

Background: The zinc-binding domain of DNA primase has been implicated in template recognition.
Results: Residues around cysteines within the zinc-binding domain coordinate template binding but not sequence recognition.
Conclusion: The integrity and functional conformation of the zinc-binding domain are important for DNA primase function.
Significance: The zinc-binding domain of DNA primase is essential for template binding and primer synthesis.
Keywords: Bacteriophage, DNA Primase, DNA Replication, DNA-Protein Interaction, RNA Synthesis, Bacteriophage T7, Template DNA Recognition, Homolog Scanning
Abstract
The zinc-binding domain (ZBD) of prokaryotic DNA primases has been postulated to be crucial for recognition of specific sequences in the single-stranded DNA template. To determine the molecular basis for this role in recognition, we carried out homolog-scanning mutagenesis of the zinc-binding domain of DNA primase of bacteriophage T7 using a bacterial homolog from Geobacillus stearothermophilus. The ability of T7 DNA primase to catalyze template-directed oligoribonucleotide synthesis is eliminated by substitution of any five-amino acid residue-long segment within the ZBD. The most significant defect occurs upon substitution of a region (Pro-16 to Cys-20) spanning two cysteines that coordinate the zinc ion. The role of this region in primase function was further investigated by generating a protein library composed of multiple amino acid substitutions for Pro-16, Asp-18, and Asn-19 followed by genetic screening for functional proteins. Examination of proteins selected from the screening reveals no change in sequence-specific recognition. However, the more positively charged residues in the region facilitate DNA binding, leading to more efficient oligoribonucleotide synthesis on short templates. The results suggest that the zinc-binding mode alone is not responsible for sequence recognition, but rather its interaction with the RNA polymerase domain is critical for DNA binding and for sequence recognition. Consequently, any alteration in the ZBD that disturbs its conformation leads to loss of DNA-dependent oligoribonucleotide synthesis.
Introduction
A problem encountered by the replisome is the simultaneous replication of antiparallel DNA strands by the unidirectional polymerizing activity of DNA polymerases. This dilemma is resolved by different modes of DNA synthesis for the leading and lagging strands. Unlike the continuous synthesis of the leading strand, the lagging strand is synthesized discontinuously in the form of Okazaki fragments. The Okazaki fragments reside within a replication loop that allows for the overall synthesis of both strands in the same direction with all events occurring within the replisome. Synthesis of the Okazaki fragments however necessitates a supply of oligoribonucleotides on the single-stranded lagging strand extruded by the helicase during leading strand synthesis. DNA primases are designated to fulfill this function by catalyzing the synthesis of oligoribonucleotides (1).
Unlike their eukaryotic counterparts, prokaryotic DNA primases (DnaG family) synthesize primers from specific sites on single-stranded DNA (ssDNA).2 Amino acid sequence alignment of DNA primases from bacteria and bacteriophages has identified six conserved motifs (2). The first motif at the N terminus of the primase is a zinc-binding domain (ZBD) that consists of four cysteines (Cys4) or three cysteines and one histidine (Cys-His-Cys2) that coordinate a zinc ion. The other five motifs reside in an RNA polymerase domain (RPD) located in the C-terminal portion of the primase where binding and the catalytic condensation of NTPs occur. Motifs 4–6 form a TOPRIM-fold that contains two magnesium ions that participate in a two metal-mediated phosphodiester formation reaction similar to most polymerases (3). Together with the TOPRIM-fold, motifs 2 and 3 are believed to create the template-binding site. Although the RPD is responsible for the template-directed coupling of the NTPs, the ZBD plays a critical role in the binding of the primase to specific trinucleotide sequences in the template defined as primase recognition sites (1). Each prokaryotic primase has its own recognition sequence for primer synthesis, distinct from others. The “cryptic” nucleotide at the 3′-side of the trinucleotide is not copied into RNA products, but its presence is essential for recognition of the sequence by the primase. NTPs complementary to the next nucleotides at the 5′-side of the template are then added to generate the dinucleotide, and it is further extended to the oligonucleotide of a functional length that can be used by the DNA polymerase as a primer. Primers synthesized de novo must be stabilized by the primase and then handed off to DNA polymerase for the initiation of DNA synthesis.
One of the well characterized prokaryotic primases is the primase-helicase encoded by gene 4 of bacteriophage T7. The C-terminal domain of the gene 4 protein is a member of superfamily 4 hexameric helicases. The helicase domain binds ssDNA and unwinds parental DNA at the expense of dTTP hydrolysis. The primase domain, located in the N-terminal half of the gene 4 protein, recognizes the sequence 5′-GTC-3′ in ssDNA. At this trinucleotide recognition site, the T7 DNA primase catalyzes the synthesis of the diribonucleotide pppAC and further extends it to functional tetraribonucleotides, and pppACCC, pppACCA, and pppACAC provided cognate sequences are available in the template (4, 5). The crystal structure of the T7 primase revealed a modular organization in which the ZBD and the RPD are connected by a flexible linker (6). The ZBD of T7 primase contains a four-stranded, antiparallel β-sheet (Fig. 1). Four cysteines, located at loop regions connecting β-strands, coordinate a zinc ion to form a structure typical of members of the zinc-ribbon family. The RPD of T7 primase contains an N-terminal subdomain consisting of four β-strands (motifs 2 and 3) and the TOPRIM-fold (motifs 4–6). Substitution of conserved residues in these signature motifs results in loss of ability to catalyze phosphodiester bond formation and thus synthesis of oligoribonucleotides illustrating their critical roles in catalysis (7, 8). The flexible linker region connecting the ZBD and RPD modulates contacts between the two subdomains during primer synthesis (9). Within the hexameric structure composed of full-length gene 4 primase-helicase, a ZBD from one subunit can interact with the RPD from the adjacent subunit to catalyze synthesis of oligoribonucleotides in a trans mode (10), suggesting a regulatory role of primase during DNA synthesis (11).
FIGURE 1.

ZBD of T7 and G. stearothermophilus (Gst) DNA primases. A, x-ray crystallographic structures of the ZBDs of DNA primases from bacteriophage T7 (Protein Data Bank code 1NUI) and G. stearothermophilus (Protein Data Bank code 1D0Q). Despite the differences, the overall structures of the two ZBDs near the zinc are similar. Zinc atoms shown by a gray sphere are coordinated by side chains of cysteines and histidine located in two loop regions flanked by β-strands. Residues coordinating the zinc in T7 and G. stearothermophilus primases are shown in magenta and orange, respectively. Primase recognition sequences for each primase are shown at the bottom of structures. B, superimposed structures of zinc-binding region. C, comparison of amino acid sequences of ZBDs of T7 and G. stearothermophilus primases. Amino acid sequences were initially aligned by ClustalW (35) and manually modified to minimize drastic substitution in homolog-scanning analysis. Residues underlined are in β-strand regions. ZBDs were divided into five segments (H1 to H5), and each segment of T7 primase was replaced with the corresponding portion of the G. stearothermophilus primase to generate five hybrid gene 4 proteins. Residue numbers for T7 ZBD are: H1 (Phe-11–Ile-15), H2 (Pro-16–Cys-20), H3 (Gly-21–Gly-25), H4 (Asn-26–Ser-30), and H5 (Asp-31–Phe-35).
The importance of the ZBD in DNA-dependent oligoribonucleotide synthesis was initially noticed in studies with the 56-kDa gene 4 protein, a truncated form of the 63-kDa gene 4 protein, that arises from in-frame translation of the full-length gene 4 (12). The 56-kDa gene 4 protein lacks the N-terminal 63 amino acid residues corresponding to the ZBD and is not capable of catalyzing primer synthesis in a template-dependent manner (13). However, the 56-kDa gene 4 protein, like the full-length protein, does synthesize random diribonucleotide in the absence of template, albeit at a very slow rate. Replacement of a single cysteine in the ZBD with a serine results in loss of the zinc ion and abolishes template-directed synthesis (14). Furthermore, site-directed mutagenesis has identified amino acid residues critical to the recognition of 5′-GTC-3′ (15).
A persistent goal has been to elucidate the molecular mechanism by which the T7 primase recognizes a trinucleotide sequence in ssDNA. Prokaryotic DNA primases, each having a distinct ZBD, synthesize primers of different sequences. Consequently, it has been suggested that sequence specificity is dictated by interactions between amino acid residues in the ZBD and the trinucleotide sequence in DNA (1). Previously, we attempted to modify the specificity by either swapping the entire ZBD (16) or a portion of the zinc-spanning region with equivalent parts of homologs (17). These studies demonstrated that the integrity of ZBD is important in the recognition of specific sequences, but they failed to reveal the basis for the recognition.
Recent reports of the structures and recognition sequences of DNA primases have provided an approach to obtain additional information on the role of the ZBD in template recognition and binding (18–20). X-ray crystallographic structures show that the ZBDs of the T7 primase and of the primase of Geobacillus stearothermophilus (formerly known as Bacillus stearothermophilus) are similar in their overall structures (6, 21). With the exception of some misaligned regions at the N terminus of the G. stearothermophilus primase and the C terminus of the T7 primase, the two enzymes exhibit a similar folding in the zinc-spanning region (Fig. 1). Both primases have four antiparallel β-sheets forming two loops in which cysteines (or a histidine) coordinate a zinc ion. Despite these resemblances, T7 primase recognizes 5′-GTC-3′ in the template, whereas G. stearothermophilus primase recognizes either 5′-TTA-3′ or 5′-CTA-3′ (22). In an attempt to identify a region critical to the function of the ZBD, we have constructed altered T7 primases containing homologous portion of the G. stearothermophilus primase and examined their ability to catalyze template-directed synthesis of oligoribonucleotides and to deliver them to DNA polymerase. In addition, a collection of proteins consisting of a variety of amino acid substitutions in the vicinity of zinc-coordinating residues was generated and analyzed to examine their interaction with the template DNA. Our results lead us to conclude that the zinc-binding motif is per se not responsible for sequence recognition. However, its interaction with the RNA polymerase domain is critical for DNA binding and hence for sequence recognition.
EXPERIMENTAL PROCEDURES
Materials
Oligonucleotides were obtained from Integrated DNA Technology and Dharmacon. Plasmid DNA purification kits and nickel-nitrilotriacetic acid resins were from Qiagen. Restriction endonucleases, Deep Vent® polymerase, and T4 DNA ligase were purchased from New England Biolabs. Radiolabeled nucleotides were purchased from PerkinElmer Life Sciences. Sensor chips and reagents for surface plasmon resonance studies were from GE Healthcare.
Mutagenesis, Overproduction, and Purification of Recombinant Proteins
Alterations at selective positions of the ZBD were introduced into plasmids expressing either the entire T7 gene 4 (pET24-gp4 or pET28-gp4) or the T7 DNA primase coding region (pET24-g4P) following procedures described previously (7). A DNA fragment containing alterations in gene 4 was generated by PCR and incubated with NdeI and BstBI. The insert DNA purified on an agarose gel was ligated into the plasmid treated with the same restriction enzymes. After the presence of the alteration was confirmed by DNA sequence analysis of the entire gene 4 coding region, the resulting plasmids were transformed into Escherichia coli BL21(DE3). Proteins were overproduced by inducing cells with 1 mm isopropyl β-d-1-thiogalactopyranoside at 37 °C for 3 h after the culture reached A600 of around 1. A full-length gene 4 protein containing no His tag was purified as described previously (7) using DEAE-anion exchange column chromatography instead of ATP-agarose affinity chromatography. The ATP-agarose affinity chromatography was omitted to avoid use of EDTA, a potential chelator for zinc ion during the protein elution step. Proteins containing a His tag at the N terminus were purified using nickel-nitrilotriacetic acid affinity chromatography followed by DEAE-anion exchange column chromatography (23). All purified proteins were over 90% pure as judged on SDS-polyacrylamide gel.
Complementation of Phage Growth
E. coli DH5α (0.3 ml) harboring a plasmid that expresses T7 gene 4 was cultured until an A600 of around 1 and mixed with serially diluted T7Δ4 phage lacking gene 4 (0.1 ml) in 3 ml of soft agar. The mixture was plated on the top of LB plate, and the plaques that emerged after incubation at 37 °C for 5 h were counted. Additional deletions around gene 4 in T7Δ4 (T7 nucleotide 11,481–13,621) prevent homologous recombination between the phage and the gene 4-expressing plasmid.
Template-directed Oligoribonucleotide Synthesis
Reaction mixture contained 40 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 10 mm DTT, 50 mm potassium glutamate, 0.1 mm each of ATP and [α-32P]CTP (0.1 μCi/μl), indicated oligomer DNA containing a primase recognition site (5′-GGGTC-3′ or 5′-TGGTC-3′) or 10 nm M13 ssDNA, and the indicated amount of gene 4 protein or primase. After incubation at 37 °C for 30 min, the reaction was terminated by the addition of 2 μl of sequencing dye, and reaction products were separated on a 25% denaturing polyacrylamide sequencing gel containing 3 m urea. The gel was dried for autoradiography, and the amount of CMP incorporated into the major product, the tetraribonucleotide, was determined using a Fuji BAS 1000 Bioimaging analyzer.
RNA-primed DNA Synthesis
T7 DNA polymerase cannot initiate synthesis on ssDNA. Gene 4 primase allows T7 DNA polymerase to initiate synthesis on ssDNA by synthesizing tetraribonucleotides for use as primers by the polymerase or by delivering preformed oligoribonucleotides to the polymerase. The ability of T7 gene 4 primase to support DNA synthesis was examined in the following assay. The reaction contained 10 nm M13 ssDNA, 0.3 mm each of [α-3H]dGTP (10 cpm/pmol), dCTP, dATP, and dTTP, 200 nm T7 gene 4 protein, 10 nm T7 DNA polymerase, and either a mixture of 0.1 mm each of NTPs or the indicated amount of preformed tetraribonucleotide ACCA in 40 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 10 mm DTT, and 50 mm potassium glutamate. The reaction was incubated at 37 °C for 10 min and terminated by the addition of EDTA to a final concentration of 20 mm. The reaction products were spotted onto a DE81 membrane (Whatman), and the membrane was washed three times with 10 ml of 0.3 m ammonium formate, pH 8.0, to remove unincorporated radioactive nucleotide. The amount of DNA synthesis was determined by measuring the radioactive products retained on the membrane.
Construction of Plasmid Library and Selection of Functional Gene 4 Protein
A library of plasmids containing multiple amino acid substitutions for Pro-16, Asp-18, and Asn-19 was constructed by introducing mixed codons into the coding region using a random mutagenic primer (5′-GT GTA TTT CTT TAC CAC ATT NNK TGT NNK NNK TGT GGG AGT AGT GAT GGG-3′ where N = any base; K = G or T; mutated codon shown in boldface). The mixture of E. coli cells containing the plasmid library was infected with T7 phage Δ4-1 lacking T7 nucleotides 11,563–11,806 (24). The presence of the T7 nucleotide 11,333–11,950 in the plasmid allows for homologous recombination with the phage genome to produce phages containing various codons at the three positions in the ZBD. Upon plating the lysate on E. coli lacking a gene 4 expressing plasmid, only phages that acquire a functional gene 4 coding region as the result of homologous recombination produce plaques. E. coli cells (4 × 105) harboring the plasmid library were infected with the T7 phage Δ4-1 (2 × 104 pfu), and the resulting lysate was collected. Plaques were isolated, and the sequence of their gene 4 coding region (T7 nucleotide 11,565 to 12,200) was determined by DNA sequence analysis.
Measurement of DNA Binding Affinity by Surface Plasmon Resonance
The binding affinity of T7 primase to ssDNA was determined using surface plasmon resonance as described previously (25). Single-stranded 25-mer DNA biotinylated at the 5′ end was immobilized on a streptavidin-coated sensor chip. Binding of protein was detected by flowing the indicated primase (10 μm) over the chip in a binding buffer (10 mm Tris-HCl, pH 7.5, 50 mm potassium glutamate, 10 mm MgCl2, and 1 mm DTT) at a flow rate of 40 μl/min. The surface of the chip was regenerated by injecting 100 μl of 1 m NaCl and 50 mm NaOH at a flow rate of 100 μl/min to remove bound proteins. Binding signal was determined by subtracting the nonspecific signal from a control flow cell in which biotin was immobilized instead of biotinylated DNA.
RESULTS
Homolog-scanning Mutagenesis of ZBD of T7 DNA Primase
The structures of the ZBDs of T7 DNA primase and G. stearothermophilus primase are similar especially near the zinc atom as seen in their superimposed structures (Fig. 1, A and B). An alignment of amino acid sequences shows that their secondary structures are also conserved (Fig. 1C). Yet the two primases recognize different trinucleotide sequences for primer synthesis as follows: 5′-GTC-3′ for T7 primase and 5′-TTA-3′ or 5′-CTA-3′ for G. stearothermophilus primase. To identify a region responsible for this divergent template recognition, we used homolog-scanning mutagenesis of T7 primase using G. stearothermophilus primase (26). A portion of the ZBD of T7 DNA primase in the vicinity of the zinc ion (Phe-11 to Phe-35) was divided into five segments based on the similarity of secondary structure to that of G. stearothermophilus primase. Each of the segments was replaced with the homologous region of the ZBD of G. stearothermophilus, generating five hybrid T7 gene 4 proteins containing a portion of the ZBD of G. stearothermophilus (Fig. 1).
Complementation of T7Δ4 Growth
The effect of the alterations on the in vivo function of gene 4 protein was examined by a phage growth complementation assay. In this assay, the viability of T7 phage lacking the entire gene 4 (T7Δ4) depends on the expression of gene 4 from a plasmid harbored in E. coli. As shown in Table 1, compared with wild-type protein, all hybrid proteins exhibit a 102 to 107-fold decrease in their ability to support growth of T7Δ4. Plaque sizes obtained with the hybrid proteins were also significantly smaller than those observed with the wild-type gene 4 protein (data not shown). In particular, alterations of a segment containing Cys-17 and Cys-20 that coordinate the zinc (H2) and a segment neighboring another zinc-coordinating cysteine, Cys-36 (H5), result in a more severe defect than the other alterations (H1, H3, and H4).
TABLE 1.
Ability of gene 4 hybrid proteins to complement growth of T7 phage lacking gene 4
The T7 phage lacking gene 4 (T7Δ4) was plated on E. coli DH5α containing a plasmid expressing each of the recombinant genes 4 described in Fig. 1 (H1, H2, H3, H4 and H5). The plaques were counted and complementation ability of gene 4 protein was expressed as relative plating efficiency normalized against the value obtained with the wild-type gene 4 protein. Data were from at least duplicate experiments.
| Gene 4 protein | Efficiency of plating |
|---|---|
| Wild type | 1 |
| H1 | 0.03 |
| H2 | 2 × 10−7 |
| H3 | 0.03 |
| H4 | 0.06 |
| H5 | 1 × 10−4 |
Oligoribonucleotide Synthesis Activity
To determine defects that lead to the loss of in vivo function, the hybrid proteins were purified and examined in biochemical assays. In all assays, the 56-kDa gene 4 protein lacking ZBD was included for comparison. At first, the ability of gene 4 protein to catalyze template-directed oligoribonucleotide synthesis was examined using a short ssDNA template containing the sequence 5′-GGGTC-3′. All hybrid proteins as well as the 56-kDa gene 4 protein fail to synthesize oligoribonucleotide (Fig. 2A). Because alterations of the ZBD could change specificity of the primase recognition site, we used M13 ssDNA that should contain all possible recognition sequences for primer synthesis. Again, none of the hybrid gene 4 proteins synthesize any oligoribonucleotide on M13 ssDNA (Fig. 2B). The multiple products observed with wild-type protein reflect the use of minor recognition sites within the M13 DNA sequence (4). Use of radiolabeled NTPs other than CTP did also not yield any oligoribonucleotides synthesized by the hybrid proteins (data not shown).
FIGURE 2.
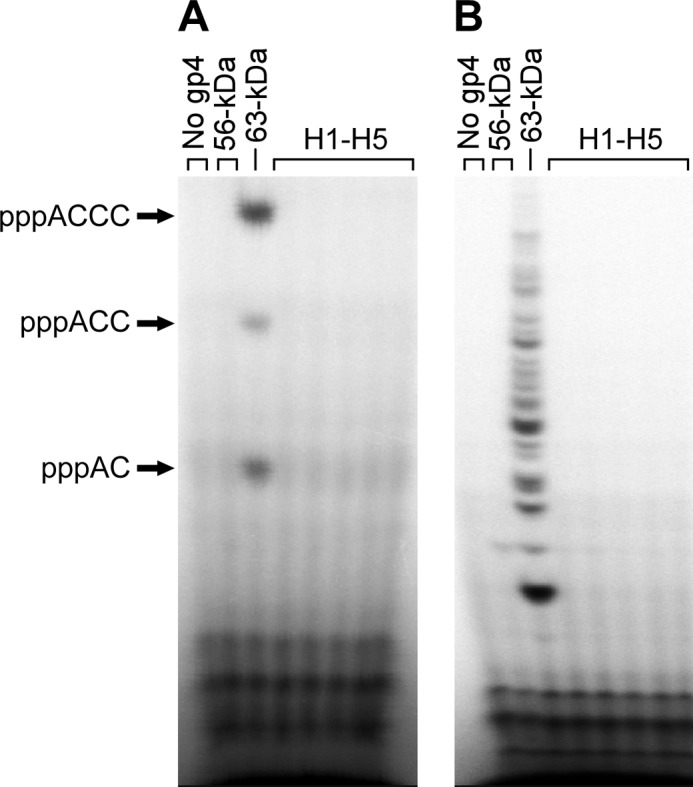
Template-directed oligoribonucleotide synthesis by hybrid gene 4 primases. A, gene 4 protein (200 nm) was incubated with 36 μm 15-mer ssDNA containing a primase recognition sequence 5′-GGGTC-3′ and 0.1 mm each of ATP and [α-32P]CTP (0.1 μCi/μl) in a reaction buffer (40 mm Tris-HCl, pH 7.5, 10 mm DTT, 10 mm MgCl2, and 50 mm potassium glutamate). After incubation for 30 min at 37 °C, reaction products were separated on a 25% denaturing polyacrylamide gel and detected by autoradiography. Major reaction products are indicated at the left side of the gel. B, gene 4 protein (200 nm) was incubated with 10 nm M13 ssDNA, 0.1 mm each of all four NTPs, and [α-32P]CTP (0.1 μCi/μl) in the buffer as described in A. dTTP was supplied at a final concentration of 0.5 mm to facilitate translocation of gene 4 protein on the ssDNA template.
The loss of ability to catalyze the template-directed synthesis of oligoribonucleotides could result from defective function of either the ZBD or RPD (10). However, all the hybrid DNA primases, like the 56-kDa gene 4 protein, catalyze the template-independent synthesis of random diribonucleotides (supplemental Fig. S1). Therefore, the inability to mediate template-directed oligoribonucleotide synthesis appears to arise from a defect in the ZBD.
RNA-primed DNA Synthesis
Oligoribonucleotides synthesized by DNA primase are delivered to DNA polymerase to initiate lagging strand synthesis. This function of the primase was examined in an RNA-primed DNA synthesis assay under two different conditions. In the first experiment, a mixture of all four NTPs was supplied as precursors for oligoribonucleotides. Wild-type gene 4 protein supports DNA synthesis by T7 DNA polymerase on M13 ssDNA by providing oligoribonucleotides that serve as primers to initiate DNA synthesis (Fig. 3A). Not surprisingly, in view of their inability to synthesize template-directed oligoribonucleotide synthesis, no DNA synthesis was observed with any of the hybrid gene 4 proteins (Fig. 3A). The lack of DNA synthesis confirms the absence of oligoribonucleotides because DNA synthesis would amplify their presence. In the second experiment, preformed synthetic ACCC was supplied to gene 4 protein to bypass the step of oligoribonucleotide synthesis. In the absence of gene 4 protein, T7 DNA polymerase is unable to use a tetraribonucleotide as a primer (7, 8). However, as shown in Fig. 3B, wild-type gene 4 protein supports DNA synthesis by T7 DNA polymerase proportional to the amount of ACCC supplied (Fig. 3B). The ability of gene 4 protein to stabilize and transfer preformed oligoribonucleotides to the polymerase efficiently is dependent on the correct recognition sequence (27). Although the 56-kDa protein is much less efficient than wild-type protein, it can also support DNA synthesis in the presence of ACCC. Most of the hybrid proteins showed defects more or less similar to the 56-kDa protein except for two-hybrid proteins containing substitution for H2 (gp4-H2) and H5 (gp4-H5). These proteins support the minimal amount of DNA synthesis over the entire range of the oligoribonucleotide examined. These hybrid proteins were not as good in supporting T7Δ4 growth in the complementation assay. Therefore, these data indicate that alterations of the ZBD in all the hybrid proteins result in loss (or reduction) of the primase function to deliver oligoribonucleotide to DNA polymerase.
FIGURE 3.
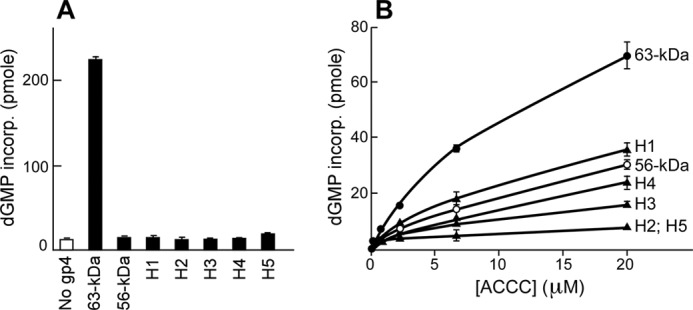
RNA-primed DNA synthesis in the presence of hybrid gene 4 proteins. The ability of gene 4 protein to support DNA synthesis by T7 DNA polymerase was determined by measuring the incorporation of [α-3H]dGMP into DNA. A, gene 4 protein (200 nm) was incubated with 10 nm M13 ssDNA, 0.1 mm each of all four NTPs, 0.3 mm each of all four dNTP, [α-3H]dGTP (10 cpm/pmol), and 10 nm T7 DNA polymerase in a reaction buffer (40 mm Tris-HCl, pH 7.5, 10 mm DTT, 10 mm MgCl2, and 50 mm potassium glutamate). After incubation for 10 min at 37 °C, reaction products were spotted on a DE81 filter, and incorporation of dGMP was determined by measuring radioactivity remained on the filter after extensive washing as described under “Experimental Procedures.” B, reaction was similar to A except that the indicated amount of preformed ACCC was used instead of the four NTPs.
We have previously shown that modifications of the ZBD can lead to loss of the zinc and deformation of the ZBD resulting in inactive primases (14, 28). Therefore, the inability of the hybrid primases to recognize a specific sequence for primer synthesis and to deliver primer to DNA polymerase might arise from conformational changes that result in loss of the zinc. We examined the zinc content of the hybrid primases using inductively coupled plasma-mass spectrometry (supplemental Table S1). Hybrid proteins that retain marginal activity to deliver preformed primer to DNA polymerase (gp4-H1, gp4-H3, and gp4-H4) show significantly reduced levels of zinc compared with wild-type protein. Thus, the loss of zinc explains why these three hybrid primases deliver preformed oligoribonucleotides at a reduced level similar to that observed with the 56-kDa protein lacking the entire ZBD. In contrast, gp4-H2 and gp4-H5 that are unable to deliver preformed oligoribonucleotides to T7 DNA polymerase have normal amounts of zinc. This finding suggests that the modifications in these hybrid primases do not completely disrupt conformation of the ZBD, yet they destroy the ability of the ZBD to properly interact with the template and RPD.
Activity of Helicase Domain
The helicase domain of gene 4 protein is located in the C-terminal half of the polypeptide. The helicase domain assembles on ssDNA as a hexamer and uses the energy derived from the hydrolysis of dTTP to translocate unidirectionally on the ssDNA. All the hybrid gene 4 proteins catalyze the ssDNA-dependent hydrolysis of dTTP in a concentration-dependent manner, confirming that the helicase domain is not affected by alterations in the ZBD (supplemental Fig. S2).
Construction of Library and Selection of Functional Gene 4 Proteins
The results from the homolog-scanning analysis described above show that all alterations of the ZBD as small as five amino acid residues lead to a defect in template-directed synthesis and delivery of oligoribonucleotide. The complementation analysis of the hybrid primases and the RNA-primed DNA synthesis assays indicate that alterations in the H2 segment spanning Pro-16 to Cys-20 result in the most severe defect in primase function relative to the other hybrid primases. The two cysteine residues, Cys-17 and Cys-20, in this portion of the ZBD are involved in coordination of a zinc ion and are strictly conserved in prokaryotic primases. Most bacteria have the same amino acid sequence, Cys-Pro-Phe-His, in this segment, whereas bacteriophages have various residues between the two cysteines, Cys-Xaa-Xaa-Cys (2). Therefore, this portion of the ZBD might be one of the most important regions for primase function, including template-directed oligoribonucleotide synthesis and its delivery to DNA polymerase.
To investigate the role of this region in primase function in more detail, we created a plasmid library that consists of random amino acid substitutions at the three variable positions (residue numbers 16, 18, and 19) by introducing mixed codons. E. coli cells containing the plasmid library were infected with a T7 phage lacking a portion of gene 4 coding the first 80 amino acids of the gene 4 protein (T7Δ4-1). Homologous recombination between the plasmid and T7Δ4-1 generated T7 phages that have various amino acid compositions at positions 16, 18, and 19 in gene 4 protein. T7 phages that acquire a functional gene 4 from recombination are viable and can be selectively isolated in the absence of gene 4 expression from a plasmid. When an E. coli pool containing the plasmid library (4 × 105 cells) was infected with T7Δ4-1 at a multiplicity of infection of 0.05, 42 phage plaques emerged. In a control in which the same number of bacterial cells containing wild-type gene 4-expressing plasmid was used, 100 plaques were observed, reflecting a recombination efficiency of 0.5%. Amino acid sequence analysis of recombinant phages reveals that 10 of 42 phages encode wild-type gene 4 protein (Table 2). The other 32 sequences were aligned based on characteristics of side chain of the residue at position of 18 because this position appears to play the most critical role in the function of ZBD as discussed later.
TABLE 2.
Gene 4 primases selected from the functional recombinant phage library and their properties
| Property at position 18 | Position (wild-type) |
Occurrence (42 in total) | Selected for analysis | T7Δ4 plating efficiencya | ||||
|---|---|---|---|---|---|---|---|---|
| Pro-16 | Cys-17 | Asp-18 | Asn-19 | Cys-20 | ||||
| Negatively charged | ||||||||
| Asp | Pro | Asp | Asn | 10 | WT | 1 | ||
| Gln | Asp | Val | 1 | |||||
| Ala | Asp | Gly | 1 | |||||
| Arg | Asp | Gly | 1 | |||||
| Ser | Asp | Gly | 1 | H2-SDG | 1.66 | |||
| Thr | Asp | Met | 1 | |||||
| Glu | Lys | Glu | Arg | 1 | H2-KER | 1.42 | ||
| Lys | Glu | Trp | 1 | |||||
| Gly | Glu | Asp | 1 | |||||
| Ser | Glu | Ile | 1 | |||||
| Thr | Glu | Gly | 1 | |||||
| Polar/no charge | ||||||||
| Asn | Glu | Asn | Glu | 1 | H2-ENE | 1 | ||
| Lys | Asn | Gly | 1 | |||||
| Gln | Gly | Gln | Thr | 1 | ||||
| Gly | Gln | Glu | 1 | |||||
| Ser | Ser | Ser | Gly | 1 | H2-SSG | 0.83 | ||
| Arg | Ser | Gly | 1 | H2-RSG | 1.25 | |||
| Ser | Ser | Trp | 1 | |||||
| Ser | Ser | Asp | 1 | |||||
| Thr | Lys | Thr | Glu | 1 | ||||
| Asn | Thr | Gly | 1 | |||||
| Aliphatic | ||||||||
| Gly | Asn | Gly | His | 1 | ||||
| Asn | Gly | Val | 1 | |||||
| Gly | Gly | Cys | 1 | |||||
| Ala | His | Ala | Gly | 2 | ||||
| V | Glu | Val | Asn | 1 | ||||
| L | Lys | Leu | Ser | 1 | ||||
| Met | Lys | Met | Ser | 1 | ||||
| Gly | Met | Asn | 1 | |||||
| Positively charged | ||||||||
| His | Lys | His | Arg | 2 | H2-KHR | 0.18 | ||
| Glu | His | Asp | 1 | |||||
| His | C20H | 0.03 | ||||||
a The relative efficiency of plating was normalized against the value obtained with the wild-type gene 4 protein.
Analysis of Altered Gene 4 Primases Containing Substitution at Positions 16, 18, and 19
Among the functional gene 4 proteins selected from the homologous recombination described above, we chose six representative proteins based on the amino acid at each position and purified them for biochemical analysis. Most of the altered proteins differ from one another by a single residue to facilitate analysis of the role. We also included gp4-C20H in which histidine is substituted for cysteine at position 20 to examine its effect on primase; most bacterial primases contain a histidine at this position.
Complementation of T7Δ4 Growth and Zinc Content
Examination of their ability to complement growth of T7Δ4 phage lacking gene 4 confirms that the altered proteins behave similar to wild-type protein except for gp4-H2-KHR and gp4-C20H. gp4-H2-KHR with Lys, His, and Arg at positions 16, 18, and 19, respectively, exhibits a 5-fold reduction in the complementation assay (Table 2). gp4-C20H with histidine replacing cysteine at position 20 is ∼30-fold less efficient than wild-type protein in the complementation assay. All of the altered proteins have a zinc content equivalent to wild-type protein except for gp4-C20H, which has essentially no zinc (supplemental Table S1). Taken together with the phage complementation data, it appears that histidine cannot replace the cysteine at position 20 in the T7 primase. This finding is relevant to the defect observed with the hybrid primase gp4-H2.
Oligoribonucleotide Synthesis Activity
Because the altered primases were selected from a positive screening for functional gene 4 proteins, they must have at least a minimal primase activity. The ability of the primases to synthesize oligoribonucleotides was examined using a 60-mer ssDNA containing a primase recognition site (5′-TGGTC-3′). As expected, all the altered primases catalyze the synthesis of ACCA (Fig. 4A). However, when oligoribonucleotide synthesis is measured in an assay with varying amounts of template, some proteins were observed to synthesize oligoribonucleotides more efficiently than does the wild-type protein. For example, gp4-H2-KHR, RSG, and SSG have apparent binding constants 10–30-fold lower than the wild-type (Fig. 4, B and C). The other proteins exhibit similar or slightly higher values than wild-type protein. A comparison of the amino acid sequence of the altered proteins reveals an approximate correlation between the net charge of the residues in this segment and the binding constant. The more positively charged residues present in the segment, the better binding to the ssDNA template and the more oligoribonucleotides produced at low concentrations of template. For example, gp4-H2-KHR containing the most basic residues has the highest DNA binding affinity, whereas gp4-H2-ENE containing the most acidic residues shows the lowest affinity (Fig. 4C).
FIGURE 4.

Oligoribonucleotide synthesis by gene 4 primases selected from functional recombinant phage library. A, gel analysis of oligoribonucleotides synthesized on a 60-mer template. The experiment is similar to the one described in Fig. 2. Gene 4 protein (200 nm) was incubated with 10 μm 60-mer ssDNA containing a primase recognition sequence 5′-TGGTC-3′ in the presence of ATP and [α-32P]CTP. Major reaction products are indicated at the left side of the gel. B, template-directed synthesis of oligoribonucleotides. Reactions similar to A were carried out using various concentrations of the template. The amounts of [α-32P]CMP (0.1 μCi/μl) incorporated into the major product, pppACCA, were determined by phosphorimager analysis after denaturing gel electrophoresis, and the values were plotted against template concentration. C, apparent binding of gene 4 protein to the 60-mer template was calculated from the data obtained in B. Vmax and KDNA values were calculated by the nonlinear regression method.
Sequence Recognition
To determine whether alterations in the H2 segment lead to any modification in sequence-specific recognition, we carried out oligoribonucleotide synthesis using M13 ssDNA as examined earlier for the hybrid primases (Fig. 5). No significant difference in sequence recognition by any of the altered primases was found as compared with the wild-type primase.
FIGURE 5.
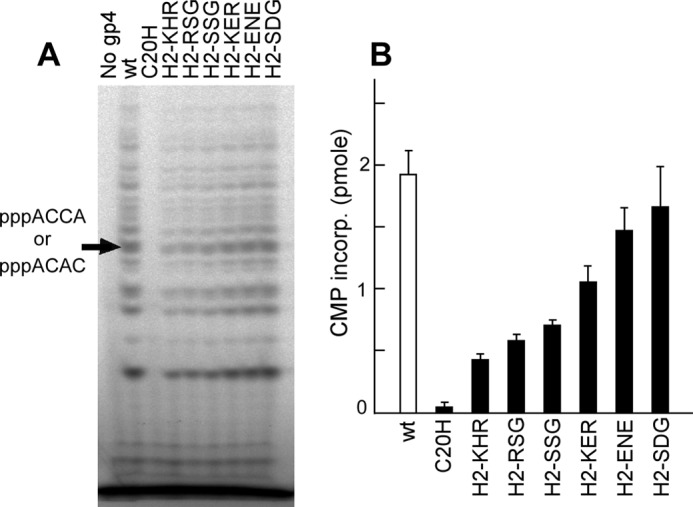
Oligoribonucleotide synthesis on M13 ssDNA by gene 4 primases selected from functional recombinant phage library. A, gel analysis of oligoribonucleotide synthesis. Gene 4 protein (200 nm) was incubated with 10 nm M13 ssDNA, 0.1 mm each of all four NTPs, and [α-32P]CTP (0.1 μCi/μl) as described in Fig. 2. dTTP was supplied at a final concentration of 0.5 mm. Major products, pppACCC and pppACAC, are indicated at the left side of the gel. B, quantitative analysis of the major products (pppACCA and pppACAC) by gene 4 proteins. Amount of CMP incorporated into pppACCA and pppACAC by the indicated protein was determined by phosphorimager analysis after denaturing gel electrophoresis.
Interaction with Long DNA Templates
In the above experiments, a trend was observed in the amount of oligoribonucleotides synthesized by the altered proteins. Altered primases that show better binding to the short DNA template compared with wild-type primase (Fig. 4) synthesize less oligoribonucleotides on the longer M13 template. One might expect the altered primase with better DNA binding to produce more oligoribonucleotides on the M13 ssDNA. However, unlike synthesis on short templates, oligoribonucleotide synthesis on the long M13 DNA requires translocation of gene 4 protein by the helicase domain in search of primase recognition sites. Tight binding on a primase recognition site might prevent the protein from translocating to another primase recognition site, resulting in less oligoribonucleotides being synthesized. This contention is consistent with reduced activity of the altered proteins to support RNA-primed DNA synthesis in which translocation activity is also necessary to search for primase recognition sites (supplemental Fig. S3). However, these altered proteins do not show any significant difference in helicase activity as judged by dTTP hydrolysis and unwinding of a mini-replication fork in the absence of primer synthesis (supplemental Fig. S4). Therefore, the decreased oligoribonucleotide synthesis on long DNA templates by the altered proteins consistently suggests tight DNA binding through alteration in the ZBD.
DNA Binding of Primase Domain
Binding of the gene 4 protein to DNA is predominantly dictated by the helicase domain (29). To confirm that the enhanced DNA binding observed with the altered proteins comes from alteration in the primase, we introduced the same alterations in the ZBD into a primase fragment containing the primase domain but lacking the helicase domain of the gene 4 protein. This truncated protein contains only the primase domain and thus eliminates any influence from the helicase domain. Both primase concentration-dependent and template concentration-dependent oligoribonucleotide synthesis assays show that the primase fragment containing the substitution of KHR (primase-H2-KHR) catalyzes the synthesis of oligoribonucleotides more efficiently than does the wild-type primase fragment (Fig. 6). Similar to the activity observed with the full-length proteins, the primase fragment containing the substitution of ENE (primase-H2-ENE) produces fewer oligoribonucleotides than does the wild-type primase fragment. The results confirm that alterations in template binding seen with full-length proteins result from changes within the primase domain.
FIGURE 6.

Oligoribonucleotide synthesis activity of altered primase fragments lacking the helicase domain. A, oligoribonucleotide synthesis as a function of primase concentration. The indicated amounts of wild-type, primase-H2-KHR, and primase-H2-ENE were incubated with 0.5 μm 25-mer ssDNA containing a primase recognition sequence 5′-TGGTC-3′ in the presence of ATP and [α-32P]CTP as described in Fig. 2. The amount of CMP incorporated into the major product pppACCA was quantitated and plotted against the primase concentration. The inset shows oligoribonucleotide synthesis at low primase concentrations. B, oligoribonucleotide synthesis as a function of template concentration. Oligoribonucleotide synthesis catalyzed by wild-type, primase-H2-KHR, and primase-H2-ENE (0.5 μm) was measured with various concentrations of the 25-mer template containing a primase recognition sequence 5′-TGGTC-3′ along with ATP and [α-32P]CTP as described in Fig. 4.
In the above experiments, the activity of the primases is inferred to be a result of the binding of the primase to its recognition sequence, and the affinity for the primases was calculated based on the kinetic results. DNA binding affinity of the primases was also examined directly using surface plasmon resonance (Fig. 7). In this assay, a 25-mer ssDNA template either containing (5′-TGGTC-3′) or lacking (5′-TGGTG-3′) a primase recognition site was immobilized on a chip through biotin-streptavidin interaction, and the primase was flowed over the chip (25). The sequence 5′-TGGTG-3′ lacks the essential cryptic cytosine; the cytosine is replaced with guanosine. As reported previously (25), the wild-type primase binds to a template containing a primase recognition site slightly better than a template lacking the recognition sequence in the absence of NTP, although the binding to either is minimal. However, sequence-specific recognition of a template containing the primase recognition site is observed in the presence of ATP and CTP that are required for oligoribonucleotide synthesis (Fig. 7A). The primase fragment containing the alteration of KHR differs in two respects to wild-type primase fragment (Fig. 7B). First, primase-H2-KHR exhibits significantly increased binding to the template containing the recognition sequence (about 700 response units) even in the absence ATP and CTP. Second, the binding is less dependent on the presence of the recognition sequence. The altered primase binds almost equally to templates that either contain or lack the recognition sequence. Regardless of the sequence in the template, the presence of ATP and CTP further enhances binding. In contrast, primase-H2-ENE does not bind to ssDNA in a sequence-dependent manner, and its binding is not affected by the presence of ATP and CTP (Fig. 7C). Despite the nonspecific binding to the template lacking a primase recognition sequence, no primer synthesis by the altered proteins was observed without a primase recognition sequence (data not shown).
FIGURE 7.

DNA binding of wild-type primase fragment, primase-H2-KHR, and primase-H2-ENE determined by surface plasmon resonance. An equal amount (1000 response units (RU)) of a 25-mer ssDNA either containing (5′-TGGTC-3′) or lacking (5′-TGGTG-3′) a primase recognition sequence was immobilized on a streptavidin-coated BIAcore sensor chip through the 5′-biotinylated end. T7 primase (10 μm) was flowed over the chip in the presence or absence of 250 μm each of ATP and CTP in a binding buffer (10 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 1 mm DTT, and 50 mm potassium glutamate), and the binding to DNA was monitored. Sensograms represent the binding of T7 primase to ssDNA containing the primase recognition sequence in the presence (red) or absence (pink) of the NTPs and the binding to ssDNA lacking the sequence in the presence (blue) or absence (green) of the NTPs.
DISCUSSION
Zinc-binding motifs play diverse roles in biological events from gene regulation to protein folding (30). Many zinc-binding motifs fulfill their functions through binding to specific sequences of DNA. Most DNA primases contain zinc-binding motifs. A small catalytic subunit of the two-component primases in archaea and eukaryotes contains a zinc knuckle consisting of Cys-His-Cys2 or Cys2-His2 (31). Despite structural information from several primases, the precise role of the zinc-binding motifs in primer synthesis remains unclear (32). Different from the primases in archaea and eukaryotes, primases encoded by bacteria and bacteriophages catalyze the template-directed synthesis of oligoribonucleotides from specific sequences in the DNA. Their modular structures reveal a ZBD composed of Cys-His-Cys2 or Cys4 separated from the RPD (6, 33). Because the RPD alone exhibits a catalytic activity independent of template sequence, it has been speculated that the ZBD of the primase is responsible for the sequence-specific recognition of the template (13). Interestingly, each of the prokaryotic primases that share overall structural similarity recognizes distinct sequences in the template. In this report, we have investigated the molecular basis of template recognition by modifying the ZBD of T7 DNA primase through homolog-scanning mutagenesis.
Role of Zinc-binding Domain in Sequence Recognition
Most substitutions (H1, H3, and H4) examined in the homolog-scanning analysis result in loss of the zinc and thus disrupt the integrity of the ZBD. These DNA primases in which the ZBD has been distorted behave similar to the 56-kDa gene 4 protein lacking the entire ZBD. It appears that the ZBD has a unique conformation that is stabilized through elaborate interactions of the residues, particularly those that retain the zinc. A similar disruption of the ZBD was previously observed by the single amino acid replacement of His-33 with alanine (28). However, replacement of a region coordinating the zinc ion with a homologous region (H2 and H5) does not result in loss of the metal ion. This observation suggests that this portion of the primase has evolved to maintain the metal and thus structural integrity of ZBD. However, maintaining the zinc ion is not sufficient for sequence recognition by the primase. Altered proteins containing these substitutions fail to catalyze template-directed synthesis of oligoribonucleotides or to deliver preformed oligoribonucleotides to DNA polymerase. These results indicate that a suitable conformation of the ZBD is required for interactions with the DNA template and RPD for proper function.
Data from a previous structural study along with data from an alanine-scanning mutagenesis enabled the identification of residues critical for primase function on one face of the ZBD (34). This face of the ZBD might interact with RPD directly or through template DNA. However, no structural change from Asp-31 and His-33 upon the addition of DNA was detected. These residues were previously shown to be critical for sequence specificity, suggesting that more than a single residue in the ZBD determines the specificity (15, 34).
Requirement of Amino Acid Residue in the H2 Region for DNA Binding
The small number of recombinant phages arising from the screen for functional gene 4 limits our interpretation for amino acid requirement in the H2 region of the ZBD of T7 primase. Nonetheless, a direct comparison of the altered proteins does provide insight into preference at specific positions. For example, gp4-H2-SSG containing serine at position 18 (underlined) exhibits higher binding to DNA than does gp4-H2-SDG harboring aspartic acid at the same position. Similarly, a comparison of kinetic parameters between gp4-H2-KHR and gp4-H2-KER indicates positively charged histidine is better than negatively charged glutamic acid for template binding. The equivalent activity of gp4-H2-SDG and wild-type primase, both of which contain the same aspartic acid at this position, implicates this position as an important factor in DNA binding. Therefore, a trend emerges where a positively charged residue at position 18 leads to better binding with DNA.
In contrast, the residue at position 16 (underlined) does not make a significant difference in DNA binding as observed with gp4-H2-SSG and gp4-H2-RSG. The preference for a positively charged residue at position 18 can be further extended to the overall charge arising from residues at positions 16, 18, and 19 in the H2 region. Alignment of primases reveals a positive correlation between the number of positively charged residues in this region and DNA binding affinity. gp4-H2-KHR with positively charged residues at the three positions exhibits much higher DNA binding affinity than does gp4-H2-ENE containing the mostly negatively charged residues. Although structural analysis of the primase-template interaction will ultimately resolve the precise contacts necessary for DNA binding, it is tempting to propose that this region of ZBD is in proximity to the template as augmented in gp4-H2-KHR. The same pattern of oligoribonucleotide synthesis by proteins with alterations in this region also suggests that this region is not responsible for recognition of specific sequences in the template. Although we observed predominantly changes in the charged (47%) and aliphatic (50%) residues at positions 16 and 19, respectively, no significant tendency was found, implying that it could be biased by the small number of phages in the library.
Single substitution of histidine for cysteine at position 20, a distinctive difference in the ZBDs of bacteria and bacteriophages, results in the loss of the zinc in the T7 primase. This result is surprising because histidine was anticipated to coordinate the metal ion within a flexible loop as in the bacterial ZBD. The incompatibility of histidine suggests that the architecture of the T7 ZBD restricts the position for cysteine.
Implication on Interactions between ZBD and RPD
Several lines of data indicate that the altered ZBDs interact with the RPD in a manner different from that observed with the wild-type ZBD. First, both primase-H2-KHR and primase-H2-ENE do not bind to DNA in a sequence-specific manner as measured by surface plasmon resonance. Because neither the ZBD nor the RPD alone can support DNA binding (25), the changes in DNA binding must result from interactions between the ZBD and RPD. Second, the requirement for ATP and CTP binding in RPD for efficient binding is altered by changes in segment H2 of the ZBD. The binding of primase-H2-KHR to its recognition sequence is enhanced by the presence of ATP and CTP, but primase-H2-ENE is not. Third, the ZBD containing H2-KHR alone does not form a stable structure. Analysis of ZBD-H2-KHR by circular dichroism spectroscopy reveals a disordered conformation as indicated by the disappearance of characteristic peaks shown in the wild-type, and the altered ZBD forms a dimer in solution (supplemental Fig. S5). Indeed, no zinc was detected by inductively coupled plasma-mass spectrometry. The absence of zinc raises the possibility that ZBD-H2-KHR alone is not stable due to the lack of critical contacts with the RPD. These data suggest that changes in DNA binding by alterations in the ZBD are mediated through interactions with the RPD.
Thus, it seems that a more likely approach to change sequence specificity would be the introduction of amino acid changes in the RPD as well as the ZBD. A comparison of several bacterial primases whose template specificities were recently identified supports this argument. G. stearothermophilus primases and the primase of Staphylococcus aureus synthesize primers from 5′-(C/T)TA-3′, whereas the primase of Aquifex aeolicus recognizes 5′-CCC-3′ (18, 19, 22). Amino acid sequence alignment of the three primases shows their ZBDs to be more conserved than the RPDs. In particular, regions coordinating the zinc ion (Cys-Pro-Phe-His and Cys-Phe-Gly-Cys) are identical for all three primases. Residues common in G. stearothermophilus and S. aureus primases but not in A. aeolicus primase are more abundant in the RPD than in the ZBD, implying a significant contribution of the RPD to sequence recognition.
Coordination between Primase and Helicase for DNA Binding
One of the gene 4 proteins containing an alteration in the ZBD, gp4-H2-KHR, exhibits about 30-fold higher sequence-specific DNA binding affinity than the wild-type primase as inferred from oligoribonucleotide synthesis on a short template. However, this tight association with ssDNA through the primase domain of the altered protein inhibits translocation activity of the helicase. As a result, oligoribonucleotide synthesis on long templates is impaired resulting in a reduced ability of the altered gene 4 protein to complement T7Δ4 lacking gene 4. This finding suggests that both primase and helicase activity must be coordinated, particularly in the gene 4 protein in which the two domains coexist. This altered gene 4 protein was also less efficient than wild-type protein in strand-displacement DNA synthesis and in a reconstituted replisome where leading and lagging strand synthesis are coordinated (data not shown). The architecture of the ZBD in DNA primase ensures moderate template binding for synthesis of oligoribonucleotides as well as for their transfer to DNA polymerase for use as primers without interfering with helicase movement.
Acknowledgments
We thank F. William Studier for providing T7 phages used for recombination assay. We are also grateful to Steve Moskowitz (Advanced Medical Graphics) for preparing the figures.
This work was supported, in whole or in part, by National Institutes of Health Grant GM 54397 (to C. C. R.).

This article contains supplemental Figs. S1–S5 and Table 1.
- ssDNA
- single-stranded DNA
- ZBD
- zinc-binding domain
- RPD
- RNA polymerase domain.
REFERENCES
- 1. Frick D. N., Richardson C. C. (2001) DNA primases. Annu. Rev. Biochem. 70, 39–80 [DOI] [PubMed] [Google Scholar]
- 2. Ilyina T. V., Gorbalenya A. E., Koonin E. V. (1992) Organization and evolution of bacterial and bacteriophage primase-helicase systems. J. Mol. Evol. 34, 351–357 [DOI] [PubMed] [Google Scholar]
- 3. Aravind L., Leipe D. D., Koonin E. V. (1998) Toprim-a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases, and RecR proteins. Nucleic Acids Res. 26, 4205–4213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tabor S., Richardson C. C. (1981) Template recognition sequence for RNA primer synthesis by gene 4 protein of bacteriophage T7. Proc. Natl. Acad. Sci. U.S.A. 78, 205–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mendelman L. V., Richardson C. C. (1991) Requirements for primer synthesis by bacteriophage T7 63-kDa gene 4 protein. Roles of template sequence and T7 56-kDa gene 4 protein. J. Biol. Chem. 266, 23240–23250 [PubMed] [Google Scholar]
- 6. Kato M., Ito T., Wagner G., Richardson C. C., Ellenberger T. (2003) Modular architecture of the bacteriophage T7 primase couples RNA primer synthesis to DNA synthesis. Mol. Cell 11, 1349–1360 [DOI] [PubMed] [Google Scholar]
- 7. Lee S. J., Richardson C. C. (2001) Essential lysine residues in the RNA polymerase domain of the gene 4 primase-helicase of bacteriophage T7. J. Biol. Chem. 276, 49419–49426 [DOI] [PubMed] [Google Scholar]
- 8. Lee S. J., Richardson C. C. (2005) Acidic residues in the nucleotide-binding site of the bacteriophage T7 DNA primase. J. Biol. Chem. 280, 26984–26991 [DOI] [PubMed] [Google Scholar]
- 9. Qimron U., Lee S. J., Hamdan S. M., Richardson C. C. (2006) Primer initiation and extension by T7 DNA primase. EMBO J. 25, 2199–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee S. J., Richardson C. C. (2002) Interaction of adjacent primase domains within the hexameric gene 4 helicase-primase of bacteriophage T7. Proc. Natl. Acad. Sci. U.S.A. 99, 12703–12708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee J. B., Hite R. K., Hamdan S. M., Xie X. S., Richardson C. C., van Oijen A. M. (2006) DNA primase acts as a molecular brake in DNA replication. Nature 439, 621–624 [DOI] [PubMed] [Google Scholar]
- 12. Dunn J. J., Studier F. W. (1983) Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 166, 477–535 [DOI] [PubMed] [Google Scholar]
- 13. Bernstein J. A., Richardson C. C. (1988) A 7-kDa region of the bacteriophage T7 gene 4 protein is required for primase but not for helicase activity. Proc. Natl. Acad. Sci. U.S.A. 85, 396–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mendelman L. V., Beauchamp B. B., Richardson C. C. (1994) Requirement for a zinc motif for template recognition by the bacteriophage T7 primase. EMBO J. 13, 3909–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kusakabe T., Hine A. V., Hyberts S. G., Richardson C. C. (1999) The Cys-4 zinc finger of bacteriophage T7 primase in sequence-specific single-stranded DNA recognition. Proc. Natl. Acad. Sci. U.S.A. 96, 4295–4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hine A. V., Richardson C. C. (1994) A functional chimeric DNA primase: the Cys-4 zinc-binding domain of bacteriophage T3 primase fused to the helicase of bacteriophage T7. Proc. Natl. Acad. Sci. U.S.A. 91, 12327–12331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kusakabe T., Richardson C. C. (1996) The role of the zinc motif in sequence recognition by DNA primases. J. Biol. Chem. 271, 19563–19570 [DOI] [PubMed] [Google Scholar]
- 18. Koepsell S. A., Larson M. A., Griep M. A., Hinrichs S. H. (2006) Staphylococcus aureus helicase but not Escherichia coli helicase stimulates S. aureus primase activity and maintains initiation specificity. J. Bacteriol. 188, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Larson M. A., Bressani R., Sayood K., Corn J. E., Berger J. M., Griep M. A., Hinrichs S. H. (2008) Hyperthermophilic Aquifex aeolicus initiates primer synthesis on a limited set of trinucleotides comprised of cytosines and guanines. Nucleic Acids Res. 36, 5260–5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Larson M. A., Griep M. A., Bressani R., Chintakayala K., Soultanas P., Hinrichs S. H. (2010) Class-specific restrictions define primase interactions with DNA template and replicative helicase. Nucleic Acids Res. 38, 7167–7178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pan H., Wigley D. B. (2000) Structure of the zinc-binding domain of Bacillus stearothermophilus DNA primase. Structure 8, 231–239 [DOI] [PubMed] [Google Scholar]
- 22. Thirlway J., Soultanas P. (2006) In the Bacillus stearothermophilus DnaB-DnaG complex, the activities of the two proteins are modulated by distinct but overlapping networks of residues. J. Bacteriol. 188, 1534–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu B., Lee S. J., Richardson C. C. (2009) An in trans interaction at the interface of the helicase and primase domains of the hexameric gene 4 protein of bacteriophage T7 modulates their activities. J. Biol. Chem. 284, 23842–23851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rosenberg A. H., Griffin K., Washington M. T., Patel S. S., Studier F. W. (1996) Selection, identification, and genetic analysis of random mutants in the cloned primase/helicase gene of bacteriophage T7. J. Biol. Chem. 271, 26819–26824 [PubMed] [Google Scholar]
- 25. Lee S. J., Zhu B., Hamdan S. M., Richardson C. C. (2010) Mechanism of sequence-specific template binding by the DNA primase of bacteriophage T7. Nucleic Acids Res. 38, 4372–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cunningham B. C., Jhurani P., Ng P., Wells J. A. (1989) Receptor and antibody epitopes in human growth hormone identified by homolog-scanning mutagenesis. Science 243, 1330–1336 [DOI] [PubMed] [Google Scholar]
- 27. Kusakabe T., Richardson C. C. (1997) Gene 4 DNA primase of bacteriophage T7 mediates the annealing and extension of ribo-oligonucleotides at primase recognition sites. J. Biol. Chem. 272, 12446–12453 [DOI] [PubMed] [Google Scholar]
- 28. Akabayov B., Lee S. J., Akabayov S. R., Rekhi S., Zhu B., Richardson C. C. (2009) DNA recognition by the DNA primase of bacteriophage T7: a structure-function study of the zinc-binding domain. Biochemistry 48, 1763–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frick D. N., Richardson C. C. (1999) Interaction of bacteriophage T7 gene 4 primase with its template recognition site. J. Biol. Chem. 274, 35889–35898 [DOI] [PubMed] [Google Scholar]
- 30. Laity J. H., Lee B. M., Wright P. E. (2001) Zinc finger proteins. New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 11, 39–46 [DOI] [PubMed] [Google Scholar]
- 31. Iyer L. M., Koonin E. V., Leipe D. D., Aravind L. (2005) Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins. Structural insights and new members. Nucleic Acids Res. 33, 3875–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lao-Sirieix S. H., Nookala R. K., Roversi P., Bell S. D., Pellegrini L. (2005) Structure of the heterodimeric core primase. Nat. Struct. Mol. Biol. 12, 1137–1144 [DOI] [PubMed] [Google Scholar]
- 33. Corn J. E., Pease P. J., Hura G. L., Berger J. M. (2005) Cross-talk between primase subunits can act to regulate primer synthesis in trans. Mol. Cell 20, 391–401 [DOI] [PubMed] [Google Scholar]
- 34. Kato M., Ito T., Wagner G., Ellenberger T. (2004) A molecular handoff between bacteriophage T7 DNA primase and T7 DNA polymerase initiates DNA synthesis. J. Biol. Chem. 279, 30554–30562 [DOI] [PubMed] [Google Scholar]
- 35. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]