

Background: CXCL12/CXCR4 and hedgehog pathways, predominantly acting in paracrine fashion, play important roles in pancreatic cancer pathobiology.
Results: CXCL12/CXCR4 signaling regulates the expression of hedgehog ligand, the sonic hedgehog, in pancreatic cancer cells.
Conclusion: Our findings indicate a novel molecular link between CXCL12/CXCR4 and hedgehog pathways.
Significance: Our data provide a molecular basis for an active bidirectional tumor-stromal interaction in pancreatic cancer.
Keywords: Chemokines, CXCR4, Hedgehog, MAP Kinases (MAPKs), NF-κB, Pancreatic Cancer, Protein Kinases
Abstract
Recent evidence suggests a major role of tumor-stromal interactions in pancreatic cancer pathobiology. The chemokine CXCL12 (stromal cell-derived factor 1 (SDF-1)), abundantly produced by stromal cells, promotes progression, metastasis, and chemoresistance of pancreatic cancer cells. On the other hand, pancreatic tumor cell-derived sonic hedgehog (SHH) acts predominantly on stromal cells to induce desmoplasia and, thus, has a paracrine effect on tumorigenesis and therapeutic outcome. In this study, we examined the association between these two proteins of pathological significance in pancreatic cancer. Our data demonstrate that CXCL12 leads to a dose- and time-dependent up-regulation of SHH in pancreatic cancer cells. CXCL12-induced SHH up-regulation is specifically mediated through the receptor CXCR4 and is dependent on the activation of downstream Akt and ERK signaling pathways. Both Akt and ERK cooperatively promote nuclear accumulation of NF-κB by inducing the phosphorylation and destabilization of its inhibitory protein, IκB-α. Using dominant negative IκB-α, a SHH promoter (deletion mutant) reporter, and chromatin immunoprecipitation assays, we demonstrate that CXCL12 exposure enhances direct binding of NF-κB to the SHH promoter and that suppression of NF-κB activation abrogates CXCL12-induced SHH expression. Finally, our data demonstrate a strong correlative expression of CXCR4 and SHH in human pancreatic cancer tissues, whereas their expression is not observed in the normal pancreas. Altogether, our data reveal a novel mechanism underlying aberrant SHH expression in pancreatic cancer and identify a molecular link facilitating bidirectional tumor-stromal interactions.
Introduction
Significant progress has been made in our understanding of the biology of pancreatic cancer over the past decade. However, it still remains as one of the most lethal malignancies (1–3). Currently, it is the fourth leading cause of cancer-related mortality in the United States, and its incidence is increasing with every coming year (4). Emerging data now suggest that tumor-stromal interactions play a major role during early and late stages of pancreatic cancer development (5, 6). Indeed, tumor cells remodel the surrounding stroma over the course of malignant progression and establish mutual functional association to cooperatively promote their growth. Despite this recognition, we are still in the beginning phase of understanding this essential “give and take” relationship between pancreatic tumor and stromal cells at the molecular level.
Hedgehog signaling has been shown to play important roles in the pathobiology of pancreatic cancer (7). Accumulating evidence indicates that ligand-dependent activation of the hedgehog pathway generally occurs in the stromal compartment through a tumor-derived hedgehog signal (sonic hedgehog (SHH)4 ligand) that phenotypically culminates into desmoplasia (8). It was shown recently that extensive desmoplasia is one of the underlying causes of pancreatic cancer chemoresistance (9). Inhibition of the hedgehog pathway depleted tumor-associated stromal tissue, enhanced intratumoral vascularity and drug accumulation, and thus led to disease stabilization. Although these effects were transient, they clearly highlighted the importance of tumor stroma and paracrine hedgehog signaling.
The CXCL12/CXCR4 pathway is critical for many normal cellular processes, including hematopoiesis, organogenesis, and vascularization, and is also utilized by the cancer cells to promote the processes of metastasis, growth, and survival (10, 11). CXCR4 expression is elevated in majority of pancreatic cancers and preinvasive neoplastic lesions, suggesting its role in the pathogenesis and progression of pancreatic neoplasia (12, 13). CXCL12, the sole ligand for CXCR4, is also abundantly produced by tumor-associated stromal cells and promotes pancreatic cancer progression and metastasis through CXCR4 activation (14, 15). CXCR4 expression by pancreatic cancer stem cells is also essential for their invasive and metastatic properties (16). Importantly, in a recent study, we have demonstrated that CXCL12-CXCR4 signaling, through activation of survival pathways, confers gemcitabine resistance to pancreatic cancer cells (17).
The aforementioned findings clearly suggest important roles for CXCL12/CXCR4 and hedgehog pathways in pancreatic cancer pathobiology. In this study, we have examined the molecular association between these two pathways of pathological significance in pancreatic cancer. Our findings demonstrate, for the first time, that the CXCL12/CXCR4 signaling axis regulates SHH expression in pancreatic cancer cells. Our data show that CXCL12 specifically signals through CXCR4 to activate Akt and ERK, which then phosphorylate IκB-α. This phosphorylation directs IκB-α to degradation pathways, causing a release and nuclear translocation of NF-κB, which then directly binds to the SHH promoter. We also present data demonstrating a correlative expression of CXCR4 and SHH in pancreatic cancer clinical specimens, whereas their expression is not detected in normal pancreatic tissues. Thus, our findings suggest a novel molecular link between paracrine-acting chemokine and hedgehog pathways, and they are indicative of an active bidirectional tumor-stromal interaction.
EXPERIMENTAL PROCEDURES
Antibodies, siRNAs, and Plasmids
Antibodies against ERK1/2 (rabbit monoclonal), pERK1/2 (mouse monoclonal), IκB-α (mouse monoclonal), p-IκB-α (Ser-32/36) (rabbit polyclonal), and NF-κB/p65 (rabbit monoclonal) were procured from Cell Signaling Technology (Beverly, MA). Antibodies against CXCR4 (rabbit polyclonal for immunoblot assay), CXCR4 (mouse monoclonal for surface CXCR4 neutralization), and SHH (rabbit monoclonal) were from Abcam (Cambridge, MA) and Millipore (Temecula, CA), respectively. Antibodies against Akt (rabbit monoclonal), p-Akt (rabbit monoclonal), and CXCR7 (rabbit polyclonal) were from Epitomics (Burlingame, CA). β-Actin (mouse monoclonal) antibody was purchased from Sigma-Aldrich (St. Louis, MO). Horseradish peroxidase-conjugated secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). All non-target (ON-TARGET plus Non-targeting pool) and target-specific (ON-TARGET plus SMART pool) siRNAs and transfection reagent (DharmaFECT) were from Dharmacon (Lafayette, CO). SHH promoter reporter plasmids (pGL3-SHH) were described previously (18). The IκB-α dominant negative vector set (pCMV-IκB-α and pCMV-IκB-αM) was from Clontech Laboratories (Mountain View, CA), and pGL4.32[luc2P/NF-κB-RE/Hygro] and pRL-TK plasmids were from Promega (Madison, WI). pcDNA3.1 was from Invitrogen. HA protein kinase B (PKB) T308D S473D pcDNA3 from the Jim Woodgett Laboratory and pBabe-Puro-MEK-DD from the William Hahn Laboratory were procured through Addgene (Cambridge, MA) (plasmid numbers 14751 and 15268, respectively).
Cell Lines, Culture Conditions, and Pancreatic Tissue Specimens
Pancreatic cancer cell lines (MiaPaCa, HPAF, and ASPC1) were purchased from the ATCC, and the Colo357 cell line was provided by Dr. Subhash Chauhan (University of South Dakota/Sanford Health). All cell lines were maintained as monolayer cultures in RPMI 1640 medium (Invitrogen) supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA) and 100 μm each of penicillin and streptomycin (Invitrogen) in a humidified atmosphere of 5% CO2 at 37 °C. Cell line validation was done by sequence tandem repeat (STR) genotyping and presence of defined markers (MUC1, MUC4, Vimentin, and DPC4). Cells were continuously monitored for their typical morphology and intermittently tested for mycoplasma using a MycoSensorPCR assay kit (Stratagene, catalog no. 302109) according to the protocol of the manufacturer. Frozen pancreatic tissue samples (normal and malignant) were obtained through the Cooperative Human Tissue Network at the University of Alabama at Birmingham under an Institutional Review Board-approved protocol.
Treatments and Transfections
Cells were cultured in complete medium in 6- or 12- well plates until they reached 50–60% confluence. Subsequently, cells were incubated in serum-free medium overnight, followed by treatments with different doses of CXCL12 (R&D Systems, Minneapolis, MN) under similar conditions for various time intervals (as indicated in the pertinent figure legends). To dissect the role of specific signaling pathways, cells were pretreated for 1 h with 20 μm LY294002 (PI3K inhibitor) and 25 μm PD98059 (ERK inhibitor) (Cell Signaling Technology) alone or in combination. For neutralization of CXCL12 binding, cells were pretreated for 60 min with CXCR4-neutralizing antibody. For the knockdown of CXCR4 and CXCR7, cells were cultured in 6-well plates and transiently transfected with 50 nm of non-target or target-specific siRNAs using DharmaFECT according to the protocol of the manufacturer. To achieve constitutive activation of Akt and ERK, cells were transiently transfected with constitutively active mutant plasmids of Akt and MEK alone or in combination. Empty plasmids were used in control transfections. FuGENE (Roche) was used as the transfection reagent for plasmid transfection.
RNA Isolation and RT-PCR
Total RNA was isolated using RNeasy purification kit (Qiagen), and subsequently cDNA was synthesized using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Carlsbad, CA) following the instructions of the manufacturer. Quantitative real-time PCR was performed in 96-well plates using SYBR Green MasterMix (Applied Biosystems) on an iCycler system (Bio-Rad). The following PCR primer pairs were used: SHH, 5′-ACCGAGGGCTGGGACGAAGA-3′ (forward) and 5′-ATTTGGCCGCCACCGAGTT-3′ (reverse) and GAPDH, 5′-gctgtgtggcaaagtccaa-g-3′ (forward) and 5′-ggtcaggctcctggaagata-3′ (reverse). The thermal conditions for real-time PCR assays were as follows: cycle 1, 95 °C for 10 min; cycle 2, (×40), 95 °C for 10 s and 58 °C for 45 s. Threshold cycle (CT) values for SHH were normalized against CT values for GAPDH, and a relative fold change in expression with respect to a reference sample was calculated by the 2−ΔΔCt method.
ELISA Assay
Culture supernatants of CXCL12-treated pancreatic cancer cells were collected at different time intervals (24–72 h), centrifuged, and subjected to ELISA using an human SHH-specific ELISA kit (Abcam) according to the protocol of the manufacturer.
Nuclear and Cytoplasmic Fractionation
The preparation of cytoplasmic and nuclear extracts was performed using the nuclear extract kit (Active Motif, Carlsbad, CA). In brief, cells were washed following treatment with 1 ml ice-cold PBS/phosphatase inhibitors (Roche), lysed in 500 μl of hypotonic buffer, and then centrifuged at 14,000 × g for 30 s at 4 °C. After collecting supernatant (cytoplasmic fraction), pellets were resuspended in 50 μl of complete lysis buffer and centrifuged at 14,000 × g for 10 min at 4 °C. Supernatants (nuclear fraction) were stored at −80 °C.
Immunoblot Analysis
Immunoblotting was performed as described earlier (19). In brief, total or fractionated protein lysates (20–60 μg) were resolved by electrophoresis on 10% SDS-PAGE and transferred onto PVDF membranes. Blots were subjected to a standard immunodetection procedure using specific antibodies against various proteins and a SuperSignal West Femto Maximum sensitivity substrate kit (Thermo Scientific, Logan, UT). The signal was detected using an LAS-3000 image analyzer (Fuji Photo Film Co., Tokyo, Japan).
Promoter-Reporter Assay
Cells were transiently cotransfected with 1 μg of pGL4.32 (luc2P/NF-κB-RE/Hygro) or pGL3-SHH along with 0.25 μg pRL-TK (a control reporter plasmid containing a Renilla reniformis luciferase gene downstream of the thymidine kinase (TK) promoter). 24 h after transfection, cells were treated with CXCL12 for 24 h as described earlier and harvested in reporter lysis buffer (Promega, Madison, WI). Firefly and Renilla luciferase activities were measured using a dual-luciferase assay kit (Promega) according to the manufacturer instructions. The luciferase activity normalized as a ratio of firefly luciferase to Renilla luciferase units. To further confirm the role of NF-κB in the CXCL12/CXCR4-induced SHH expression, we mutated the putative NF-κB binding region in the minimal CXCL12-responsive reporter construct (−102 to −1) using a Quikchange XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). The primer sequences used were as follows: forward, 5′-ctgggtggggagcggtgtg-tgacaccccgcagccgcggcgggcaagg-3′ and reverse, 5′-ccttgcccgccgcgg-ctgcggggtgtcacacaccgctccccacccag-3′. NF-κB transcriptional activity after CXCL12 treatment was measured using wild-type or mutant- reporter plasmids as described above.
ChIP Assay
Pancreatic cancer cells (untreated or treated with CXCL12) were subjected to ChIP analyses using a ChIP-IT enzymatic kit (Active Motif). In brief, DNA-protein cross-linking was done with paraformaldehyde (37%) followed by enzymatic DNA shearing. Anti-NF-κB/p65 antibody was added to the sheared DNA-protein (chromatin) complexes. A normal rabbit IgG was added to serve as a background control antibody. Following immunoprecipitation, cross-linking was reversed, the proteins were digested by proteinase K, and the DNA was isolated for PCR analyses. The primer set used was as follows: SHH, 5′-ACCAAACTCCGATGTGTTCC-3′ (forward) and 5′-GTGCGCTCTCTCTTTTGCTTC-3′ (reverse). Input DNA (cross-linked chromatin without immunoprecipitation) and negative control antibody-precipitated DNA were used as positive and negative controls, respectively.
RESULTS
CXCL12 Induces the Expression of SHH in Pancreatic Cancer Cells
The effect of CXCL12 on SHH expression was studied in four pancreatic cancer cell lines (HPAF, Colo357, AsPC1, and MiaPaCa). We observed a dose-dependent up-regulation of SHH at both transcript and protein levels upon CXCL12 treatment of pancreatic cancer cells (Fig. 1A). A 2.0- to 3.0-fold induction of SHH was detected with the lowest dose of 25 ng/ml, which increased further (over 10-fold) with higher doses of CXCL12. In a time course assay, we observed an induction of SHH as early as 4 h after treatment at both mRNA and protein levels (Fig. 1B). The SHH expression increased with exposure time, and maximum up-regulation of SHH was observed after 24 h of exposure to CXCL12, which decreased thereafter. As SHH is a secreted protein, we also measured its levels in culture media of CXCL12-treated pancreatic cancer cells. Our data demonstrate a significant accumulation of secreted SHH in the media over the 1- to 3-day period (supplemental Fig. 1). Together, these data demonstrate that CXCL12 up-regulates the expression of SHH in a dose- and time-dependent manner in pancreatic cancer cells.
FIGURE 1.

CXCL12 induces the expression of SHH in human pancreatic cancer cells. Subconfluently grown pancreatic cancer cells in 6-well plates were treated with different doses of CXCL12 (0–200 ng/ml) for 24 h (A) or with a constant dose (100 ng/ml) of CXCL12 for indicated (0–48 h) time intervals (B). Total RNA and protein were extracted and analyzed for SHH expression by quantitative RT-PCR and immunoblot analysis, respectively. GAPDH (quantitative RT-PCR) and β-actin (immunoblot analysis) were used as internal controls. CXCL12 induced dose- and time-dependent expression of SHH at both transcript and protein levels. Error bars represent the mean of triplicates ± S.D. *, p < 0.05; **, p < 0.01.
CXCR4, Not CXCR7, Mediates CXCL12-induced Expression of SHH
CXCL12 can produce its stimulatory effects by binding to its two receptors, CXCR4 and CXCR7 (10, 11). Therefore, we next examined the expression of both these receptors in pancreatic cancer cell lines. Expression of both CXCR4 and CXCR7 was observed in all four pancreatic cancer cell lines (data not shown). Subsequently, we examined which of these two chemokine receptors mediate SHH induction upon CXCL12 treatment. For this, we silenced the expression of CXCR4 and CXCR7 through RNA interference in two pancreatic cancer cell lines (MiaPaCa and Colo357) prior to treatment with CXCL12. CXCR4- and CXCR7-targeted siRNAs led to specific and effective silencing of gene expression (Fig. 2). Furthermore, we observed that CXCL12-induced SHH expression was abrogated only in cells transfected with CXCR4-targeted siRNAs, whereas CXCR7 silencing did not have any suppressive effect on SHH induction (Fig. 2). Interestingly, we also observed an increased expression of CXCR4 in CXCL12-treated pancreatic cells, indicating a positive feedback regulation. A similar observation has been made earlier in glioma cells (20). To further ascertain the role of CXCR4 in mediating CXCL12-induced SHH up-regulation, we treated the cells with CXCR4-neutralizing antibody prior to the induction with CXCL12. We observed that pretreatment of pancreatic cancer cells with CXCR4-neutralizing antibody abolished CXCL12-induced SHH up-regulation (supplemental Fig. 2). Together, these data demonstrate that CXCR4, not CXCR7, mediates CXCL12-induced up-regulation of SHH in pancreatic cancer cells.
FIGURE 2.
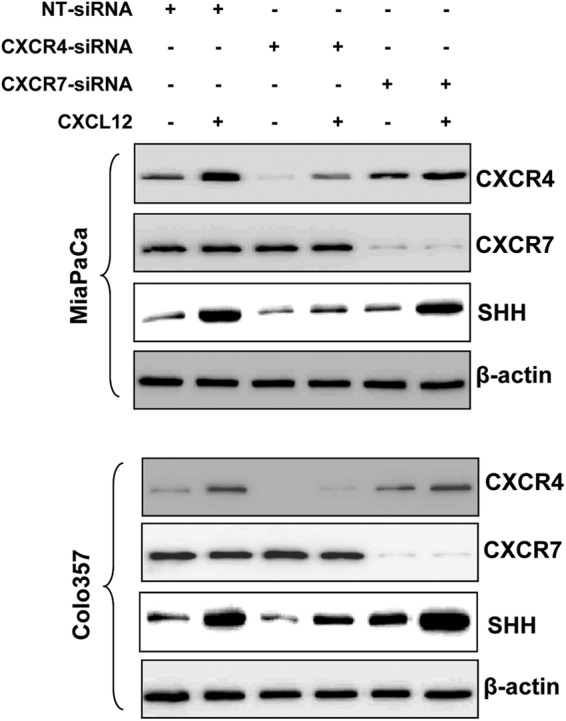
CXCL12-induced expression of SHH is mediated through CXCR4 in pancreatic cancer cells. MiaPaCa and Colo357 cells were transiently transfected with non-target (NT) or CXCR4- and CXCR7-targeted siRNAs. Following 24 h of transfection, cells were treated with CXCL12 (100 ng/ml) for another 24 h period. Total protein was isolated and subjected to immunoblot analysis to assess the expression of CXCR4, CXCR7, and SHH. β-actin was used as an internal control. Data indicate specific silencing of CXCR4 and CXCR7 by target-specific siRNAs, and clearly demonstrate that CXCL12-induced SHH expression is specifically mediated through CXCR4 in pancreatic cancer cells.
Inhibition of CXCL12-induced Akt and ERK Activation Abrogates SHH Up-regulation
We next set out to examine the effector signaling pathways involved in CXCL12-induced SHH expression. CXCL12 induces receptor phosphorylation, which leads to conformational changes and subsequent activation of a heterotrimeric G protein complex bound to the receptor intracellular domain, thus initiating downstream signaling (11). We and others have reported earlier that treatment of pancreatic cancer cells with CXCL12 elicits the activation of Akt and ERK (17, 21, 22). Activation of Akt and ERK by CXCL12 occurs as early as 1 min and starts diminishing by 30 min post-treatment (supplemental Fig. 3A). Therefore, to examine the role of these signaling nodes in CXCL12-induced expression of SHH, we treated the pancreatic cancer cells with specific pharmacological inhibitors of Akt (LY294002) and ERK (PD98059) prior to stimulation with CXCL12. Immunoblot analyses of the extracted proteins demonstrated an effective and specific inhibition of CXCL12-induced phosphorylation of Akt and ERK by their respective pharmacological inhibitors (Fig. 3, upper panel). Furthermore, we observed that inhibition of either Akt or ERK led to abrogation of CXCL12-induced SHH expression, whereas their combined inhibition led to a more potent suppression (Fig. 3, lower panel). Together, these findings indicate that both the Akt and ERK pathways cooperatively mediate CXCL12-induced SHH up-regulation.
FIGURE 3.

CXCL12-induced SHH expression in pancreatic cancer cells is dependent on the downstream activation of Akt and ERK. Pancreatic cancer cells (MiaPaCa and Colo357) were pretreated with Akt inhibitor (LY294002, 20 μm) or ERK inhibitor (PD98059, 25 μm) for 1 h, followed by treatment with CXCL12 (100 ng/ml) for either 15 min or 24 h. Total protein was isolated, and expression of Akt, p-Akt, ERK, p-ERK (after 15 min of CXCL12 exposure), and SHH (after 24 h exposure) was examined by immunoblot analysis. β-actin was used as a loading control. Data indicate the selective efficacy of inhibitors and demonstrate that the induction of SHH upon CXCL12 exposure occurs through Akt and ERK pathways.
CXCL12-induced Activation of Akt and ERK Promotes Nuclear Accumulation and Transcriptional Activity of NF-κB
To further explore downstream of Akt and ERK, we focused on NF-κB, which has been shown earlier to be activated by CXCL12 (17) and to be implicated in transcriptional regulation of SHH (23). We observed a time-dependent enhanced nuclear accumulation of NF-κB in pancreatic cancer cells upon CXCL12 treatment that correlated with a concomitant decrease in its cytoplasmic levels (supplemental Fig. 3B). Similarly, we also observed an increased transcriptional activity of the NF-κB-responsive promoter in CXCL12-treated pancreatic cancer cells (data not shown). When we treated pancreatic cancer cells (MiaPaCa and Colo357) with Akt and ERK inhibitors, alone or in combination, prior to exposing them to CXCL12, a decreased nuclear accumulation of NF-κB was observed (Fig. 4A). These data were corroborated with increased cytoplasmic levels of NF-κB in Akt- and/or ERK inhibitor-treated pancreatic cancer cells (Fig. 4A). To further substantiate the role of Akt and ERK in NF-κB translocation, we transfected the pancreatic cancer cells with active mutants of Akt and MEK (an upstream kinase of ERK). We observed an enhanced nuclear localization of NF-κB in activated Akt- and/or MEK-expressing cells that was also correlated with increased SHH expression (supplemental Fig. 4). To examine what causes the reduction in NF-κB nuclear accumulation upon activation of Akt and/or ERK, we analyzed the cytoplasmic extracts of treated pancreatic cancer cells for determination of IκB-α levels. IκB is a biological inhibitor of NF-κB that keeps it sequestered in the cytoplasm in an inactive complex (24). We observed that CXCL12 treatment led to a drastic decrease in IκB-α level, which was associated with a concomitant increase in its phosphorylation, thus indicating the destabilization of IκB-α after exposure to CXCL12. This effect was abrogated in cells that were pretreated with Akt and/or ERK inhibitors prior to stimulation with CXCL12 (Fig. 4A). To further investigate the functional consequence of this observation, we examined whether inhibition of Akt and/or ERK also suppressed CXCL12-induced NF-κB transcriptional activity. For this, we examined the CXCL12-induced transcriptional activation of NF-κB-responsive promoter in Akt and ERK-inhibited pancreatic cancer cells. We observed that inhibition of Akt and/or ERK overrode the CXCL12-induced increase in the transcriptional activity of the NF-κB-responsive promoter (Fig. 4B). Together, these data confirm a role for both Akt and ERK pathways in CXCL12-induced NF-κB activation.
FIGURE 4.

CXCL12-induced activation of Akt and ERK promotes nuclear accumulation and transcriptional activity of NF-κB in pancreatic cancer cells. A, pancreatic cancer cells were pretreated with Akt inhibitor (LY294002, 20 μm) or ERK inhibitor (PD98059, 25 μm) for 1 h, followed by treatment with CXCL12 (100 ng/ml). Total, nuclear (Nuc), and cytoplasmic (Cyto) extracts were prepared after 8 h of CXCL12 treatment to examine effects on NF-κB/p65, p-IκB-α, and IκB-α. Effects on various proteins were examined by immunoblot analysis. Laminin (for nuclear fraction) and α-tubulin (for cytoplasmic fraction) were used as loading controls. B, cells were transiently cotransfected with NF-κB-responsive or control reporter plasmids for 24 h. Subsequently, cells were pretreated (for 1 h) with Akt and ERK inhibitors and stimulated with CXCL12 for the next 24 h. Firefly and Renilla luciferase activities were examined in the treated cells as a measure of NF-κB transcriptional activity and transfection efficiency, respectively. Data are presented as the normalized luciferase units (firefly/Renilla luciferase). Error bars represent the mean of triplicates ± S.D. *, p ≤ 0.01.
Suppression of NF-κB Inhibits CXCL12-induced Up-regulation of SHH
Having observed CXCL12-induced Akt-/ERK-mediated activation of NF-κB, we next evaluated the role of NF-κB in SHH up-regulation. For this, we utilized a degradation-resistant IκB-α mutant that carries serine-to-alanine mutations at the 32 and 36 positions and thus cannot be phosphorylated in response to upstream kinase stimuli. Pancreatic cancer cells were transfected with either WT or mutant (MUT) IκB-α, and the effect on cellular distribution of NF-κB in response to CXCL12 treatment was examined. Although CXCL12 induced considerable phosphorylation and degradation of IkB-α in IκB-α-WT transfected cells, IκB-α-MUT-transfected cells exhibited limited phosphorylation and degradation of IκB-α (Fig. 5A). As a consequence, NF-κB remained sequestered in the cytoplasm in IκB-α-MUT-transfected cells, whereas substantial nuclear localization of NF-κB was observed in IκB-α-WT-transfected and CXCL12-treated pancreatic cancer cells. When we examined the effect of NF-κB activation blockade on CXCL12 induced up-regulation of SHH, we observed that it was significantly inhibited in pancreatic cancer cells transfected with IκB-α-MUT (Fig. 5B). To further confirm the role of NF-κB in transcriptional up-regulation of SHH by CXCL12, we utilized SHH promoter (deletion mutant)-reporter plasmids carrying various sequences upstream of the SHH open reading frame (18). We observed that the region between −102 to −1 of the SHH promoter, which contains a NF-κB binding site, was essential for CXCL12-induced transactivation (Fig. 6, A and B). We next mutated the putative NF-κB binding site in the minimal responsive promoter by site-directed mutagenesis and found that this led to the abrogation of CXCL12-induced transcriptional activity (supplemental Fig. 5). Finally, we found that NF-κB directly bound to the SHH promoter and exhibited greater binding in CXCL12-treated cells (Fig. 6C). Together, these findings confirm that the SHH up-regulation by CXCL12 occurs through Akt- and ERK-mediated NF-κB activation.
FIGURE 5.

Suppression of NF-κB/p65 activation abrogates CXCL12-induced SHH expression in pancreatic cancer cells. Pancreatic cancer cells grown to subconfluence in 6-well plates were transfected with IκB-α-wild-type or IκB-α-mutant plasmid. After 24 h, cells were treated with CXCL12 (100 ng/ml) for the next 8 h (for NF-κB, IκB-α, and p-IκB-α) and 24 h (for SHH). Total, nuclear (Nuc), and cytoplasmic (Cyto) extracts were prepared, and effects on NF-κB/p65, p-IκB-α (S32/36), and IκB-α (A) and SHH (B) were determined by immunoblot analyses. Laminin (nuclear fraction), α-Tubulin (cytoplasmic fraction), and β-actin (total protein) were used as loading controls.
FIGURE 6.

Direct binding of NF-κB to the SHH promoter is essential for CXCL12-induced SHH expression. A, pancreatic cancer cells were cotransfected with plasmids containing a firefly luciferase gene downstream of the SHH promoter deletion mutants and a control Renilla luciferase plasmid. After 24 h of transfection, cells were treated with CXCL12 (100 ng/ml) for another 24 h. Firefly and Renilla luciferase activities were analyzed, and data are presented as the normalized fold difference with respect to untreated cells (mean ± S.D.; n = 3; *, p ≤ 0.01). B, the SHH promoter region (-1 to −102) containing an NF-κB-binding site depicting the sequence and location of primers used in the ChIP assay. C, pancreatic cancer cells were treated with CXCL12 (100 ng/ml), and proteins and DNA were cross-linked with formaldehyde. Cross-linked chromatin was sheared and immunoprecipitated (IP) with an anti-NF-κB/p65 or nonspecific IgG. Immunoprecipitated chromatin was subjected to PCR amplification using SHH promoter-specific primers, and amplified products were resolved by electrophoresis.
CXCR4 and SHH Exhibit Correlative Expression in Pancreatic Cancer Tissues
To determine the association of CXCR4 and SHH in clinical cases, we examined their expression along with CXCR7 in protein lysates from an available limited set of normal (n = 7) and cancerous (n = 21) pancreatic tissues. Our study demonstrated no expression of CXCR4 or SHH in normal pancreatic samples, whereas a low expression of CXCR7 was detected in two of seven tissues (Fig. 7A). On the other hand, a variable expression (low to high) of CXCR4 was detected in all pancreatic cancer tissues, whereas one case each was negative for CXCR7 (C12) and SHH (C8) (Fig. 7A). We next performed densitometry to estimate the level of CXCR4, CXCR7, and SHH expression, and resulting data were subjected to Pearson correlation coefficient analysis. Our analysis demonstrated a strong correlation between CXCR4 and SHH (r = 0.863, p ≤ 0.0001), whereas no significant correlation was observed between CXCR7 and SHH (0.16, p = 0.4889) (Fig. 7B). These findings are suggestive of a clinical association between CXCL12/CXCR4 and hedgehog pathways in pancreatic cancer.
FIGURE 7.
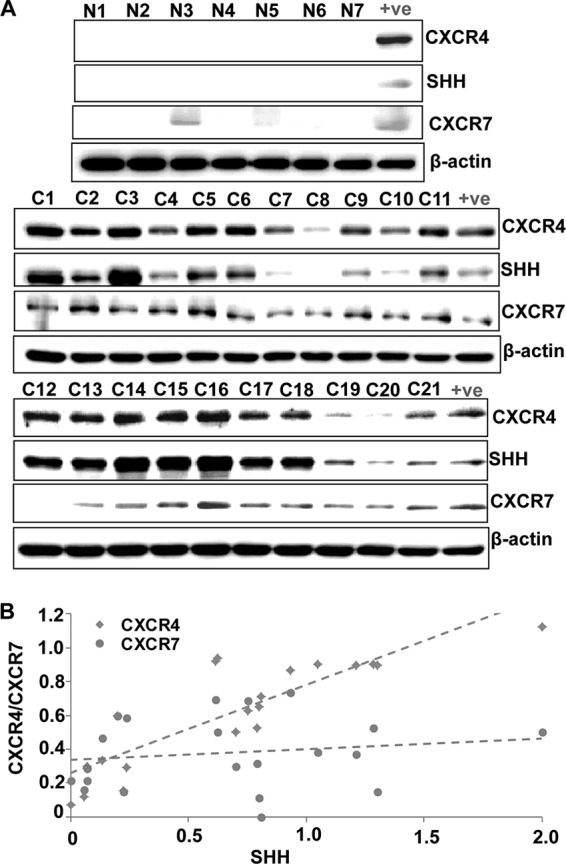
Expression of CXCR4 and SHH correlates in pancreatic cancer tissues. A, expression of CXCR4, CXCR7, and SHH was examined in normal (N1-N7) and pancreatic cancerous (C1-C21) tissues at protein level by Western blot analysis. There was no expression of CXCR4 or SHH in normal pancreatic samples, whereas a low expression of CXCR7 was detected in two of seven normal tissues. A variable expression (low to high) of CXCR4, SHH, and CXCR7 was detected in the majority or all pancreatic cancer tissues. β-actin was used as a loading control. B, intensities of the immunoreactive bands were quantified by densitometry. Normalized densitometric values of CXCR4, CXCR7, and SHH expression were subjected to Pearson correlation coefficient (R) analysis. Our analysis demonstrated a strong correlation between CXCR4 and SHH (r = 0.863, p < 0.0001), whereas no significant correlation was observed between CXCR7 and SHH (r = 0.16, p = 0.4889).
DISCUSSION
There has been a renewed interest in defining the importance of microenvironmental control in cancer pathobiology. It is now well recognized that the microenvironment plays important roles in cancer progression, metastasis, and therapeutic resistance (25, 26). This has led to a major shift in scientific efforts to identify and characterize the molecular factors and pathways that facilitate interactions between neoplastic cells and the microenvironment. The chemokine CXCL12 has been implicated in chemotaxis, cell survival, and/or proliferation by acting through several divergent pathways (11, 17). In this study, we have delineated a molecular pathway through which stromal cell-derived CXCL12 induces SHH expression in pancreatic cancer cells (Fig. 8). CXCL12 signals through CXCR4 and activates the Akt and ERK pathways, which then induce phosphorylation and destabilization of IκB-α. As a consequence, NF-κB is released, translocated to the nucleus, binds to the SHH promoter, and thus directly up-regulates SHH expression.
FIGURE 8.

A proposed molecular model of paracrine interactions between CXCL12/CXCR4 and hedgehog pathways. CXCL12 secreted by stromal cells binds to its receptor, CXCR4, present on pancreatic cancer cells. The downstream signaling thus initiated causes activation of Akt and ERK, leading to phosphorylation and destabilization of IκB-α. As a consequence, NF-κB is released and accumulated in the nucleus. Nuclear NF-κB directly binds to the SHH promoter and induces SHH expression. This proposed model establishes the role of the Akt/ERK-IKK-IkB-NF-κB cascade in CXCL12-induced SHH expression.
Hedgehog signaling is activated upon binding of hedgehog ligands (SHH and others) to their receptor (patched, PTCH), which relieves the inhibitory effect of patched on another transmembrane protein, smoothened (SMO) (27, 28). This initiates a series of downstream signaling events culminating into the nuclear translocation of Gli transcription factors and regulation of their target genes (27, 28). Aberrant hedgehog activation under malignant condition occurs through a variety of mechanisms, including gene mutations in the receptor and effector signaling molecules, aberrant expression of ligands, and non-canonical up-regulation of Gli transcription factors (29–35). SHH is expressed in preinvasive neoplastic and malignant pancreatic ductal epithelial cells of the pancreas but is absent in the normal pancreas (34). Recent findings have clearly suggested a role of SHH in the initiation and progression of pancreatic cancer. It either acts through autocrine signaling, causing selective activation of cancer stem cells, and/or through a paracrine mechanism impacting the stromal cells (8, 36–38). Therapeutic applicability of the latter mechanism was tested recently in a multicenter study using KPC (KrasG12D/+; Trp53R172H/+; Pdx-1-Cre) mice (9). It was shown that inhibition of hedgehog signaling with a small molecule antagonist improves drug delivery at the tumor site by impacting tumor stroma and vasculature. In another study, inhibition of hedgehog signaling prolonged the survival in a genetically engineered mouse and abrogated systemic metastasis (37). Therefore, observed up-regulation of SHH by CXCL12 in this study has unfolded a novel mechanism for the pathobiological actions of CXCL12 in pancreatic cancer and, thus, holds significant translational value.
In most cases, there is multiplicity of chemokine ligands and/or their receptors (10). CXCL12/SDF-1 binds to the receptors CXCR4 and CXCR7 to initiate its downstream signaling (39). On the other hand, CXCR4 has CXCL12 as its only ligand, whereas CXCR7 can bind to another chemokine, CXCL11 (10). Knockout studies in mice have identified critical functions of CXCL12, CXCR4, and CXCR7 in hematopoiesis, vascularization, and cardiac development (40–42). Although not much is known about CXCL12 signaling downstream of CXCR7, it is believed that both CXCR4 and CXCR7 may confer diverse functions. In fact, it was initially thought that CXCR7 primarily acts as a CXCL12 scavenger, thus negatively regulating CXCL12-CXCR4 signaling (43, 44). It has been shown that CXCR7 forms a heterodimer with CXCR4 and changes the conformation of the CXCR4/G-protein complexes to repress its signaling (45). Our data clearly demonstrate that up-regulation of SHH by CXCL12 is specifically mediated through CXCR4, thus further highlighting the functional significance of this signaling node in pancreatic cancer cells.
The binding of CXCL12 to CXCR4 induces diverse signaling pathways that act independently or cross-talk to promote a variety of cellular and molecular responses (11). Importantly, we and others have characterized significant roles of the ERK and Akt pathways in promoting pancreatic cancer proliferation, survival, invasion, and chemoresistance through direct and indirect mechanisms (15, 17, 22, 46). Dependence of SHH up-regulation by CXCL12 on ERK and Akt in our study further adds to the diversity of phenotypic responses influenced by these pathways. These responses were mediated by NF-κB, which serves as an intermediary for various inflammatory responses and is aberrantly activated in pancreatic cancer (47, 48). ERK- and Akt-dependent activation of NF-κB correlated with enhanced phosphorylation and degradation of IκB-α, an inhibitor of NF-κB that keeps it sequestered in the cytoplasm. A similar mechanism of NF-κB activation by Akt and ERK has been reported earlier by others, which likely involves phosphorylation of IκB kinase (IKK), a kinase that phosphorylates the IκB inhibitor protein (21, 49). A role of NF-κB has also been reported previously in SHH regulation through direct binding and transactivation of its promoter. Therefore, our data are consistent with these observations and provide further support for the pathobiological significance of NF-κB in pancreatic cancer.
In summary, we have identified a novel role of the CXCL12-CXCR4 signaling axis in SHH up-regulation in pancreatic cancer. This is particularly significant, as other studies have ascribed important pathobiological roles to both the CXCR4 and hedgehog pathways in pancreatic cancer (7, 22). Data from human clinical specimens and transgenic mouse models of spontaneous pancreatic cancer progression have suggested a role for these signaling nodes in early development and progression of pancreatic cancer (13, 34). Interestingly, in pancreatic cancer, both CXCL12 and SHH act predominantly in a paracrine manner. Abundant levels of CXCL12 produced by the stromal cells influence the growth and malignant behavior of the pancreatic tumor cells, whereas SHH secreted by the tumor cells promotes desmoplasia (8). Extensive desmoplasia is a hallmark of pancreatic cancer and is proposed to be a restraining factor in the outcome of chemotherapy (9). On the other hand, we have recently shown that the CXCL12-CXCR4 signaling axis protects pancreatic cancer cells from drug toxicity by potentiating intrinsic survival mechanisms (17). The findings from this study further emphasize the importance of this signaling node and suggest that it can confer chemoresistance not only by directly impacting the tumor cells but also indirectly through SHH-induced pancreatic fibrosis. In conclusion, our data provide novel insight into the mechanisms that allow tumor-stromal interactions and highlight the potential of the CXCL12-CXCR4 pathway as a therapeutic target in pancreatic cancer.
This study was supported by National Cancer Institute (NCI) Grants CA137513, CA167137 (to A. P. S.), and NCI P50 SPORE CA101955 (to W. E. G.), and by internal funding support from University of South Alabama Mitchell Cancer Institute (USAMCI).

This article contains supplemental Figs. 1–5.
- SHH
- sonic hedgehog
- ERK
- extracellular regulated kinase
- SDF-1
- stromal cell-derived factor-1
- PKB
- protein kinase B
- TK
- thymidine kinase
- IKK
- IκB kinase
- STR
- short tandem repeats.
REFERENCES
- 1. Bardeesy N., DePinho R. A. (2002) Pancreatic cancer biology and genetics. Nat. Rev. Cancer. 2, 897–909 [DOI] [PubMed] [Google Scholar]
- 2. Ding Y., Cravero J. D., Adrian K., Grippo P. (2010) Modeling pancreatic cancer in vivo. From xenograft and carcinogen-induced systems to genetically engineered mice. Pancreas 39, 283–292 [DOI] [PubMed] [Google Scholar]
- 3. Morris J. P., 4th, Wang S. C., Hebrok M. (2010) KRAS, Hedgehog, Wnt, and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 10, 683–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Siegel R., Naishadham D., Jemal A. (2012) Cancer statistics, 2012. CA-Cancer J. Clin. 62, 10–29 [DOI] [PubMed] [Google Scholar]
- 5. Erkan M., Reiser-Erkan C., Michalski C. W., Kleeff J. (2010) Tumor microenvironment and progression of pancreatic cancer. Exp. Oncol. 32, 128–131 [PubMed] [Google Scholar]
- 6. Neesse A., Michl P., Frese K. K., Feig C., Cook N., Jacobetz M. A., Lolkema M. P., Buchholz M., Olive K. P., Gress T. M., Tuveson D. A. (2011) Stromal biology and therapy in pancreatic cancer. Gut 60, 861–868 [DOI] [PubMed] [Google Scholar]
- 7. Kelleher F. C. (2011) Hedgehog signaling and therapeutics in pancreatic cancer. Carcinogenesis 32, 445–451 [DOI] [PubMed] [Google Scholar]
- 8. Bailey J. M., Swanson B. J., Hamada T., Eggers J. P., Singh P. K., Caffery T., Ouellette M. M., Hollingsworth M. A. (2008) Sonic Hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 14, 5995–6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Olive K. P., Jacobetz M. A., Davidson C. J., Gopinathan A., McIntyre D., Honess D., Madhu B., Goldgraben M. A., Caldwell M. E., Allard D., Frese K. K., Denicola G., Feig C., Combs C., Winter S. P., Ireland-Zecchini H., Reichelt S., Howat W. J., Chang A., Dhara M., Wang L., Rückert F., Grützmann R., Pilarsky C., Izeradjene K., Hingorani S. R., Huang P., Davies S. E., Plunkett W., Egorin M., Hruban R. H., Whitebread N., McGovern K., Adams J., Iacobuzio-Donahue C., Griffiths J., Tuveson D. A. (2009) Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun X., Cheng G., Hao M., Zheng J., Zhou X., Zhang J., Taichman R. S., Pienta K. J., Wang J. (2010) CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 29, 709–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Teicher B. A., Fricker S. P. (2010) CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 16, 2927–2931 [DOI] [PubMed] [Google Scholar]
- 12. Maréchal R., Demetter P., Nagy N., Berton A., Decaestecker C., Polus M., Closset J., Devière J., Salmon I., Van Laethem J. L. (2009) High expression of CXCR4 may predict poor survival in resected pancreatic adenocarcinoma. Br. J. Cancer 100, 1444–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thomas R. M., Kim J., Revelo-Penafiel M. P., Angel R., Dawson D. W., Lowy A. M. (2008) The chemokine receptor CXCR4 is expressed in pancreatic intraepithelial neoplasia. Gut 57, 1555–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsuo Y., Ochi N., Sawai H., Yasuda A., Takahashi H., Funahashi H., Takeyama H., Tong Z., Guha S. (2009) CXCL8/IL-8 and CXCL12/SDF-1α cooperatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 124, 853–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marchesi F., Monti P., Leone B. E., Zerbi A., Vecchi A., Piemonti L., Mantovani A., Allavena P. (2004) Increased survival, proliferation, and migration in metastatic human pancreatic tumor cells expressing functional CXCR4. Cancer Res. 64, 8420–8427 [DOI] [PubMed] [Google Scholar]
- 16. Hermann P. C., Huber S. L., Herrler T., Aicher A., Ellwart J. W., Guba M., Bruns C. J., Heeschen C. (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1, 313–323 [DOI] [PubMed] [Google Scholar]
- 17. Singh S., Srivastava S. K., Bhardwaj A., Owen L. B., Singh A. P. (2010) CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells. A novel target for therapy. Br. J. Cancer 103, 1671–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caserta T. M., Kommagani R., Yuan Z., Robbins D. J., Mercer C. A., Kadakia M. P. (2006) p63 overexpression induces the expression of Sonic Hedgehog. Mol. Cancer Res. 4, 759–768 [DOI] [PubMed] [Google Scholar]
- 19. Arora S., Bhardwaj A., Srivastava S. K., Singh S., McClellan S., Wang B., Singh A. P. (2011) Honokiol arrests cell cycle, induces apoptosis, and potentiates the cytotoxic effect of gemcitabine in human pancreatic cancer cells. PLoS ONE 6, e21573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. do Carmo A., Patricio I., Cruz M. T., Carvalheiro H., Oliveira C. R., Lopes M. C. (2010) CXCL12/CXCR4 promotes motility and proliferation of glioma cells. Cancer Biol. Ther. 9, 56–65 [DOI] [PubMed] [Google Scholar]
- 21. Lu D. Y., Tang C. H., Yeh W. L., Wong K. L., Lin C. P., Chen Y. H., Lai C. H., Chen Y. F., Leung Y. M., Fu W. M. (2009) SDF-1α up-regulates interleukin-6 through CXCR4, PI3K/Akt, ERK, and NF-κB-dependent pathway in microglia. Eur. J. Pharmacol. 613, 146–154 [DOI] [PubMed] [Google Scholar]
- 22. Shen X., Artinyan A., Jackson D., Thomas R. M., Lowy A. M., Kim J. (2010) Chemokine receptor CXCR4 enhances proliferation in pancreatic cancer cells through AKT- and ERK-dependent pathways. Pancreas 39, 81–87 [DOI] [PubMed] [Google Scholar]
- 23. Kasperczyk H., Baumann B., Debatin K. M., Fulda S. (2009) Characterization of Sonic Hedgehog as a novel NF-κB target gene that promotes NF-κB-mediated apoptosis resistance and tumor growth in vivo. FASEB J. 23, 21–33 [DOI] [PubMed] [Google Scholar]
- 24. Perkins N. D. (2012) The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 12, 121–132 [DOI] [PubMed] [Google Scholar]
- 25. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer. The next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 26. Hanahan D., Coussens L. M. (2012) Accessories to the crime. Functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322 [DOI] [PubMed] [Google Scholar]
- 27. Evangelista M., Tian H., de Sauvage F. J. (2006) The Hedgehog signaling pathway in cancer. Clin. Cancer Res. 12, 5924–5928 [DOI] [PubMed] [Google Scholar]
- 28. Ng J. M., Curran T. (2011) The Hedgehog's tale. Developing strategies for targeting cancer. Nat. Rev. Cancer 11, 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dennler S., André J., Alexaki I., Li A., Magnaldo T., ten Dijke P., Wang X. J., Verrecchia F., Mauviel A. (2007) Induction of Sonic Hedgehog mediators by transforming growth factor β. Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 67, 6981–6986 [DOI] [PubMed] [Google Scholar]
- 30. Hahn H., Wicking C., Zaphiropoulous P. G., Gailani M. R., Shanley S., Chidambaram A., Vorechovsky I., Holmberg E., Unden A. B., Gillies S., Negus K., Smyth I., Pressman C., Leffell D. J., Gerrard B., Goldstein A. M., Dean M., Toftgard R., Chenevix-Trench G., Wainwright B., Bale A. E. (1996) Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85, 841–851 [DOI] [PubMed] [Google Scholar]
- 31. Ji Z., Mei F. C., Xie J., Cheng X. (2007) Oncogenic KRAS activates Hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 282, 14048–14055 [DOI] [PubMed] [Google Scholar]
- 32. Nolan-Stevaux O., Lau J., Truitt M. L., Chu G. C., Hebrok M., Fernández-Zapico M. E., Hanahan D. (2009) GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 23, 24–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Raffel C., Jenkins R. B., Frederick L., Hebrink D., Alderete B., Fults D. W., James C. D. (1997) Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 57, 842–845 [PubMed] [Google Scholar]
- 34. Thayer S. P., di Magliano M. P., Heiser P. W., Nielsen C. M., Roberts D. J., Lauwers G. Y., Qi Y. P., Gysin S., Fernández-del Castillo C., Yajnik V., Antoniu B., McMahon M., Warshaw A. L., Hebrok M. (2003) Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425, 851–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xie J., Murone M., Luoh S. M., Ryan A., Gu Q., Zhang C., Bonifas J. M., Lam C. W., Hynes M., Goddard A., Rosenthal A., Epstein E. H., Jr., de Sauvage F. J. (1998) Activating Smoothened mutations in sporadic basal cell carcinoma. Nature 391, 90–92 [DOI] [PubMed] [Google Scholar]
- 36. Feldmann G., Dhara S., Fendrich V., Bedja D., Beaty R., Mullendore M., Karikari C., Alvarez H., Iacobuzio-Donahue C., Jimeno A., Gabrielson K. L., Matsui W., Maitra A. (2007) Blockade of Hedgehog signaling inhibits pancreatic cancer invasion and metastases. A new paradigm for combination therapy in solid cancers. Cancer Res. 67, 2187–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feldmann G., Fendrich V., McGovern K., Bedja D., Bisht S., Alvarez H., Koorstra J. B., Habbe N., Karikari C., Mullendore M., Gabrielson K. L., Sharma R., Matsui W., Maitra A. (2008) An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol. Cancer Ther. 7, 2725–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yauch R. L., Gould S. E., Scales S. J., Tang T., Tian H., Ahn C. P., Marshall D., Fu L., Januario T., Kallop D., Nannini-Pepe M., Kotkow K., Marsters J. C., Rubin L. L., de Sauvage F. J. (2008) A paracrine requirement for Hedgehog signalling in cancer. Nature 455, 406–410 [DOI] [PubMed] [Google Scholar]
- 39. Duda D. G., Kozin S. V., Kirkpatrick N. D., Xu L., Fukumura D., Jain R. K. (2011) CXCL12 (SDF1α)-CXCR4/CXCR7 pathway inhibition. An emerging sensitizer for anticancer therapies? Clin. Cancer Res. 17, 2074–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Agarwal U., Ghalayini W., Dong F., Weber K., Zou Y. R., Rabbany S. Y., Rafii S., Penn M. S. (2010) Role of cardiac myocyte CXCR4 expression in development and left ventricular remodeling after acute myocardial infarction. Circ. Res. 107, 667–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sierro F., Biben C., Martínez-Muñoz L., Mellado M., Ransohoff R. M., Li M., Woehl B., Leung H., Groom J., Batten M., Harvey R. P., Martínez-A C., Mackay C. R., Mackay F. (2007) Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. U.S.A. 104, 14759–14764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tachibana K., Hirota S., Iizasa H., Yoshida H., Kawabata K., Kataoka Y., Kitamura Y., Matsushima K., Yoshida N., Nishikawa S., Kishimoto T., Nagasawa T. (1998) The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393, 591–594 [DOI] [PubMed] [Google Scholar]
- 43. Boldajipour B., Mahabaleshwar H., Kardash E., Reichman-Fried M., Blaser H., Minina S., Wilson D., Xu Q., Raz E. (2008) Control of chemokine-guided cell migration by ligand sequestration. Cell 132, 463–473 [DOI] [PubMed] [Google Scholar]
- 44. Naumann U., Cameroni E., Pruenster M., Mahabaleshwar H., Raz E., Zerwes H. G., Rot A., Thelen M. (2010) CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 5, e9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Levoye A., Balabanian K., Baleux F., Bachelerie F., Lagane B. (2009) CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 113, 6085–6093 [DOI] [PubMed] [Google Scholar]
- 46. Wang Z., Ma Q., Liu Q., Yu H., Zhao L., Shen S., Yao J. (2008) Blockade of SDF-1/CXCR4 signalling inhibits pancreatic cancer progression in vitro via inactivation of canonical Wnt pathway. Br. J. Cancer 99, 1695–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bassères D. S., Baldwin A. S. (2006) Nuclear factor κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene 25, 6817–6830 [DOI] [PubMed] [Google Scholar]
- 48. Ben-Neriah Y., Karin M. (2011) Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 12, 715–723 [DOI] [PubMed] [Google Scholar]
- 49. Bai D., Ueno L., Vogt P. K. (2009) Akt-mediated regulation of NF-κB and the essentialness of NF-κB for the oncogenicity of PI3K and Akt. Int. J. Cancer 125, 2863–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]