

Background: Cell migration is involved in altering the cell and matrix interface on the cell surface.
Results: βig-h3 is cleaved by MMP-9, and its cleavage results in changes in its binding properties, cell adhesion, cell migration, FAK/Src signals, and chemoattractant effects.
Conclusion: MMP-9-cleaved βig-h3 modulates tumor cell and macrophage migration.
Significance: The MMP-9-mediated βig-h3 processing mechanism is crucial for understanding cell migration.
Keywords: Adhesion, Cell Migration, Chemotaxis, Matrix Metalloproteinase (MMP), Protein Processing, βig-h3, Extracellular Matrix, FAK/Src Phosphorylation, Matrix Metalloproteinase-9
Abstract
Cell migration is critically involved in inflammation, cancer, and development. In this study, transforming growth factor-β-induced protein (βig-h3) was identified as a substrate of matrix metalloproteinase-9 (MMP-9) by site-directed mutagenesis. βig-h3 has two cleavage sites with the consensus sequence Pro-Xaa-Xaa-Hy-(Ser/Thr) (Hy is a hydrophobic amino acid) (PGSFT beginning at amino acid 135 and PPMGT beginning at amino acid 501). Using recombinant human βig-h3 and MMP-9, βig-h3 from βig-h3-transfected HEK293F cells, and MMP-9 from MMP-9-transfected HEK293F cells, human macrophages, and neutrophils, we found that MMP-9 proteolytically cleaves βig-h3. Cleavage leads to the loss of its adhesive property and its release from extracellular matrix proteins, collagen IV, and fibronectin. Spheroids formed by increased cell-cell interactions were observed in βig-h3-transfected HEK293F cells but not in vehicle-transfected HEK293F cells. In human glioma U87MG cells, MMP-9 constitutive overexpression resulted in endogenous βig-h3 cleavage. βig-h3 cleavage by MMP-9 led to increased cell invasion, and βig-h3 knockdown also resulted in increased cell invasion. The βig-h3 fragment cleaved by MMP-9 could bind to the surface of macrophages, and it may play a role as a peptide chemoattractant by inducing macrophage migration via focal adhesion kinase/Src-mediated signal activation. Thus, intact βig-h3 is responsible for cell migration inhibition, cell-cell contact, and cell-extracellular matrix interaction. Experimental evidence indicates that MMP-9-cleaved βig-h3 plays a role in MMP-9-mediated tumor cell and macrophage migration.
Introduction
The extracellular matrix (ECM)3 is composed of various macromolecules such as collagen, laminin, vitronectin, secreted protein acidic and rich in cysteine (SPARC), and fibronectin that form a highly organized structure among cells. Changes in ECM composition alter the matrix structure and consequently modify matrix-matrix and matrix-cell interactions (1, 2).
Matrix metalloproteinases (MMPs) are ECM-degrading proteinases with biological functions in development, tissue remodeling in inflammatory diseases, and cancer metastasis (3–6). MMPs in a growing tumor promote metastasis through proteolysis of the ECM and activation of signals that are important for tumor cell migration (7). MMPs alter cell function through the release of molecules cleaved from the ECM (8–12). MMPs also cleave non-ECM-binding proteins such as intracellular adhesion molecule-1 (ICAM-1), monocyte chemoattractant protein-3, Fas ligand, and Notch (13–16).
MMP-9, like other MMPs, belongs to a matrixin family of metalloendopeptidases and is synthesized as a 92-kDa zymogen, which is converted to an active enzyme with a molecular mass of 82 kDa (3, 17). MMP-9 is induced by proinflammatory cytokines, and it plays a role in inflammation in various diseases (18–20). It is inhibited by endogenous tissue inhibitors of metalloproteinases (TIMPs) (21, 22). Until now, reported MMP-9 substrates included collagen IV or V, fibronectin, ICAM-1, plasminogen, and interleukin-2 (IL-2) receptor (17). MMP-9-dependent release of vascular endothelial growth factor (VEGF) acts as a chemoattractant for osteoclast recruitment and invasion in development, although the actual substrate has not been identified (23). MMP-9-null mice have reduced development of carcinomas (24).
Transforming growth factor-β-induced protein (also named βig-h3, Bigh3, TGF-β-induced protein h3, keratoepithelin, and RGD-CAP) is a secretory protein that is induced by transforming growth factor-β (ΤGF-β) (25–27). The βig-h3 protein has 683 amino acids and contains an N-terminal cysteine-rich domain and four internal repeat fasciclin (FAS1) domains. The FAS1 domains include YH and RGD motifs, both of which interact with integrins α1β1, α3β1, and αvβ5 (28–32). βig-h3 is ubiquitously expressed at high levels in various normal human tissues but at low levels in many tumor cell lines, including lung, breast, colon, prostate, leukemia, and kidney (33). βig-h3 is produced in fibroblasts at significant levels but is undetectable in one kidney cell line, 293 cells (33). βig-h3 protein suppresses human breast tumor progression and malignant transformation of cells (27, 34–38). Mutation or altered expression of βig-h3 has been linked to the pathogenesis of human corneal dystrophy and osteogenesis (39). However, the underlying molecular mechanism of βig-h3 effects is not well understood. Although MMPs are responsible for extracellular matrix modulation, the mechanism of MMP-9 action is also not clear.
In this study, we found that MMP-9 induction coincided with βig-h3 degradation in IL-1β- or TNF-α-stimulated glioma cells. We also found that βig-h3 is a substrate of MMP-9 and that βig-h3 processing by MMP-9 results in reduced binding affinity to ECM proteins such as fibronectin and collagen IV. βig-h3 processing alters cell invasion and adhesion-related FAK/Src signals in glioma cells. Intact βig-h3 is responsible for cell-cell contact, cell-ECM interaction, and cell migration inhibition. However, its cleavage by MMP-9 leads to increased cell migration. The cleaved fragment of βig-h3 plays a chemoattractant role in macrophage migration via its regulation of adhesion-related FAK/Src signals.
EXPERIMENTAL PROCEDURES
Materials
Recombinant human interleukin-1β (IL-1β) and TNF-α were purchased from R&D Systems. Recombinant human TIMP-1 was obtained from ProSpec. Commercially available recombinant human active MMP-9 catalytic subunit (ProSpec), recombinant human full-length pro-MMP-9 monomer (Calbiochem), recombinant human βig-h3 (Sino Biological), recombinant human βig-h3 fragment (ProSpec, βig-h3–3rd), βig-h3-mutant from βig-h3-overexpressing 293F cells, and pro-MMP-9 from MMP-9-overexpressing 293F cells were used.
Cell Lines
Human embryonic kidney (HEK) 293F cells and human glioblastoma cell line U87MG were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Invitrogen). The human astroglioma cell line CRT-MG was cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% FBS. The cells were maintained at 37 °C in a humidified incubator with a 5% CO2 atmosphere. Cells were confirmed to be mycoplasma-free by PCR. The human monocyte cell line U937 was cultured in RPMI 1640 medium supplemented with 10% FBS and differentiated into macrophages with phorbol 12-myristate 13-acetate (PMA; Calbiochem) treatment (100 μm) for 24 h. The neutrophils were purified from heparinized human peripheral blood by density sedimentation with Ficoll-Paque (GE Healthcare) followed by red blood cell (RBC) lysis with RBC lysis buffer. Isolated neutrophils were cultured in DMEM supplemented with 10% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin. Purity of neutrophils was confirmed by Hema 3 staining.
Plasmids, Transfection, and the Collection of Cell Culture Conditioned Media
βig-h3 and MMP-9 (ImaGenes GmhH, Berlin, Germany) were cloned into pcDNA3.1-myc/His (Invitrogen). The correct sequence of the cloned genes was verified by sequencing. The cloned pcDNA3.1-βig-h3-myc, pcDNA3.1-βig-h3 mutants (P135E, P501E, and P135E/P501E)-myc, and pcDNA3.1-myc (vehicle) were stably transfected into HEK293F cells with Lipofectamine 2000 (Invitrogen). After transfectants were established, cells were cultured with Opti-MEM (Invitrogen) supplemented with a protease inhibitor mixture (Sigma) for 24 h. The conditioned media were collected, centrifuged at 12,000 × g for 10 min, and filtered (0.2 μm, Millipore) to remove cell debris.
Site-directed Mutagenesis
The P135E, P501E, and P135E/P501E mutations in the pcDNA3.1-βig-h3-myc were generated by PCR using the following primers (the mutated codon is underlined) and by using a site-directed mutagenesis kit (iNtRON, Daejon, Korea): P135E forward 5′-GAG ATG GAG GGG GAG GGC AGC TTC ACC-3′ and the P135E reverse 5′-GGT GAA GCT GCC CTC CCC CTC CAT CTC-3′; P501E forward 5′-CGG GTG CTG ACC GAG CCA ATG GGG ACT-3′ and P501E reverse 5′-AGT CCC CAT TGG CTC GGT CAG CAC CCG-3′. The correct sequence and orientation of all cloned genes were verified by sequencing.
βig-h3 Fragment Cloning and Overexpression
The coding sequences of human βig-h3 containing amino acid residues 1–135, 136–501, 502–683, 1–501, and 136–683 were cloned into pcDNA3.1-myc/His (Invitrogen). The correct sequence of the cloned genes was verified by sequencing. Each clone has the coding sequence of the human βig-h3 fragment (amino acid residues 1–135, 136–501, 502–683, 1–501, and 136–683), which is expected to be generated by MMP-9 treatment. “1st,” “2nd,” “3rd,” “1st + 2nd,” and “2nd + 3rd” represent each clone containing amino acid residues 1–135, 136–501, 502–683, 1–501, and 136–683, as indicated in the diagram of Fig. 2C. The cloned pcDNA3.1-βig-h3 fragments (1st + 2nd, 2nd + 3rd, 1st, 2nd, and 3rd)-myc were stably transfected into HEK293F cells with Lipofectamine 2000 (Invitrogen). After transfectants were established, each overexpressed βig-h3 fragment, which was examined by immunoblotting using anti-Myc antibody or anti-βig-h3 antibody, was used as a control for comparing with βig-h3 fragments cleaved by MMP-9.
FIGURE 2.

Cleavage of βig-h3 by recombinant human MMP-9. A, βig-h3 overexpression in HEK293F cells. βig-h3 expression in conditioned media from vehicle (vehicle)-transfected or βig-h3 (βig-h3/293F)-transfected HEK293F cells was examined by immunoblotting using antibodies specific for βig-h3 or Myc (n = 3). B, active recombinant human MMP-9 catalytic subunit and its activity. SDS-PAGE (left panel), immunoblotting analysis (middle panel), and gelatin zymography (right panel) are shown. M is a prestained molecular mass marker (n = 3). C, in vitro cleavage of βig-h3 and βig-h3 mutants by active recombinant human MMP-9 catalytic subunit. Conditioned media (15 μl, 2 μg of total protein) from vehicle (vehicle)-transfected, wild-type βig-h3 (βig-h3/293F)-transfected, or βig-h3 mutant (P135E/P501E (P135/501E), P135E, P501E)-transfected HEK293F cells were treated with recombinant human MMP-9 (rhMMP-9, 0.25 μm), in reaction buffer with 10 mm CaCl2 at 37 °C for 24 h. Immunoblotting was performed using antibodies specific for βig-h3, Myc, and MMP-9 (n = 5). D, full-length recombinant human pro-MMP-9 protein (pro) and APMA-activated MMP-9 (active) identified by immunoblotting analysis using antibody specific for MMP-9. E, in vitro cleavage of βig-h3 and βig-h3 mutants by recombinant human full-length MMP-9. Active MMP-9 was converted from full-length recombinant human pro-MMP using APMA (0.5 mm) and was then purified as described under “Experimental Procedures.” Active MMP-9 (rhMMP-9, 0.05 μm) was pretreated with recombinant human TIMP-1 (rhTIMP-1, 0.25 μm) for 2 h and then was incubated with conditioned media (15 μl, 2 μg of total protein) from vehicle (vehicle)-transfected, wild-type βig-h3 (βig-h3)-transfected, or βig-h3 mutant (P135E/P501E)-transfected HEK293F cells, in reaction buffer with 10 mm CaCl2, at 37 °C for 3 h. Immunoblotting was performed using antibodies specific for βig-3 and MMP-9 (n = 5). F, time course for cleavage of βig-h3 by active recombinant human MMP-9 catalytic subunit. Τhe conditioned media (15 μl, 2 μg of total protein) from βig-h3-transfected HEK293F cells was treated with recombinant human MMP-9 (rhMMP-9 0.25 μm) in reaction buffer with 10 mm CaCl2 at 37 °C for the indicated time periods. After the treatment, immunoblotting was performed using antibodies specific for βig-h3 and Myc (n = 3). G, βig-h3 treated with recombinant MMP-9 for 1 h. Τhe conditioned media (15 μl, 2 μg of total protein) from vehicle (−)-transfected or βig-h3 (+)-transfected HEK293F cells was analyzed by SDS-PAGE (left panel). After the conditioned media (15 μl, 2 μg of total protein) from vehicle (−)- or βig-h3 (+)-transfected HEK293F cells were treated with recombinant human MMP-9 (rhMMP-9, 0.25 μm) at 37 °C for 1 h, immunoblotting was performed using antibodies specific for βig-h3 C terminus (middle panel) and Myc (right panel) (n = 3). H, dose dependence for the cleavage of wild-type βig-h3 (βig-h3) or βig-h3 mutant (P135E/P501E (P135/501E)) by full-length recombinant human MMP-9. Left and right panels, active MMP-9 was converted from full-length recombinant human pro-MMP by APMA (1 mm) and was purified as described under “Experimental Procedures.” The conditioned media (15 μl, 5 μg of total protein) from wild-type βig-h3 (βig-h3)-transfected or βig-h3 mutant (P135E/P501E)-transfected HEK293F cells was treated with various concentrations of active MMP-9 (0, 0.05, 0.25, and 0.5 μm) in reaction buffer with 10 mm CaCl2 at 37 °C for 24 h. Immunoblotting (IB) was performed using antibody specific for a different region of βig-h3 (n = 5). Middle panel, confirmation of βig-h3 fragments overexpressed in βig-h3 fragment-transfected HEK293F cells by immunoblotting. The cloned pcDNA3.1-βig-h3 fragments, pcDNA3.1–1 + 2nd (amino acids 1–501)-myc, pcDNA3.1–2 + 3rd (amino acids 136–683)-myc, and pcDNA3.1–3rd (amino acids 502–683)-myc-transfected 293F cells were transfected into HEK293F cells, respectively. The overexpressed protein was confirmed by immunoblotting using antibodies specific for Myc (n = 3). I, time course for cleavage of wild-type βig-h3 (βig-h3) or βig-h3 mutant (P135E/P501E) by full-length recombinant human MMP-9. Active MMP-9 was converted from full-length recombinant human pro-MMP by APMA (1 mm) and was purified as described under “Experimental Procedures.” Active MMP-9 (0.5 μm) was incubated with conditioned media (15 μl, 5 μg of total protein) from wild-type βig-h3 (βig-h3)- or βig-h3 mutant (P135E/P501E)-transfected HEK293F cells in reaction buffer with 10 mm CaCl2 at 37 °C for various times (0, 1, 6, and 24 h). Immunoblotting was performed using antibody specific for different region of βig-h3 (n = 5).
MMP-9 Activation
Full-length pro-MMP-9 (recombinant human MMP-9 monomer; Calbiochem; 0.1 mg/ml) or serum-free conditioned media from cultures of MMP-9 overexpressing 293F cells were incubated with p-aminophenylmercuric acetate (APMA; Calbiochem; 0.5 or 1 mm) for 24 h at 37 °C. To optimally activate pro-MMP-9 with APMA solution (20 mm APMA, in 0.1 m NaOH), APMA solution was mixed with MMP-9 at a 10:1 volume ratio (MMP-9/APMA, v/v). After incubation, the activated form of MMP-9 was confirmed by SDS-PAGE or gel zymography.
MMP-9 Purification
MMP-9 purification was performed using gelatin-Sepharose chromatography. Briefly, MMP-9 activated by APMA was applied to a column of gelatin-Sepharose equilibrated with buffer containing 50 mm Tris-HCl (pH 7.5), 0.4 m NaCl, and 5 mm EDTA. After washing, elution was performed using a buffer containing 50 mm Tris-HCl (pH 7.5), 1 m NaCl, 5 mm EDTA, and 5% (v/v) DMSO. The eluted fraction was collected, concentrated, and dialyzed against PBS supplemented with MMP inhibitor-free protease inhibitor mixture (Sigma). The purified samples were analyzed by gelatin zymography and immunoblotted to confirm the depletion of other proteins.
Gelatin Zymography
The samples were loaded under nonreducing conditions onto a 7% SDS-PAGE containing 1 mg/ml gelatin. After electrophoresis and washing with a buffer containing 20 mm Tris-HCl (pH 7.5) and 2% Triton X-100 for 1 h, the gel was incubated in MMP reaction buffer containing 20 mm Tris-HCl (pH 7.5) and 10 mm CaCl2 at 37 °C for 16 h. Gelatinolytic activity was detected by staining with Coomassie Brilliant Blue G-250 staining solution.
LC-MS/MS Analysis
Protein bands (Fig. 1E, *) were excised from the stained SDS-polyacrylamide gels and de-stained with destaining solution (25 mm ammonium bicarbonate, 50% acetonitrile). In-gel digestion of dried gel pieces was performed with sequencing grade trypsin (Promega) in 25 mm ammonium bicarbonate buffer overnight at 37 °C. The tryptic peptides were desalted using a GELoader tip (Eppendorf) packed with 1.5 μg of Poros 20 R2 resin (PerSpective Biosystems) and applied onto a C-18 RP-HPLC column (75 μm × 150 mm). An Agilent 1100 Series LC system (Agilent Technologies) was used to separate tryptic peptides, which were eluted with a 0–40% acetonitrile gradient for 60 min. The eluant was analyzed with a Finnigan LCQ Deca (ThermoQuest) equipped with a nanoelectrospray ion source. Spray voltage and tube lens voltage was 1.9 kV and 40 V, respectively. The temperature of the capillary was kept at 210 °C, and capillary voltage was 30 V. The individual spectra from LC-MS/MS were processed using TurboSEQUEST software (ThermoQuest) and searched with NCBI databases using MASCOT software (Matrix Science Ltd.). LC-MS/MS analysis was conducted by ProteomeTech.
FIGURE 1.

MMP-9 induction and identification of βig-h3. A–D, MMP-9 induction (A), cell invasion (B), FAK/Src signal activation (C), and ECM proteins (D). CRT-MG cells were treated with IL-1β (10 ng/ml), TNF-α (10 ng/ml), DETA-NONOate (DETA, 75 μm), or SNAP (0.5 mm) for 24 h. MMP-9 activity, cell invasion, FAK/Src signaling, and ECM proteins were examined (n = 3). A and B, active MMP-9 was detected by gelatin zymography (A) and cell invasion assays (B) on Matrigels. *, p < 0.05. M1 is a marker used to indicate active MMP-9 and pro-MMP-2, and M2 is a marker used to indicate pro-MMP-9 and pro-MMP-2. C and D, FAK/Src/paxillin phosphorylation (C), fibronectin, laminin, collagen IV, SPARC, GAPDH, and unknown protein (*) (D) were detected by immunoblotting (IB). E, upper panel, an unknown protein (*) and SPARC were co-immunoprecipitated (IP) with antibody specific for SPARC. Lower panel, to identify this unknown protein, liquid chromatography tandem mass spectrometry (LC/MS/MS) analysis was performed. The unknown protein was identified as βig-h3. F, βig-h3 protein level in IL-1β (10 ng/ml)-treated and TNF-α (10 ng/ml)-treated CRT-MG cells. G, βig-h3 mRNA level in IL-1β (10 ng/ml)-treated and TNF-α (10 ng/ml)-treated CRT-MG cells. CRT-MG cells were treated with IL-1β (10 ng/ml) or TNF-α (10 ng/ml) for 24 h. The level of βig-h3 expression was measured by RT-PCR and immunoblotting using antibody specific for βig-h3 (n = 3). Control represents untreated samples. (A–D, F, and G).
Solid-phase Binding Assay
Tissue culture plates (96-well) were coated with 10 μg/ml collagen IV or fibronectin and stored at 4 °C overnight. After saturation with 1% bovine serum albumin (BSA control, Sigma) for 1 h at room temperature, the plates were incubated with recombinant human βig-h3 (1 μm, Sino Biological Inc.), untreated, or pretreated with recombinant human MMP-9 (0.25 μm, ProSpec) at 4 °C for 24 h and then washed in PBS with 0.1% Tween 20. After washing, the plates were blocked with 10% goat serum in PBS. Bound βig-h3 was detected using an antibody specific for βig-h3, anti-βig-h3-c (Cell Signaling Technology), or anti-βig-h3-m (Proteintech), followed by horseradish peroxidase (HRP, Cell Signaling Technology)-conjugated anti-rabbit secondary antibody. The plates were washed, treated with substrate and then stop solution, and the absorbance was read at 450 nm. The “ratio” was obtained by dividing the βig-h3 level detected by the anti-βig-h3 antibody in the βig-h3 or βig-h3 plus MMP-9 samples by the βig-h3 level detected by anti-βig-h3 antibody in non-added samples.
Invasion Assay
Invasion assays were performed using 24-Transwell inserts with 8-μm microporous membranes with or without Matrigel (BD Biosciences) coating on the upper side (Costar). The outer membranes of the Transwell inserts were coated with gelatin (1 mg/ml). Cells (5 × 104) preincubated in 100 μl of DMEM or RPMI were placed in the upper chamber, and 600 μl of DMEM containing 2% FBS was added to the lower chamber. After 24 h, the cells remaining on the inner membrane were removed with a cotton swab. The Transwells were stained with hematoxylin (Merck) and eosin Y (Sigma). The cell number was counted using a light microscope.
Spheroid Assay
The spheroid assay was modified as described (40). In brief, 5 × 105 cells were seeded into nonadhesive culture plates and cultured with 293 serum-free media (293SFM; Invitrogen) supplemented with 1 mm glutamine (Invitrogen). Spheroids were allowed to form for 3 or 6 weeks at 37 °C and 5% CO2. Spheroid formation was observed using a light microscope (4×, 3 weeks of culture) and was visible to the naked eye. “Naked eye 1” was defined as the spheroids, which could be visible to the naked eye after 3 weeks of incubation. “Naked eye 2” was defined as the spheroids, which could be visible to the naked eye after 6 weeks of incubation.
Cell Surface Binding Assay by Flow Cytometric Analysis
PMA-differentiated human macrophage U937 cells (1 × 105) were washed in cold RPMI containing 20 mm Tris-HCl (pH 7.4), 1 mm CaCl2, and 1 mm MgCl2. The cells (50-μl aliquots) were incubated with a recombinant human βig-h3 fragment (βig-h3–3rd, amino acid residues 502–683, 25 nm, ProSpec) for 2 h at 4 °C with gentle shaking. After two washes, the cells were blocked with goat serum and incubated for 30 min on ice with anti-βig-h3 antibody or isotype IgG, followed by incubating for 30 min with FITC-conjugated secondary anti-rabbit antibody. After two washes, the cells were fixed in 1% paraformaldehyde in PBS and analyzed by flow cytometry. Flow cytometry was performed on a FACSCalibur (BD Biosciences), and data were analyzed using WinMDI software version 2.8 (The Scripps Research Institute).
Chemotaxis Assay
The chemotaxis assay was performed using 24-well chemotaxis chambers (Costar). The outer membrane of the Transwell insert was coated with gelatin (1 mg/ml). PMA-differentiated human macrophage U937 cells (5 × 104) were preincubated in 100 μl of RPMI and placed in the upper chamber. Then 600 μl of RPMI-containing lipopolysaccharide (LPS) (Sigma, 10 ng/ml), the recombinant human βig-h3 fragment (βig-h3–3rd, amino acid residues 502–683, 25 nm, ProSpec), and the recombinant human βig-h3 (βig-h3 full-length, 25 nm, Sino Biological) were added to the lower chamber. All recombinant human βig-h3 were commercially available. After 16 h, the cells remaining on the inner membrane were removed with a cotton swab. Cells on the transwell plates were fixed in 4% formaldehyde and stained with hematoxylin. The cell number was counted using a light microscope. Endotoxin in the recombinant human βig-h3 fragment was not detected.
Isolation of Cell Lysates for Immunoblotting Analysis
Cells were lysed in buffer containing 20 mm Tris-HCl, 300 mm NaCl, 5 mm EDTA, 0.1% SDS, 0.5% deoxycholate, 1% Nonidet P-40, 1 mm sodium orthovanadate, 1 mm NaF, 1 mm sodium pyrophosphate, and protease inhibitor mixture (Roche Applied Science) for 30 min on ice. The cell lysates were subjected to immunoblotting with the primary antibodies anti-GAPDH (Millipore), anti-FAK, anti-p-FAK, anti-Src, anti-p-Src, anti-paxillin, anti-p-paxillin (Cell Signaling Technology), anti-fibronectin, anti-laminin (Millipore), anti-collagen IV, and anti-SPARC (Santa Cruz Biotechnology). Protein bands were detected using ECL Plus reagent (GE Healthcare). Protein concentrations were determined by the Bradford assay (Bio-Rad).
Immunoprecipitation
Cells were lysed with Lysis Buffer (50 mm HEPES, 150 mm NaCl, 1% Nonidet P-40 (pH 8.0)) for 15 min on ice and then centrifuged at 12,000 × g and at 4 °C. Supernatants were pre-cleared for 1 h at 4 °C with G protein beads (Invitrogen). The pre-cleared samples were immunoprecipitated at 4 °C for 18 h using anti-βig-h3 or control rabbit IgG, which were coupled to G protein beads (Invitrogen). Samples were examined by immunoblotting with anti-mouse Myc antibody followed by HRP-conjugated anti-mouse IgG secondary antibody.
RNA Isolation, RT-PCR, and Semi-quantitative RT-PCR Analysis
RNA was isolated from cells using the RNeasy protect mini kit (Qiagen, Hilden, Germany). Isolated RNA was reverse-transcribed into cDNA using oligo(dT) primers (Qiagen) and then amplified using specific gene primers. All PCR products were resolved on 2% agarose gels. Oligonucleotide primers for βig-h3 and β-actin were as follows: βig-h3 5′-TCATCGATAAGGTCATCTCCA-3′ (sense) and 5′-TGGTGGCTAGGTTGTCTTTAT-3′ (antisense); β-actin, 5′-GGGTCAGAAGGATTCCTATG-3′ (sense) and 5′-CCTTAATGTCACGCACGATTT-3′ (antisense). β-Actin was used as an internal control to evaluate the expression of each molecule.
Statistical Analysis
All experiments were repeated at least three times. Data represent the means ± S.E. A value of p ≤ 0.05 was considered to be statistically significant.
RESULTS
MMP-9 Induction and Identification of βig-h3
MMP-9 is induced by inflammatory cytokines, including IL-1β and TNF-α in glioma cells (41). To understand how MMP-9 affects ECM proteins, human glioma CRT-MG cells were stimulated with IL-1β, TNF-α, or NO producers, diethylenetriamine NONOate (DETA-NONOate) and S-nitroso-l-acetyl-dl-penicillamine (SNAP). The SNAP and DETA-NONOate are known to have a common signaling pathway with IL-1β through nitric oxide (42). However, SNAP and DETA-NONOate did not affect MMP-9 induction and FAK/Src activation (Fig. 1, A and C). Therefore, increases in MMP-9 induction and FAK/Src activation induced by IL-1β may be unrelated to nitric oxide-induced signaling in glioma cells. However, both IL-1β and TNF-α cause increased MMP-9, FAK/Src/paxillin phosphorylation, and cell migration in glioma CRT-MG cells (Fig. 1, A–C).
In IL-1β or TNF-α-stimulated cells, the levels of ECM proteins, including fibronectin, laminin, collagen IV, and SPARC were unchanged, even though fibronectin and collagen IV are known as substrates of MMP-9 (Fig. 1D) (17). Interestingly, an unknown protein band was detected in immunoblots with anti-SPARC antibody (Fig. 1, D and E, upper panel, *). The identity of the protein (*) is unknown, but it is distinct, and its disappearance coincides with MMP-9 induction (Fig. 1, A and D). These results suggest that the unknown protein may be involved in MMP-9-mediated cell migration. The unknown protein was identified as βig-h3 by LC/MS/MS analysis (Fig. 1E, lower panel). Results of immunoblotting analysis revealed that βig-h3 undergoes degradation in MMP-9-induced cells (Fig. 1F). To confirm that IL-1β and TNF-α do not directly affect βig-h3 mRNA, RT-PCR was conducted. In glioma CRT-MG cells, βig-h3 mRNA expression levels were not changed by IL-1β or TNF-α treatment (Fig. 1G).
In Vitro Cleavage of βig-h3 Protein by MMP-9
To determine the relationship between MMP-9 and βig-h3, the βig-h3 sequence was examined. βig-h3 contained two putative consensus MMP-9 cleavage sites with a sequence, Pro-Xaa-Xaa-Hy-Ser/Thr, where Xaa can be any amino acid and Hy is a hydrophobic amino acid (43). The sequences in βig-h3 consistent with this motif are PGSFT and PPMGT, beginning at amino acid residues 135 and 501, respectively. To determine whether these sequences are proteolytic cleavage sites for MMP-9, wild-type βig-h3 or mutant βig-h3 with proline to glutamic acid substitutions (P135E, P501E, and P135E/P501E) were cloned into the mammalian expression vector pcDNA3.1-myc. For stable overexpression of wild-type βig-h3 and βig-h3 mutants, we selected HEK293F cells, in which the endogenous βig-h3 protein is undetectable (33). βig-h3 expression in βig-h3-transfected HEK293F cells was confirmed by immunoblot analysis of the cell culture-conditioned media with antibodies specific for Myc or βig-h3 (Fig. 2A). There was no endogenous βig-h3 expression in the vehicle-transfected HEK293F cells (Fig. 2A). Commercially available recombinant human active MMP-9 catalytic subunit was used for examining the cleavage of βig-h3, because its activation process was unnecessary (Fig. 2B). When βig-h3, secreted from βig-h3-transfected HEK293F cells, was treated with recombinant human MMP-9 containing the catalytic subunit, the βig-h3 protein band disappeared, suggesting cleavage by MMP-9 (Fig. 2C). In contrast, when the βig-h3 mutant protein (P135E/P501E), with mutations in both MMP-9 cleavage sites, was treated with MMP-9, no cleavage was observed. The βig-h3 mutant proteins containing a mutation in a single cleavage site (P135E and P501E) were more susceptible to cleavage than the double mutant βig-h3 (P135E/P501E) (Fig. 2C).
To confirm if full-length MMP-9 could cleave βig-h3, the full-length commercially available recombinant human MMP-9 was tested. APMA is known for its ability to enhance the autocleavage of the proenzyme to the active and lower molecular mass form. Active and lower molecular mass form MMP-9 was confirmed by immunoblotting using anti-MMP-9 antibody (Fig. 2D). When βig-h3 secreted from βig-h3-transfected HEK293F cells was treated with the full-length MMP-9, the βig-h3 protein band also disappeared, indicating cleavage by MMP-9 (Fig. 2E). In addition, pretreatment with the MMP-9 inhibitor, TIMP-1, prevented the loss of βig-h3. However, when the βig-h3 protein (P135E/P501E) with mutations in both MMP-9 cleavage sites was treated with APMA-activated full-length MMP-9, there was no change (Fig. 2E).
A time course study of βig-h3 degradation by MMP-9 was conducted and is shown in Fig. 2F. In the absence of MMP-9 treatment, there was no βig-h3 degradation (Fig. 2F). After MMP-9 treatment for 1 h, the cleaved βig-h3 fragment was detected using anti-Myc antibody (Fig. 2G, right panel). Because the cleaved βig-h3 fragment was not detected by anti-βig-h3 antibody, which recognizes the C terminus of the protein (Fig. 2G, middle panel), antibody specific for different regions of the βig-h3 protein were used for detecting both uncleaved and cleaved products (Fig. 2, H and I). βig-h3 fragments cleaved by active full-length MMP-9 increased in a time- and concentration-dependent manner, whereas uncleaved βig-h3 decreased (Fig. 2, H and I, left panels). However, when the βig-h3 protein (P135E/P501E) with mutations in both MMP-9 cleavage sites was treated with activated full-length MMP-9, there was no change (Fig. 2, H and I, right panels).
These fragments corresponded in size to the proteins overexpressed from the cloned pcDNA3.1-βig-h3 fragments pcDNA3.1–1st + 2nd (amino acid residues 1–501)-myc, pcDNA3.1–2nd + 3rd (amino acid residues 136–683)-myc, and pcDNA3.1–3rd (amino acid residues 502–683)-myc in transfected 293F cells (Fig. 2H, middle panel). The fragments containing amino acid residues 1–135 or amino acid residues 136–501 were not overexpressed in transfected 293F cells because the transfected cells were detached, and their cell viability decreased during the establishment of transfection.
Full-length MMP-9 Purified from Different Sources Also Cleaved βig-h3 Protein
To confirm that full-length MMP-9 secreted from different sources can cleave βig-h3 protein, MMP-9 from MMP-9-transfected HEK293F cells, differentiated U937 macrophages, and human blood neutrophils were isolated and examined. MMP-9 transfected HEK293F cells were established by stable transfection with the MMP-9 expression vector (MMP-9-pcDNA3.1-myc) into HEK293F cells (Fig. 3A). MMP-9 expression was confirmed by immunoblotting of the cell culture media with antibodies specific for Myc or MMP-9. Unexpectedly, pro-MMP-9 was overexpressed and secreted from MMP-9-transfected HEK293F cells (Fig. 3, A–D). The protein was detected by gelatin zymography that HEK293F cells expressed low levels of endogenous, active MMP-9. However, the level was very low when compared with overexpressed pro-MMP-9 (Fig. 3B). To induce the conversion of pro-MMP-9 to the lower molecular mass active form of MMP-9, the conditioned media of MMP-9-transfected HEK293F cells were incubated with APMA (Fig. 3D). The active form of MMP-9 observed after APMA treatment was confirmed by gelatin zymography (Fig. 3E, upper panel). The active MMP-9 was purified by affinity chromatography using gelatin-Sepharose to purify it from other proteins (Fig. 3E, upper panel). The purified active MMP-9 cleaved βig-h3, as indicated by immunoblotting analysis (Fig. 3E, lower panel). In addition, the MMP-9 inhibitor TIMP-1 pretreatment inhibited βig-h3 loss (Fig. 3F).
FIGURE 3.

βig-h3 cleavage by purified MMP-9 from different sources. A and B, MMP-9 expression and activity in MMP-9-transfected HEK293F cells. MMP-9 expression in the conditioned media from vehicle (vehicle)-transfected or MMP-9 (MMP-9/293F)-transfected HEK293F cells were examined by immunoblotting using antibodies specific for MMP-9 and Myc (A) and gelatin zymography (B). M is a marker for active MMP-9 and pro-MMP-2 (n = 3). C, purified MMP-9 from MMP-9-transfected HEK293F cells. MMP-9 from the conditioned media of vehicle (vehicle)- or MMP-9 (MMP-9/293F)-transfected HEK293F cells purified by gelatin-Sepharose chromatography were detected by SDS-PAGE. T represents the total conditioned media from the vehicle-transfected or MMP-9 (MMP-9/293F)-transfected HEK293F cells, and P represents purified MMP-9 from the conditioned media (n = 3). D, conversion of pro-MMP-9 to active MMP-9 by APMA treatment. After the conditioned media (15 μl, 2 μg of total protein) from MMP-9-transfected HEK293F cells was treated with 1 mm APMA at 37 °C for 24 h, immunoblotting was performed using an antibody specific for MMP-9 (n = 3). E and F, in vitro cleavage of βig-h3 by purified MMP-9 and the inhibitory effect of TIMP-1. E, upper panel, after the conditioned media from MMP-9-transfected HEK293F cells were treated with 1 mm APMA at 37 °C for 24 h, active MMP-9 was purified by gelatin-Sepharose chromatography, and enzyme activity was detected by gelatin zymography. The pro-MMP-9 (total) in the conditioned media from MMP-9-transfected HEK293F cells was detected (n = 3). Lower panel, after the conditioned media (15 μl, 2 μg of total protein) from vehicle (vehicle)-transfected or βig-h3 (βig-h3/293F)-transfected HEK293F cells was treated with purified human MMP-9 (purified MMP-9/293F) at 37 °C for 24 h, immunoblotting was performed using antibodies specific for βig-h3, Myc, and MMP-9 (n = 3). F, purified human active MMP-9 (purified MMP-9/293F) was pretreated with recombinant human TIMP-1 (rhTIMP-1, 0.25 μm) for 2 h and then was incubated with conditioned media (15 μl, 2 μg of total protein) from βig-h3 (βig-h3/293F)-transfected HEK293F cells at 37 °C for 3 h. After incubation, immunoblotting was performed using antibodies specific for βig-h3 and MMP-9 (n = 3). G, in vitro βig-h3 cleavage by MMP-9 purified from PMA-differentiated U937 cells. Upper panel, MMP-9 from PMA-differentiated U937 cells was purified by gelatin-Sepharose chromatography, and its activity was detected by gelatin zymography. Active MMP-9 was detected in conditioned media from PMA-differentiated U937 cells. Lower panel, after the conditioned media (15 μl, 2 μg of total protein) from vehicle (vehicle)-transfected or βig-h3 (βig-h3/293F)-transfected HEK293F cells was treated with purified MMP-9 (purified MMP-9/macrophages) at 37 °C for 24 h, immunoblotting was performed using antibodies specific for βig-h3, Myc, and MMP-9 (n = 3). H, in vitro βig-h3 cleavage by MMP-9 purified from human blood neutrophils. Upper panel, MMP-9 from human blood neutrophils was purified by gelatin-Sepharose chromatography, and its activity was detected by gelatin zymography. Active MMP-9 was detected in conditioned media from human blood neutrophils (total) (n = 3). Lower panel, after the conditioned media (15 μl, 2 μg of total protein) from vehicle (vehicle)- or βig-h3 (βig-h3/293F)-transfected HEK293F cells were treated with purified MMP-9 (purified MMP-9/neutrophils) at 37 °C for 24 h, immunoblotting was performed using antibodies specific for βig-h3, Myc, and MMP-9. In the upper panels, total represents unpurified MMP-9 and purified represents purified MMP-9. M1 is a pre-stained molecular weight marker, and M2 is a marker to indicate active MMP-9 and MMP-2 (n = 3).
MMP-9 production is primarily observed in neutrophils and macrophages in humans (44–46). PMA-differentiated U937 macrophages or human blood neutrophils release large amounts of active MMP-9 (Fig. 3, G and H). MMP-9 from neutrophils without TIMP were more effective than from other cells (47). To obtain MMP-9 from macrophages, human monocytes (U937) were differentiated with PMA, which is known to stimulate macrophage differentiation from monocytes (48). Active MMP-9 from the culture media of PMA-differentiated U937 macrophages or from human blood neutrophils was purified by gelatin affinity-Sepharose chromatography and detected by gelatin zymography (Fig. 3, G and H, upper panel). Treatment with this purified MMP-9 caused βig-h3 degradation (Fig. 3, G and H, lower panel). Overall, these results suggest that βig-h3 contains a cleavage site, which is recognized by MMP-9.
MMP-9 Cleavage Alters the Adhesiveness of βig-h3 to ECM and to Cell to Cell Interactions
We examined the ability of βig-h3 to bind ECM proteins in a solid-phase assay using commercially available recombinant human βig-h3 with the ECM proteins collagen IV and fibronectin. To determine whether the binding of βig-h3 to ECM proteins is influenced by MMP-9, we used a solid-phase assay to quantitate the amount of βig-h3 bound to collagen IV and fibronectin with or without MMP-9 pretreatment. Two antibodies specific for recombinant human βig-h3 were used for detection of βig-h3; one recognizes the C terminus of βig-h3 (anti-βig-h3-c) and the other recognizes the middle region of βig-h3 (anti-βig-h3-m). Untreated βig-h3 strongly bound to collagen IV and fibronectin, whereas βig-h3 pretreated with MMP-9 had decreased binding to collagen IV (Fig. 4A) and fibronectin (Fig. 4B).
FIGURE 4.
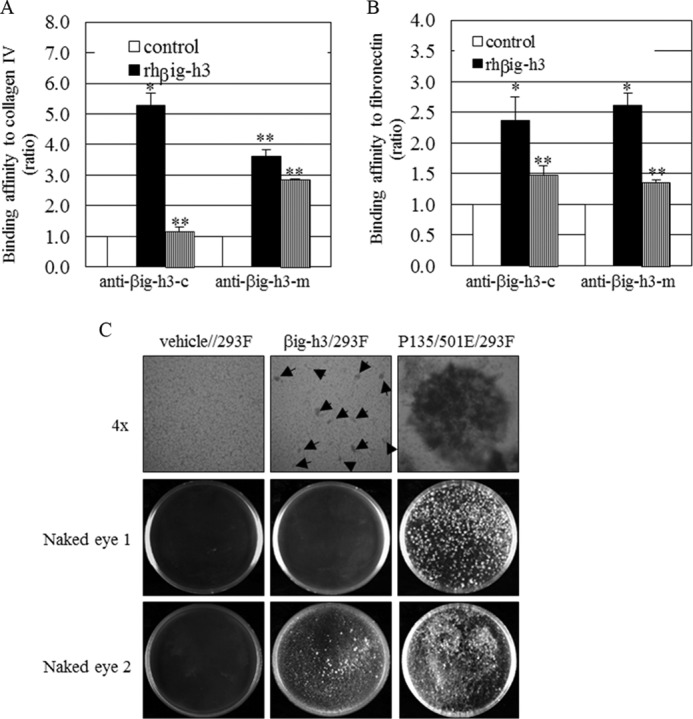
βig-h3 cleavage, adhesiveness, and cell-cell interaction. A and B, βig-h3 adhesiveness to collagen IV (A) and fibronectin (B) after MMP-9 addition. Recombinant human βig-h3 (rhβig-h3, 1 μm) was pretreated or untreated with recombinant human MMP-9 (rhMMP-9, 0.25 μm), and a solid-phase binding assay was used to determine the adhesiveness of βig-h3 to collagen IV and fibronectin (*, p < 0.05; **, p < 0.01, and n = 3). The ratio refers to the βig-h3 level detected by anti-βig-h3 antibody, in βig-h3 or βig-h3 plus MMP-9 samples, divided by the βig-h3 level detected by anti-βig-h3 antibody, without additions. C, cell-cell interactions in the presence of βig-h3, and the endogenous MMP-9 effects. Spheroids were formed in vehicles (vehicle/293F), in βig-h3 (βig-h3/293F)-transfected, or the double mutant βig-h3 (P139/501E/293F)-transfected HEK293F cells within 3 weeks, and spheroid formation was visible under light microscopy (4×, upper panel) or with the naked eye (Naked eye 1). Spheroids were more visible with the naked eye at 6 weeks (Naked eye 2). Arrows marking the βig-h3-transfected cells (βig-h3/293F) indicate small aggregates (C) (n = 3).
In addition, when the anti-βig-h3-c antibody that recognizes the βig-h3 C terminus was used, the effect of MMP9 on βig-h3 binding to collagen IV was greater. This effect was less when the anti-βig-h3-m antibody recognizing the βig-h3 middle region was used. We suggest that the βig-h3 C-terminal region would be involved in binding to collagen to a greater extent.
These results further suggest that βig-h3 adheres to collagen IV and fibronectin. MMP-9 cleaves βig-h3, and this leads to βig-h3 release from collagen IV and fibronectin, thereby weakening the binding between cells.
To determine whether βig-h3 can act as a bridge between cells, a spheroid assay was performed. The average size of cell spheroids can vary from very small aggregates to larger aggregates of several thousand cells with a diameter of several hundred micrometers (40). Seeding of suspended HEK293F cells transfected with either wild type or mutant βig-h3 (P135E/P501E) led to the formation of multicellular aggregates, whereas vehicle-transfected cells did not form spheroids (Fig. 4C). Cells transfected with the double mutant βig-h3 (P135E/P501E) formed dramatically larger aggregates resulting in the formation of large spheroids (Fig. 4C, right panels). Interestingly, although wild-type βig-h3-transfected HEK293F cells did not form large aggregates, very small aggregates were observed (Fig. 4C, arrows in middle panels). This was in stark contrast to the large aggregates observed in βig-h3 mutant (P135E/P501E)-transfected HEK293F cells. This suggests that wild-type βig-h3 is susceptible to cleavage by endogenous MMP-9 that has accumulated during long term culture, whereas the βig-h3 mutant is not susceptible. After further culturing for 6 weeks, spheroids were more visible to the naked eye. Spheroids from wild-type βig-h3-transfected HEK293F cells were also more detectable and those of the double mutant βig-h3 (P139/501E)-transfected HEK293F cells were larger (Fig. 4C, Naked eye 2). These results suggest that uncleaved βig-h3 helps cells to form aggregates.
Intact βig-h3 Inhibits Cell Migration but βig-h3 Cleavage or Knockdown Leads to Increased Cell Migration
We next examined whether the endogenous βig-h3 secreted from cells is cleaved by MMP-9 in these cells. Human glioma U87MG cells secrete endogenous βig-h3 and do not secrete MMP-9. To confirm that MMP-9 cleaves the endogenous βig-h3, MMP-9 cDNA (MMP-9-pcDNA3.1-myc) was stably transfected into U87MG cells (Fig. 5A). MMP-9 constitutively overexpressed in U87MG cells leads to βig-h3 cleavage (Fig. 5, A and B). Furthermore, the βig-h3 cleavage caused by MMP-9 overexpression led to increased cell invasion (Fig. 5D). However, there were no differences in cell invasion and βig-h3 protein bands between untreated and IL-1β-stimulated U87MG cells, in which MMP-9 was not induced by IL-1β stimulation (Fig. 5D).
FIGURE 5.

Endogenous βig-h3 cleavage by MMP-9 in glioma cells. A and B, βig-h3 and MMP-9 in MMP-9-transfected U87MG cells. A, βig-h3 and MMP-9 expression in the vehicle (vehicle/U87MG)-transfected or MMP-9 (MMP-9/U87MG)-transfected U87MG cells were examined by immunoblotting. B, MMP-9 activity was detected by gelatin zymography. M is a marker for active MMP-9 and pro-MMP-2 (n = 3). C, co-immunoprecipitation of MMP-9 with βig-h3 from the vehicle (vehicle/U87MG)-transfected or MMP-9 (MMP-9/U87MG)-transfected U87MG cells. After the lysates from the vehicle (vehicle/U87MG)-transfected or MMP-9 (MMP-9/U87MG)-transfected U87MG cells were immunoprecipitated (IP) with anti-βig-h3 (+) or control rabbit IgG (−), they were subjected to immunoblotting (IB) using mouse antibody for Myc (MMP-9) (n = 4). D, MMP-9 overexpressed in cells alters cell invasion. Cell invasion in vehicle-transfected, MMP-9-transfected, or IL-1β (10 ng/ml)-treated U87MG cells was examined on Matrigels. βig-h3 and MMP-9 proteins from IL-1β-treated human glioma U87MG cells were examined by immunoblotting and zymography. The control represents untreated samples (n = 3). E, βig-h3 knockdown alters cell invasion. Cell invasion in control siRNA or βig-h3 siRNA-transfected U87MG cells was examined on Matrigels. The βig-h3 and MMP-9 from these cells were examined by immunoblotting and zymography. Control represents control siRNA. (*, p < 0.05; **, p < 0.01, n = 3).
To determine whether MMP-9 and βig-h3 interact on the cell surface, immunoblotting analysis of anti-βig-h3 antibody immunoprecipitates from lysates of MMP-9-transfected U87MG cells was performed. The association of βig-h3 and MMP-9 in these immunoprecipitates was observed, although the quantity of the associated complex between βig-h3 and MMP-9 was small (Fig. 5C). These results suggest that βig-h3 may provide a docking site as well as a cleavage site for MMP-9 on the cell surface.
To confirm that the increased cell migration results from βig-h3 decrease, βig-h3 expression in U87MG cells was knocked down by βig-h3 siRNA transfection (Fig. 5E). βig-h3 knockdown in cells led to increased cell invasion even though cell migration after βig-h3 knockdown was less than when mediated by MMP-9-catalyzed βig-h3 degradation (Fig. 5, D and E). Thus, intact βig-h3 inhibits cell migration. Both MMP-9-mediated βig-h3 degradation and βig-h3 knockdown led to increased cell migration in glioma cells (Fig. 5, D and E).
βig-h3 Fragment Generated by MMP-9 Cleavage Affects Macrophage Migration
Αs βig-h3 has two cleavage sites recognized by MMP-9, three βig-h3 fragments were expected to be generated by MMP-9 treatment. The decrease of βig-h3 binding affinity to the ECM after MMP-9 cleavage was observed in a solid-phase assay (Fig. 4, A and B). The βig-h3 cleavage by MMP-9 leads to cell migration and FAK/Src activation in glioma cells (Figs. 1 and 5). However, among these fragments, the C-terminal βig-h3 fragment composed of amino acid residues 502–683 still has the RGD and YH motifs, which interact with cell surface integrins. To determine whether this βig-h3 fragment can bind to neighboring cell surfaces after its release, the cell surface binding assay was performed by flow cytometry, using the commercially available recombinant human βig-h3 fragment (βig-h3–3rd) that contains 182 amino acids (residues 502–683). This recombinant human βig-h3 fragment (βig-h3–3rd) corresponds to the C-terminal fragment of βig-h3, which is expected to be generated after MMP-9 cleavage. The recombinant human βig-h3 fragment (βig-h3–3rd) induced macrophage migration (Fig. 6A), bound to the surface of macrophages, and increased phosphorylated FAK/Src (Fig. 6, B and C). This result demonstrates that the small C-terminal βig-h3 fragment functions as a peptide chemoattractant to neighboring cells, resulting in cell migration.
FIGURE 6.
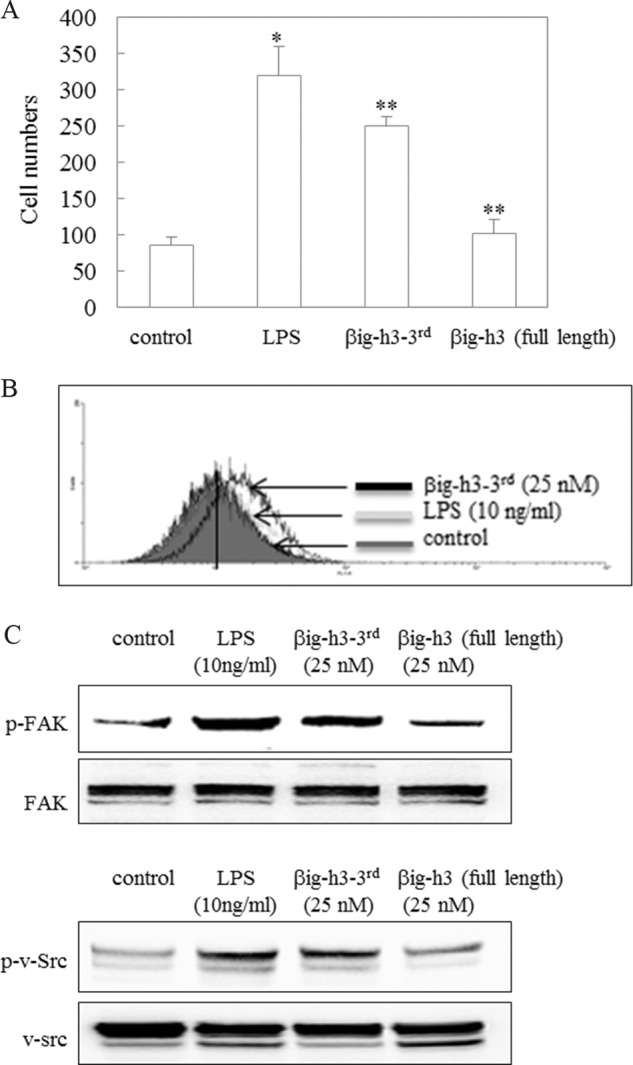
Macrophage surface binding, migration, and FAK/Src phosphorylation in the presence of recombinant human βig-h3 fragment (βig-h3–3rd). A, effect of βig-h3–3rd on macrophage migration. Macrophage migration was determined by a chemotaxis assay. After PMA-differentiated U937 macrophages were incubated with LPS (10 ng/ml), βig-h3–3rd (25 nm), or βig-h3 (full-length; 25 nm) for 20 h, the migrating macrophage numbers were counted. The control represents untreated samples. *, p < 0.05; **, p < 0.01 (n = 3). B, βig-h3–3rd binding to macrophage surfaces. βig-h3–3rd binding to the surface of PMA-differentiated U937 macrophages was determined by a surface binding assay. The control represents untreated samples (n = 3). C, enhanced FAK/Src phosphorylation by βig-h3–3rd. After PMA-differentiated U937 macrophages were incubated with LPS (10 ng/ml), βig-h3–3rd (25 nm), or βig-h3 (full length; 25 nm) for 1 h, the lysates were isolated, and FAK/Src phosphorylation was examined using antibodies specific for FAK, p-FAK, Src, and p-Src. The control represents untreated samples (n = 3).
DISCUSSION
Cell migration plays a role in inflammation, cancer, and development. MMP-9 proteolytically acts on the ECM and activates signals involved in tumor cell migration (49). In glioma, tumor invasion is inhibited by suppressing MMP-9 secretion and dephosphorylation of FAK (50). It is believed that βig-h3 may function as either an activator or an inhibitor of carcinogenesis, depending on the cell and tumor type. However, the mechanism of action of βig-h3 remains unclear.
In our study, we demonstrated that βig-h3 is a substrate of MMP-9 and identified its cleavage sites by site-directed mutagenesis. βig-h3 adheres to collagen IV and fibronectin in the ECM. Cleavage of βig-h3 by MMP-9 results in reduced binding of βig-h3 and in the release of a βig-h3 fragment from the ECM. βig-h3 has the RGD and YH motifs that interact with cell surface integrins (28, 31). Spheroids formed by increased cell-cell interactions were observed in cells overexpressing wild-type βig-h3 or in the cells with the double mutant βig-h3 (P135E/P501E). It is proposed that βig-h3 functions as a bridge between cells or between cells and the ECM. These functions of βig-h3 are directly influenced by MMP-9 cleavage.
Cell migration is involved in altering the cell-matrix interface on the cell surface. It is known that MMP-9 is involved in cell migration, but the detailed mechanism is not clear. In our study, βig-h3 was expressed in glioma cells. It was also demonstrated that βig-h3 directly binds with MMP-9 on cells and that MMP-9 overexpression leads to βig-h3 cleavage. Thus, we suggest that βig-h3 may provide both a docking site and a cleavage site for MMP-9 on the cell surface. Glioma cells expressing intact βig-h3 did not exhibit cell migration. In contrast, cells overexpressing MMP-9 in the presence of βig-h3 exhibited cleaved βig-h3 and increased cell migration. βig-h3 knockdown also led to increased cell migration. Thus, we conclude that intact βig-h3 inhibits cell migration, but degradation of βig-h3 by MMP-9 leads to increased cell migration.
MMP-9 induction in IL-1β- or TNF-α-stimulated human glioma CRT-MG cells resulted in βig-h3 cleavage, adhesion-related FAK/Src activation, and increased cell migration. However, fibronectin, collagen IV, laminin, and SPARC in these cells did not undergo degradation even though fibronectin and collagen IV are known substrates of MMP-9. We cannot exclude the possibility that the degradation of other ECM proteins besides fibronectin, collagen IV, and laminin is involved in MMP-9 induction, cell migration, and FAK/Src signal activation. However, upon MMP-9 release from the cell surface, MMP-9 activity is inhibited by TIMP (13). The proteolytic activity of MMP-9 is most effective when MMP-9 is localized on the cell surface where its activity can be modulated by eliminating its docking site (13). Thus, it is also likely that βig-h3 degradation by MMP-9 modulates cell migration and MMP-9 activity by eliminating the binding of βig-h3 as its docking site on the cell surface. βig-h3 degradation by MMP-9 leads to βig-h3 release from the cell surface, resulting in the loss of MMP-9-docking site and in MMP-9 loss from the cell surface. MMP-9, once removed from the cell surface, may have less interaction with other ECM proteins due to docking site loss. Thus, we propose that βig-h3 cleavage may be involved in decreasing the interaction of MMP-9 with ECM proteins.
MMP-9 is mainly observed in neutrophils and macrophages in humans (45–47). Macrophages enhance tumor progression and metastasis (51). Tumor-associated macrophages, following their migration into the primary tumor sites, support metastatic cancer cell extravasation (51, 52). Macrophage migration is often initiated in response to a chemotactic stimulus. In our study, intact βig-h3 did not affect cell migration, but the degradation of βig-h3 by MMP-9 leads to increased cell migration. The βig-h3 fragments cleaved by MMP-9 were easily detached from the ECM. The fragment containing the C-terminal domain could bind to the surface of macrophages and increase their migration through the activation of FAK/Src-mediated signals. These results suggest that the fragment cleaved by MMP-9 stimulates migration of the surrounding cells by binding to them. Thus, the MMP-9-dependent, processed, and released βig-h3 acts as a chemoattractant.
In this study, we found out that IL-1β and TNF-α treatment led to MMP-9 induction, FAK activation, and cell migration in glioma cells. With this correlative finding, we demonstrated that MMP-9 could cleave βig-h3 in a molecular level. Through the use of cell-based assays, we demonstrated that βig-h3 is an adhesion-related protein and that MMP-9 could cleave βig-h3 in a cell culture model. The cleavage was not degraded because the resulting fragments played a role in cell migration. Thus, in conclusion, this study provides an explanation of how MMP-9 could induce cell migration.
Furthermore, in some tumor cells it was reported that IL-1β and TNF-α secreted from macrophages could enhance tumor cell invasion (53). In our study, IL-1β and TNF-α led to MMP-9 induction in glioma cells. MMP-9 increased cell migration through βig-h3 cleavage. We hypothesize that the increased cell migration, by MMP-9-mediated βig-h3 cleavage, plays an important role in enhancing tumor cell invasion.
In conclusion, this study suggests for the first time that βig-h3 is a substrate of MMP-9 and that βig-h3 plays a regulatory role in MMP-9-mediated cell migration of tumor cells and macrophages.
Acknowledgments
We thank Dr. H. K. Koh of R&D Biolab for technical advice and Dr. J. Park of Hallym University for providing us with human CRT-MG cells. We thank Dr. B. J. Kim of ProteomeTech Inc. for assistance and technical advice with βig-h3 protein LC/MS/MS analyses.
This work was supported by the Future-based Technology Development Program through the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology Grant 2009-0081760.
- ECM
- extracellular matrix
- MMP-9
- matrix metalloproteinase 9
- βig-h3
- TGF-β-induced protein h3
- IL-1β
- Interleukin-1β
- FAK
- focal adhesion kinase
- SPARC
- secreted protein acidic and rich in cysteine
- TIMP
- tissue inhibitors of metalloproteinase
- APMA
- p-aminophenylmercuric acetate
- PMA
- phorbol 12-myristate 13-acetate
- DETA-NONOate
- diethylenetriamine NONOate
- SNAP
- S-nitroso-l-acetyl-dl-penicillamine.
REFERENCES
- 1. Aumailley M., Gayraud B. (1998) Structure and biological activity of the extracellular matrix. J. Mol. Med. 76, 253–265 [DOI] [PubMed] [Google Scholar]
- 2. Weber K. T., Sun Y., Katwa L. C. (1995) Local regulation of extracellular matrix structure. Herz 20, 81–88 [PubMed] [Google Scholar]
- 3. Visse R., Nagase H. (2003) Matrix metalloproteinases and tissue inhibitors of metalloproteinases. Structure, function, and biochemistry. Circ. Res. 92, 827–839 [DOI] [PubMed] [Google Scholar]
- 4. Egeblad M., Werb Z. (2002) New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 [DOI] [PubMed] [Google Scholar]
- 5. Seiki M. (2002) The cell surface: the stage for matrix metalloproteinase regulation of migration. Curr. Opin. Cell Biol. 14, 624–632 [DOI] [PubMed] [Google Scholar]
- 6. Handsley M. M., Edwards D. R. (2005) Metalloproteinases and their inhibitors in tumor angiogenesis. Int. J. Cancer 115, 849–860 [DOI] [PubMed] [Google Scholar]
- 7. Kessenbrock K., Plaks V., Werb Z. (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hashimoto G., Inoki I., Fujii Y., Aoki T., Ikeda E., Okada Y. (2002) Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J. Biol. Chem. 277, 36288–36295 [DOI] [PubMed] [Google Scholar]
- 9. Pilcher B. K., Dumin J. A., Sudbeck B. D., Krane S. M., Welgus H. G., Parks W. C. (1997) The activity of collagenase-1 is required for keratinocyte migration on a type I collagen matrix. J. Cell Biol. 137, 1445–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krekoski C. A., Neubauer D., Graham J. B., Muir D. (2002) Metalloproteinase-dependent predegeneration in vitro enhances axonal regeneration within acellular peripheral nerve grafts. J. Neurosci. 22, 10408–10415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Whitelock J. M., Murdoch A. D., Iozzo R. V., Underwood P. A. (1996) The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J. Biol. Chem. 271, 10079–10086 [DOI] [PubMed] [Google Scholar]
- 12. Imai K., Hiramatsu A., Fukushima D., Pierschbacher M. D., Okada Y. (1997) Degradation of decorin by matrix metalloproteinases. Identification of the cleavage sites, kinetic analyses, and transforming growth factor-β1 release. Biochem. J. 322, 809–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fiore E., Fusco C., Romero P., Stamenkovic I. (2002) Matrix metalloproteinase 9 (MMP-9/gelatinase B) proteolytically cleaves ICAM-1 and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene 21, 5213–5223 [DOI] [PubMed] [Google Scholar]
- 14. McQuibban G. A., Gong J. H., Tam E. M., McCulloch C. A., Clark-Lewis I., Overall C. M. (2000) Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 289, 1202–1206 [DOI] [PubMed] [Google Scholar]
- 15. Sawey E. T., Johnson J. A., Crawford H. C. (2007) Matrix metalloproteinase 7 controls pancreatic acinar cell transdifferentiation by activating the Notch signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 104, 19327–19332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Powell W. C., Fingleton B., Wilson C. L., Boothby M., Matrisian L. M. (1999) The metalloproteinase matrilysin proteolytically generates active soluble Fas ligand and potentiates epithelial cell apoptosis. Curr. Biol. 9, 1441–1447 [DOI] [PubMed] [Google Scholar]
- 17. McCawley L. J., Matrisian L. M. (2001) Matrix metalloproteinases. They're not just for matrix anymore! Curr. Opin. Cell Biol. 13, 534–540 [DOI] [PubMed] [Google Scholar]
- 18. Sellebjerg F., Sørensen T. L. (2003) Chemokines and matrix metalloproteinase-9 in leukocyte recruitment to the central nervous system. Brain Res. Bull. 61, 347–355 [DOI] [PubMed] [Google Scholar]
- 19. Nagase H. (1997) Activation mechanisms of matrix metalloproteinases. Biol. Chem. 378, 151–160 [PubMed] [Google Scholar]
- 20. Nygårdas P. T., Hinkkanen A. E. (2002) Up-regulation of MMP-8 and MMP-9 activity in the BALB/c mouse spinal cord correlates with the severity of experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 128, 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ries C., Petrides P. E. (1995) Cytokine regulation of matrix metalloproteinase activity and its regulatory dysfunction in disease. Biol. Chem. Hoppe Seyler 376, 345–355 [PubMed] [Google Scholar]
- 22. Brinckerhoff C. E., Matrisian L. M. (2002) Matrix metalloproteinases. A tail of a frog that became a prince. Nat. Rev. Mol. Cell Biol. 3, 207–214 [DOI] [PubMed] [Google Scholar]
- 23. Engsig M. T., Chen Q. J., Vu T. H., Pedersen A. C., Therkidsen B., Lund L. R., Henriksen K., Lenhard T., Foged N. T., Werb Z., Delaissé J. M. (2000) Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J. Cell Biol. 151, 879–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Coussens L. M., Tinkle C. L., Hanahan D., Werb Z. (2000) MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 103, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Skonier J., Bennett K., Rothwell V., Kosowski S., Plowman G., Wallace P., Edelhoff S., Disteche C., Neubauer M., Marquardt H., et al. (1994) βig-h3. A transforming growth factor-β-responsive gene encoding a secreted protein that inhibits cell attachment in vitro and suppresses the growth of CHO cells in nude mice. DNA Cell Biol. 13, 571–584 [DOI] [PubMed] [Google Scholar]
- 26. Skonier J., Neubauer M., Madisen L., Bennett K., Plowman G. D., Purchio A. F. (1992) cDNA cloning and sequence analysis of βig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-β. DNA Cell Biol. 11, 511–522 [DOI] [PubMed] [Google Scholar]
- 27. Becker J., Erdlenbruch B., Noskova I., Schramm A., Aumailley M., Schorderet D. F., Schweigerer L. (2006) Keratoepithelin suppresses the progression of experimental human neuroblastomas. Cancer Res. 66, 5314–5321 [DOI] [PubMed] [Google Scholar]
- 28. Kim J. E., Jeong H. W., Nam J. O., Lee B. H., Choi J. Y., Park R. W., Park J. Y., Kim I. S. (2002) Identification of motifs in the fasciclin domains of the transforming growth factor-β-induced matrix protein βig-h3 that interact with the αvβ5 integrin. J. Biol. Chem. 277, 46159–46165 [DOI] [PubMed] [Google Scholar]
- 29. Andersen R. B., Karring H., Møller-Pedersen T., Valnickova Z., Thøgersen I. B., Hedegaard C. J., Kristensen T., Klintworth G. K., Enghild J. J. (2004) Purification and structural characterization of transforming growth factor β-induced protein (TGFBIp) from porcine and human corneas. Biochemistry 43, 16374–16384 [DOI] [PubMed] [Google Scholar]
- 30. Doliana R., Bot S., Bonaldo P., Colombatti A. (2000) EMI, a novel cysteine-rich domain of EMILINs and other extracellular proteins, interacts with the gC1q domains and participates in multimerization. FEBS Lett. 484, 164–168 [DOI] [PubMed] [Google Scholar]
- 31. Oh J. E., Kook J. K., Min B. M. (2005) βig-h3 induces keratinocyte differentiation via modulation of involucrin and transglutaminase expression through the integrin α3β1 and the phosphatidylinositol 3-kinase/Akt signaling pathway. J. Biol. Chem. 280, 21629–21637 [DOI] [PubMed] [Google Scholar]
- 32. Ohno S., Noshiro M., Makihira S., Kawamoto T., Shen M., Yan W., Kawashima-Ohya Y., Fujimoto K., Tanne K., Kato Y. (1999) RGD-CAP ((β)ig-h3) enhances the spreading of chondrocytes and fibroblasts via integrin β(1)β(1). Biochim. Biophys. Acta 1451, 196–205 [DOI] [PubMed] [Google Scholar]
- 33. Zhao Y. L., Piao C. Q., Hei T. K. (2002) Down-regulation of βig-h3 gene is causally linked to tumorigenic phenotype in asbestos treated immortalized human bronchial epithelial cells. Oncogene 21, 7471–7477 [DOI] [PubMed] [Google Scholar]
- 34. Wen G., Partridge M. A., Li B., Hong M., Liao W., Cheng S. K., Zhao Y., Calaf G. M., Liu T., Zhou J., Zhang Z., Hei T. K. (2011) TGFBI expression reduces in vitro and in vivo metastatic potential of lung and breast tumor cells. Cancer Lett. 308, 23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao Y., Shao G., Piao C. Q., Berenguer J., Hei T. K. (2004) Down-regulation of βig-h3 gene is involved in the tumorigenesis in human bronchial epithelial cells induced by heavy-ion radiation. Radiat. Res. 162, 655–659 [DOI] [PubMed] [Google Scholar]
- 36. Shao G., Berenguer J., Borczuk A. C., Powell C. A., Hei T. K., Zhao Y. (2006) Epigenetic inactivation of βig-h3 gene in human cancer cells. Cancer Res. 66, 4566–4573 [DOI] [PubMed] [Google Scholar]
- 37. Calaf G. M., Echiburú-Chau C., Zhao Y. L., Hei T. K. (2008) BigH3 protein expression as a marker for breast cancer. Int. J. Mol. Med. 21, 561–568 [PubMed] [Google Scholar]
- 38. Nabokikh A., Ilhan A., Bilban M., Gartner W., Vila G., Niederle B., Nielsen J. H., Wagner O., Base W., Luger A., Wagner L. (2007) Reduced TGF-β1 expression and its target genes in human insulinomas. Exp. Clin. Endocrinol. Diabetes 115, 674–682 [DOI] [PubMed] [Google Scholar]
- 39. Zhang Y., Wen G., Shao G., Wang C., Lin C., Fang H., Balajee A. S., Bhagat G., Hei T. K., Zhao Y. (2009) TGFBI deficiency predisposes mice to spontaneous tumor development. Cancer Res. 69, 37–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Korff T., Augustin H. G. (1998) Integration of endothelial cells in multicellular spheroids prevents apoptosis and induces differentiation. J. Cell Biol. 143, 1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Estève P. O., Chicoine E., Robledo O., Aoudjit F., Descoteaux A., Potworowski E. F., St-Pierre Y. (2002) Protein kinase C-ζ regulates transcription of the matrix metalloproteinase-9 gene induced by IL-1β and TNF-α in glioma cells via NF-κB. J. Biol. Chem. 277, 35150–35155 [DOI] [PubMed] [Google Scholar]
- 42. Kim Y. H., Joo H. S., Kim D. S. (2010) Nitric oxide induction of IRE1-α-dependent CREB phosphorylation in human glioma cells. Nitric Oxide 23, 112–120 [DOI] [PubMed] [Google Scholar]
- 43. Kridel S. J., Chen E., Kotra L. P., Howard E. W., Mobashery S., Smith J. W. (2001) Substrate hydrolysis by matrix metalloproteinase-9. J. Biol. Chem. 276, 20572–20578 [DOI] [PubMed] [Google Scholar]
- 44. Masure S., Proost P., Van Damme J., Opdenakker G. (1991) Purification and identification of 91-kDa neutrophil gelatinase. Release by the activating peptide interleukin-8. Eur. J. Biochem. 198, 391–398 [DOI] [PubMed] [Google Scholar]
- 45. Opdenakker G., Masure S., Proost P., Billiau A., van Damme J. (1991) Natural human monocyte gelatinase and its inhibitor. FEBS Lett. 284, 73–78 [DOI] [PubMed] [Google Scholar]
- 46. Welgus H. G., Campbell E. J., Cury J. D., Eisen A. Z., Senior R. M., Wilhelm S. M., Goldberg G. I. (1990) Neutral metalloproteinases produced by human mononuclear phagocytes. Enzyme profile, regulation, and expression during cellular development. J. Clin. Invest. 86, 1496–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ardi V. C., Kupriyanova T. A., Deryugina E. I., Quigley J. P. (2007) Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 20262–20267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Daigneault M., Preston J. A., Marriott H. M., Whyte M. K., Dockrell D. H. (2010) The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS ONE 5, e8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gingis-Velitski S., Loven D., Benayoun L., Munster M., Bril R., Voloshin T., Alishekevitz D., Bertolini F., Shaked Y. (2011) Host response to short term, single-agent chemotherapy induces matrix metalloproteinase-9 expression and accelerates metastasis in mice. Cancer Res. 71, 6986–6996 [DOI] [PubMed] [Google Scholar]
- 50. Park M. J., Kim M. S., Park I. C., Kang H. S., Yoo H., Park S. H., Rhee C. H., Hong S. I., Lee S. H. (2002) PTEN suppresses hyaluronic acid-induced matrix metalloproteinase-9 expression in U87MG glioblastoma cells through focal adhesion kinase dephosphorylation. Cancer Res. 62, 6318–6322 [PubMed] [Google Scholar]
- 51. Qian B. Z., Pollard J. W. (2010) Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ramos-DeSimone N., Hahn-Dantona E., Sipley J., Nagase H., French D. L., Quigley J. P. (1999) Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J. Biol. Chem. 274, 13066–13076 [DOI] [PubMed] [Google Scholar]
- 53. Aggarwal B. B., Shishodia S., Sandur S. K., Pandey M. K., Sethi G. (2006) Inflammation and cancer. How hot is the link? Biochem. Pharmacol. 72, 1605–1621 [DOI] [PubMed] [Google Scholar]