activation and inflammation in microglia by physically modified saline. J. Biol. Chem. 287, 29529–29542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khasnavis S., Pahan K. (2012) Sodium benzoate, a metabolite of cinnamon and a food additive, up-regulates neuroprotective Parkinson disease protein DJ-1 in astrocytes and neurons. J. Neuroimmune Pharmacol. 7, 424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dasgupta S., Jana M., Zhou Y., Fung Y. K., Ghosh S., Pahan K. (2004) Antineuroinflammatory effect of NF-κB essential modifier-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J. Immunol. 173, 1344–1354 [DOI] [PubMed] [Google Scholar]
- 27. Corbett G. T., Roy A., Pahan K. (2012) Gemfibrozil, a lipid-lowering drug, up-regulates IL-1 receptor antagonist in mouse cortical neurons. Implications for neuronal self-defense. J. Immunol. 189, 1002–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saha R. N., Liu X., Pahan K. (2006) Up-regulation of BDNF in astrocytes by TNF-α. A case for the neuroprotective role of cytokine. J. Neuroimmune Pharmacol. 1, 212–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nelson J. D., Denisenko O., Bomsztyk K. (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 1, 179–185 [DOI] [PubMed] [Google Scholar]
- 30. Sohar I., Sleat D. E., Jadot M., Lobel P. (1999) Biochemical characterization of a lysosomal protease deficient in classical late infantile neuronal ceroid lipofuscinosis (LINCL) and development of an enzyme-based assay for diagnosis and exclusion of LINCL in human specimens and animal models. J. Neurochem. 73, 700–711 [DOI] [PubMed] [Google Scholar]
- 31. Sleat D. E., Gin R. M., Sohar I., Wisniewski K., Sklower-Brooks S., Pullarkat R. K., Palmer D. N., Lerner T. J., Boustany R. M., Uldall P., Siakotos A. N., Donnelly R. J., Lobel P. (1999) Mutational analysis of the defective protease in classic late-infantile neuronal ceroid lipofuscinosis, a neurodegenerative lysosomal storage disorder. Am. J. Hum. Genet. 64, 1511–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cullingford T. E., Bhakoo K., Peuchen S., Dolphin C. T., Patel R., Clark J. B. (1998) Distribution of mRNAs encoding the peroxisome proliferator-activated receptor α, β, and γ and the retinoid X receptor α, β, and γ in rat central nervous system. J. Neurochem. 70, 1366–1375 [DOI] [PubMed] [Google Scholar]
- 33. Nishizawa H., Manabe N., Morita M., Sugimoto M., Imanishi S., Miyamoto H. (2003) Effects of in utero exposure to bisphenol A on expression of RARα and RXRα mRNAs in murine embryos. J. Reprod. Dev. 49, 539–545 [DOI] [PubMed] [Google Scholar]
- 34. Hemsley K. M., Hopwood J. J. (2010) Lessons learned from animal models. Pathophysiology of neuropathic lysosomal storage disorders. J. Inherit. Metab. Dis. 33, 363–371 [DOI] [PubMed] [Google Scholar]
- 35. Bellettato C. M., Scarpa M. (2010) Pathophysiology of neuropathic lysosomal storage disorders. J. Inherit. Metab. Dis. 33, 347–362 [DOI] [PubMed] [Google Scholar]
- 36. Vuillemenot B. R., Katz M. L., Coates J. R., Kennedy D., Tiger P., Kanazono S., Lobel P., Sohar I., Xu S., Cahayag R., Keve S., Koren E., Bunting S., Tsuruda L. S., O'Neill C. A. (2011) Intrathecal tripeptidyl-peptidase 1 reduces lysosomal storage in a canine model of late infantile neuronal ceroid lipofuscinosis. Mol. Genet. Metab. 104, 325–337 [DOI] [PubMed] [Google Scholar]
- 37. Worgall S., Sondhi D., Hackett N. R., Kosofsky B., Kekatpure M. V., Neyzi N., Dyke J. P., Ballon D., Heier L., Greenwald B. M., Christos P., Mazumdar M., Souweidane M. M., Kaplitt M. G., Crystal R. G. (2008) Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum. Gene Ther. 19, 463–474 [DOI] [PubMed] [Google Scholar]
- 38. Xu S., Wang L., El-Banna M., Sohar I., Sleat D. E., Lobel P. (2011) Large volume intrathecal enzyme delivery increases survival of a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol. Ther. 19, 1842–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cabrera-Salazar M. A., Roskelley E. M., Bu J., Hodges B. L., Yew N., Dodge J. C., Shihabuddin L. S., Sohar I., Sleat D. E., Scheule R. K., Davidson B. L., Cheng S. H., Lobel P., Passini M. A. (2007) Timing of therapeutic intervention determines functional and survival outcomes in a mouse model of late infantile batten disease. Mol. Ther. 15, 1782–1788 [DOI] [PubMed] [Google Scholar]
- 40. Sleat D. E., El-Banna M., Sohar I., Kim K. H., Dobrenis K., Walkley S. U., Lobel P. (2008) Residual levels of tripeptidyl-peptidase I activity dramatically ameliorate disease in late-infantile neuronal ceroid lipofuscinosis. Mol. Genet. Metab. 94, 222–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marx N., Duez H., Fruchart J. C., Staels B. (2004) Peroxisome proliferator-activated receptors and atherogenesis. Regulators of gene expression in vascular cells. Circ. Res. 94, 1168–1178 [DOI] [PubMed] [Google Scholar]
- 42. Sands M. S., Davidson B. L. (2006) Gene therapy for lysosomal storage diseases. Mol. Ther. 13, 839–849 [DOI] [PubMed] [Google Scholar]
- 43. Juge-Aubry C. E., Gorla-Bajszczak A., Pernin A., Lemberger T., Wahli W., Burger A. G., Meier C. A. (1995) Peroxisome proliferator-activated receptor mediates cross-talk with thyroid hormone receptor by competition for retinoid X receptor. Possible role of a leucine zipper-like heptad repeat. J. Biol. Chem. 270, 18117–18122 [DOI] [PubMed] [Google Scholar]
- 44. Marcus S. L., Miyata K. S., Rachubinski R. A., Capone J. P. (1995) Transactivation by PPAR/RXR heterodimers in yeast is potentiated by exogenous fatty acid via a pathway requiring intact peroxisomes. Gene Expr. 4, 227–239 [PMC free article] [PubMed] [Google Scholar]
- 45. Krey G., Mahfoudi A., Wahli W. (1995) Functional interactions of peroxisome proliferator-activated receptor, retinoid-X receptor, and Sp1 in the transcriptional regulation of the acyl-coenzyme-A oxidase promoter. Mol. Endocrinol. 9, 219–231 [DOI] [PubMed] [Google Scholar]
- 46. Jackson T. C., Mi Z., Bastacky S. I., McHale T., Melhem M. F., Sonalker P. A., Tofovic S. P., Jackson E. K. (2007) PPAR α agonists improve renal preservation in kidneys subjected to chronic in vitro perfusion. Interaction with mannitol. Transpl. Int. 20, 277–290 [DOI] [PubMed] [Google Scholar]
- 47. Xu J., Racke M. K., Drew P. D. (2007) Peroxisome proliferator-activated receptor-α agonist fenofibrate regulates IL-12 family cytokine expression in the CNS. Relevance to multiple sclerosis. J. Neurochem. 103, 1801–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jana M., Jana A., Liu X., Ghosh S., Pahan K. (2007) Involvement of phosphatidylinositol 3-kinase-mediated up-regulation of IκBα in anti-inflammatory effect of gemfibrozil in microglia. J. Immunol. 179, 4142–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]