

Background: NS3-4A is a validated target for antiviral therapeutics whereby HCV rapidly develops resistant mutants.
Results: Peptides coordinating with a unique NS3-4A site and strongly inhibiting common resistance mutants were developed.
Conclusion: A unique “finger” structure extends binding of the inhibitory peptides to a novel druggable site.
Significance: Novel leads are of interest for the design of inhibitors refractory to known resistance mutants.
Keywords: Drug Design, Hepatitis C Virus, Phage Display, Protease Inhibitor, Protein Structure
Abstract
Hepatitis C virus (HCV) NS3-4A protease is essential for viral replication. All current small molecular weight drugs against NS3-4A are substrate peptidomimetics that have a similar binding and resistance profile. We developed inhibitory peptides (IPs) capping the active site and binding via a novel “tyrosine” finger at an alternative NS3-4A site that is of particular interest for further HCV drug development. The peptides are not cleaved due to a combination of geometrical constraints and impairment of the oxyanion hole function. Selection and optimization through combinatorial phagemid display, protein crystallography, and further modifications resulted in a 32-amino acid peptide with a Ki of 0.53 nm. Inhibition of viral replication in cell culture was demonstrated by fusion to a cell-penetrating peptide. Negligible susceptibility to known (A156V and R155K) resistance mutations of the NS3-4A protease was observed. This work shows for the first time that antiviral peptides can target an intracellular site and reveals a novel druggable site on the HCV protease.
Introduction
Some 160 million people are persistently infected with hepatitis C virus (HCV),3 with 3 to 4 million new infections each year (1, 2). HCV infections are the leading cause of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma (1). The HCV virion encodes a polyprotein of ∼3000 amino acids. The polyprotein is co- and post-translationally processed by cellular and viral proteases into mature viral proteins (3). The nonstructural (NS) proteins are processed by two viral proteases NS2-3 and NS3-4A. Although the NS2-3 auto-protease performs cleavage of NS3 from NS2, the NS3-4A protease complex catalyzes cleavage at all remaining junctions (3, 4). The NS3 protease domain located within the N-terminal region (aa 1–180) of NS3 is a chymotrypsin-like serine protease that forms a heterodimeric co-complex with NS4A, a 54-residue long important cofactor for NS3 proteolytic activity (5, 6). For activation of the NS3 protease domain, only residues 21 to 34 of NS4A are required (7). Due to its shallow, solvent-exposed substrate-binding pocket, the substrate coordination extends over the whole binding region (6, 7). This also correlates with the reported reduced ability to cleave substrates smaller than 10 amino acids (8).
The standard HCV treatment, pegylated interferon-α (INF-α) and ribavirin, achieves a sustained viral response in only half of treated patients and is associated with severe side effects (9, 10). Moreover, this regimen is expensive and is contraindicated in a substantial number of chronic HCV patients. Therefore, novel treatment options are urgently needed. The first directly acting antiviral drugs that target the viral NS3-4A protease complement the interferon-based regimen and in combination therapy increase cure rates to nearly 70% of patients infected with the most prevalent HCV genotypes in industrialized countries (11). However, resistance development and viral genotype-dependent variation in efficacy demand development of novel drugs with a higher barrier to resistance across all HCV genotypes (11, 12).
Product-based inhibitor development is so far the most successful approach for NS3-4A protease inhibitors (PIs). Based on their chemical scaffold, current PIs are divided into classes, including macrocyclic inhibitors (e.g. ITMN-191) and linear ketoamides (e.g. telaprevir and boceprevir) (12). Most of those protease inhibitors are spanning exclusively the S4–S1′ subsites of NS3, thus having a similar resistance profile (13, 14) and hence a substantial cross-resistance; for instance, R155(K/T/Q) as well as A156(V/T) mutations confer viral resistance to both ketoamide and macrocyclic inhibitors (12). Due to the low barrier to viral resistance, telaprevir and boceprevir cannot be used in monotherapy (1, 13) and do not replace the IFN-based standard therapy.
We considered that large peptides might provide an interesting alternative for rapid development of drug leads: they may lead to discovery of a priori binding at alternative sites, thus allowing novel binding mechanisms. Novel motifs having initially weak binding before further optimization may be discovered due to synergism of several motifs over a larger interactive surface. Drugs combining such motifs may well be less affected by HCV resistance mutations, which are reported to have developed on challenge with recent small peptidomimetic drugs. However, direct use of peptides as drugs with potentially high specific activity and low toxicity have not so far been seriously considered due to their low bioavailability (16). To screen for large IPs against NS3-4A, we used cosmix-plexing, a combinatorial phage display peptide library method (17). This combinatorial methodology efficiently assorts parental cassettes in combination with new cassettes carrying novel motifs. In the later stages of inhibitor optimization, we applied x-ray crystallography and rational design. This led to IPs with binding constants in the subnanomolar range bearing a novel tyrosine finger structure that showed an extended coordination to an NS3 pocket remote from the active site.
EXPERIMENTAL PROCEDURES
NS3-4A Activity Assay
The activity of the NS3-4A protease was tested with a FRET assay (18). In MTPs (μClear non-binding 96-well black plates, Greiner bio-one), 0.5–1 nm protease was diluted in buffer M2235 (19) (50 mm HEPES, pH 7.8, 100 mm NaCl, 20% (w/v) glycerol, 5 mm DTT, 0.6 mm lauryldimethylamine-N-oxide), and 5 μm substrate M2235 (Ac-Asp-Glu-Asp(EDANS)-Glu-Glu-Abu-y-(COO)Ala-Ser-Lys(DABCYL)-NH2, Bachem) was added. Respective inhibitors were incubated with NS3-4A protease for 10–15 min at room temperature prior adding of substrate to a final volume of 200 μl. Fluorescence increase was measured every minute for 30–60 min at 30 °C in a MTP reader (excitation, 360/40 nm; emission, 460/40 nm, Biotek Synergy 2). Each measurement was done in duplicate. Slopes of fluorescence increase were compared with that of a control. A nonlinear regression model (Equation 1) was used to fit curves with GraphPad Prism.
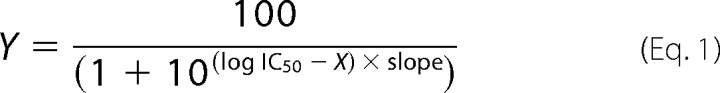 |
IC50 values were converted to apparent Ki values using Equation 2 for competitive inhibitors (20).
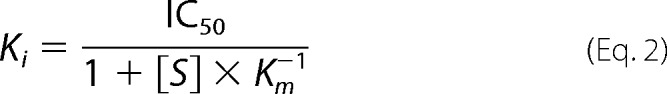 |
Affinity Selection in Microtiter Plates
For affinity selection in microtiter plates (Nunc, Maxisorp), 200 ng of anti-MBP IgG in PBS were immobilized (1 h at room temperature). Wells were washed twice with 250 μl of PBS containing 0.05% (v/v) Tween 20. Unspecific binding sites were blocked with 350 of μl 2% (w/v) milk powder in panning buffer (PB) (50 mm HEPES, pH 7.5, 0.05% (v/v) Tween 20/0.5× PBS, 0.05% (v/v) Tween 20 after cosmix-plexing) at room temperature for 60 min. Thereafter, wells were washed twice with 250 μl of PB and an excess of MBP-NS3-4A (1 μg) was immobilized at 4 °C for 90 min. Prior to selection, 200 μl of peptide phagemid particles in PB with 2% (w/v) milk powder containing 0.05% (v/v) Tween 20 were incubated with immobilized MBP to reduce unspecific binding and specific binding to either MBP or anti-MBP IgG. Wells with immobilized fusion protein were washed three times with PB, and pre-incubated peptide phage particles were added. Affinity selection was carried out on a platform shaker at room temperature for 150 min. Unbound phage was removed, and wells were washed at least 10 times (16–20 times after cosmix-plexing) with 250 μl of PB. Bound phage were eluted either by acidic or competitive elution. For acidic elution, 200 μl of elution buffer (0.1 m glycine, pH 2.2) was added and incubated for 10 min on a shaker. The eluted phage particles were removed and immediately mixed with 45 μl of neutralization buffer. For competitive elution, 200 μl of a known NS3-4A inhibitor N-1725 (Bachem) was added and incubated for 15 min on a shaker. Phage titer was determined with 10 μl of eluted phage solution. The remaining volume was used for infection of a 20-ml new grown Escherichia coli Top 10 F′ λ+ culture with an A600 of 0.5. The culture was incubated at 37 °C for 30 min without and then 30 min with agitation at 180 rpm. After incubation, the cells were pelleted (4000 rpm/10 min), resuspended in 400 μl of 2×YT medium and plated onto a 2×YT agar plate (100 μg/ml ampicillin and 6 μg/ml tetracycline, 37 °C, overnight). Cells were washed from plates and used for phagemid packaging. Single colonies from the titer determination were resuspended in 150 μl of double distilled H2O and used for colony PCR with the primer FSQJCO (5′-TTCTA CAACT TGCTT GGATT-3′). PCR products were used for sequence analysis.
Structure Determination
Purified NS3-4A protease (40–90 μm) was incubated on ice with >2-fold molar excess of peptide in elution buffer (50 mm sodium acetate, pH 6.3, 500 mm NaCl, 5 mm DTT, 0.6 mm lauryldimethylamine-N-oxide, 10% (w/v) glycerol) for 2 h. The solution was centrifuged at 15,000 rpm and 4 °C for 20 min and concentrated to ∼220 μm (∼5 mg/ml) using an Amicon Ultra-4 centrifugal filter (10 kDa; Millipore). Peptides (dissolved in dimethyl sulfoxide) were added to a final concentration of ∼500 μm. The solution was centrifuged at 15,000 rpm and 4 °C for 20 min. Dilutions with 100 μm (2 mg/ml) and 120 μm (2.5 mg/ml) complex were prepared with 0.1 m buffer of the precipitant solution and used for crystallization. Diffraction-quality crystals were obtained after several days at 19 °C by mixing equal volumes of concentrated complex solution with precipitant solution (0.1 m MES, pH 6.0, or 0.1 m sodium citrate, pH 5.1, and 2.2 m KCl, 5% (v/v) isopropanol) by hanging-drop vapor-diffusion crystallization in 24 six-well or 15 six-well plates (EasyXtal; Qiagen). Prior to flash-freezing in liquid nitrogen crystals were cryoprotected with 30% (w/v) glycerol. X-ray diffraction data were collected at 100 K on a Rigaku MicroMax 7HF copper annode equipped with a Saturn 944+ detector (home source) or at the BESSY synchrotron beamline MX-14.1. Data were indexed, integrated, and scaled with HKL2000 (21) or the XDS/XSCALE (22) package. Phases were obtained with Phaser (23) using NS3-4A coordinates (PDB code 1DXP) as a search model. The coordinates were refined with Phenix.refine (24) or Refmac5 (25) and manually checked and corrected with COOT (26). The IPs were manually built with COOT. Figures and structure alignments were prepared using PyMOL (27). Data and refinement statistics are summarized in supplemental Table S2.
RESULTS
Selection of Inhibitory Peptides
To find novel scaffolds for the design of HCV PIs, we chose an approach that did not solely rely on mimicking natural substrate analogues. Initially, the large peptide library CPL19YS-2 was added to immobilized NS3-4A protease of the HCV genotype 1b. This library (supplemental Fig. S1) contains a cassette encoding for 33-aa-long peptides with 26 variable residues and a centered His-Cys dipeptide. Each variant is presented as a single copy (monovalent) on a phagemid particle carrying the gene variant that encodes it. Some five to six reiterative cycles led to enrichment of initial binding peptide clones. Two panning strategies with acidic or competitive elution led to enrichment of one dominant clone each: K5-66 for the first and K6-10 for the later strategy. Interestingly, both selected variants show a nine-amino acid N-terminal consensus sequence VY(V/L)L(D/G) (G/D)PGY, which is mainly hydrophobic, but interrupted by charged Asp-11 or Asp-12 residues in the case of K5-66 and K6-10, respectively (Table 1). The probability of this sequence homology occurring by chance (randomly) within the region responsible for higher affinity is <1 in 109. As described below, it also corresponds to the peptide region forming the most rigid interaction in the co-crystal with the NS3-4A protease. It is thus considered to be the dominant novel motif to have been discovered in this study. Peptides K5-66 and K6-10 showed NS3-4A inhibition with IC50 values of 21 and 26 nm, respectively. Truncation of K5-66 and K6-10 revealed that their N-terminal half (comprising the consensus sequence motif) is most important for inhibition (Table 1) and leads to a 6- or 9-fold reduced inhibition, respectively. Notably, neither of the C-terminal halves of these peptides inhibit NS3-4A alone, but increase inhibition by synergy with the amino-terminal sequences.
TABLE 1.
Enriched peptides against MBP-NS3-4A protease and inhibitory effect of the peptides on NS3–4A activity
Comparison of IC50 values and Ki values of derived peptides. Constant sequence parts are colored in blue, and homologies are colored in orange. Substituted residues in later stages of optimization are colored in red. Peptide K5-66-R is a scrambled control peptide. Measurements were carried out in duplicate, the curves were fitted using GraphPad Prism with a nonlinear regression model, and the standard deviation is shown.

New Combinatorial Libraries from Pre-enriched Clones Yield a More Potent Inhibitor
Cosmix-plexing was applied (17) to generate novel diversity of N- and C-terminal regions of pre-selected populations. Isolated phagemid DNA was cleaved (supplemental Fig. S1) and recombinants submitted to further affinity selection under more stringent panning conditions. A new candidate, CP5-46, was identified showing an 8-fold higher phage-ELISA signal than K5-66 (supplemental Fig. S2A), correlating with a 2-fold lower IC50 (11 nm; Table 1 and supplemental Fig. S3). CP5-46 comprises the N-terminal part of K5-66 fused to a new C-terminal sequence. Cleavage of CP5-46 was examined with an excess of NS3-4A protease. After 16 h, neither a change of CP5-46 concentration nor any cleavage products were detectable in LC-MS (supplemental Table S1). With reference to the specificity of the inhibitors, we show that CP5-46 (≤50 μm) does not inhibit chymotrypsin, trypsin, or elastase (supplemental Fig. S2B).
The CP5-46·NS3-4A Co-complex Structure Reveals Mode of Action
The binding mode of CP5-46 to NS3-4A was determined by the crystal structure of the inhibitor·enzyme complex. To this purpose, the activating NS4A peptide (aa 21 to 34) was fused N-terminally by a diglycine linker to the NS3 protease domain (residues 2–180) (28). High quality diffracting crystals were only obtained with the shorter CP5-46-A peptide, comprising the first 21 N-terminal aa of the full-length peptide (Table 1). Crystals diffracted to 2.05 Å and show P61 symmetry (supplemental Table S2). Clear electron density was observed for peptide residues 4–21 (supplemental Fig. S4, A and B). CP5-46-A covers ∼800 Å2 of the protease surface. This represents some 9% of the total protease solvent-accessible surface (supplemental Table S6). The active site is totally occluded, and the uncleaved peptide (aa 4–20) is coordinated by 15 hydrogen bonds on the non-prime (S) and prime (S′) sites of NS3 (Fig. 1A and supplemental Table S6).
FIGURE 1.

Detailed views on the CP5-46-A/NS3-4A co-complex. A, overview on CP5-46-A (green) in complex with NS3-4A (blue/orange). Dashed red boxes indicate magnified views displayed in B–D. B, close up on NS3-4A active site (blue) with bound CP5-46-A (green). Catalytic triad residues Ser-139, His-57, and Asp-81 are colored in wheat, and other protease residues are colored in blue. A red dashed line shows the closest distance of Ser-139 and CP5-46-A backbone (Leu-9–Leu-10). H-bonds of an active site H2O are drawn with black dashed lines. C, non-primed site of the protease, CP5-46-A main chain (green) forms an anti-parallel β-strand with the backbone of NS3-4A (blue). D, primed site of NS3-4A with accommodated tyrosine finger (aa 13PGYDP17). Coordinated water molecules are shown as red spheres. The NS4A peptide is colored orange. aa 10–12 and 18–19 of CP5-46-A are shown in line representation.
Nearly fifty percent of the solvent-accessible area of CP5-46-A (1042 Å2) interacts with NS3 (supplemental Table S6), indicating tight binding, without influencing the alignment of catalytic triad residues (Fig. 1B). Several parameters seem to preclude the nucleophilic attack on the peptide backbone. The side chain of Leu-9 binds at the S1 pocket, but due to its isobutyl side chain, it sterically clashes with Phe-154 (supplemental Fig. S4C) resulting in a more remote positioning of the scissile bond. Similarly, the adjacent Leu-10, which binds to the subsite S1′, moves the peptide backbone away from the active site, although the S1′ pocket was shown to bind larger side chains than the natural P1′ serine (29). Additionally, Leu-10 causes dislocation of a nearby NS3-4A loop (Thr-38–Gln-41) (Fig. 2A). The bound peptide covers the entire active site in a lid-like fashion and holds the nearest peptide carbonyl carbons of Leu-9 or Leu-10 more than 3.5 Å away from the catalytic Ser-139 (Fig. 1B). This causes a stretched and hence unfavorable alignment for a nucleophilic attack. Interestingly, a water molecule fills the residual space (Fig. 1B). Additionally, a hydrogen bond links Leu-9 to the backbone amide of Gly-137 (supplemental Table S6), which is part of the oxyanion hole (6). This presumably diminishes its charge redistribution function, reducing the stabilization of a tetrahedral intermediate and thereby further increasing the activation energy of the reaction.
FIGURE 2.

A, structural effects upon binding of peptide CP5-46-A. CP5-46-A causes structural dislocation of a NS3-4A loop (aa Thr-38–Gln-41) close to the active site. Apo-NS3-4A protease with free active site is shown in gray (PDB code 1DXP). NS3-4A with bound peptide CP5-46-A (green) is shown in blue. B, N-terminal part of CP5-46-A binds to the non-prime (S) sites of NS3-4A. Locations of the protease subsites are labeled with semicircles (positions estimated from literature (47)). Peptide CP5-46-A is shown in stick representation with modeled surface. Salt bridge between His-19 and Asp-11 is indicated as a gray dashed line. C, comparison of the coordination of peptide CP5-46-A and the optimized peptide CP5-46A-4D5E. B-factors for the peptide residues are indicated by rainbow colors, whereas blue indicates a low and red a high B-factor (scale bar at bottom left corner). Peptides were superposed based on the NS3-4A backbone. D, illustration of the close coordination of the tyrosine finger motif. CP5-46-A residues are shown in gray, and NS3 residues are shown in orange. Inhibitory peptide residues 16–20 are not shown for the sake of clarity. Dashed lines indicate hydrogen bonds. Three coordinated water molecules close to Tyr-15 are displayed as red spheres.
The N-terminal part of CP5-46-A, which binds to the non-prime (S) sites of the protease (Fig. 2B), forms an anti-parallel β-strand with the NS3 backbone (Fig. 1C). Notably, this arrangement is conserved among all peptide-based protease substrates (30). The inhibitor and the substrate backbone (Gly-4–Leu-6) bind likewise (Fig. 3A). Binding of CP5·46-A at the non-prime (S) sites of NS3 is similar to that reported with peptidomimetic compounds such as ITMN-191 or boceprevir (Fig. 3, B and C). The macrocyclic inhibitor ITMN-191 is spanning into the S3–S1′ subsites and has a large P2 substituent extending to the S2 subsite. The linear α-ketoamide inhibitor boceprevir binds to S4-S1 subsites of NS3-4A with similar backbone coordination. Notably, the peptide backbone of CP5-46-A is slightly shifted away from the catalytic triad, although functional groups bind to the same sites on the protease surface. The P1′ and P1 cyclopropyl group of ITMN-191 (Fig. 3B) and boceprevir, respectively (Fig. 3C), are similar to the isopropyl side chain of P1′ Leu-10 or P1 Leu-9: all are hydrophobic and similarly coordinated in the protease subsites. The P2 hydrophobic functional groups of peptidomimetic inhibitors overlap closely with the P2 residue Tyr-8 of CP5-46-A. Furthermore, their P4 tert-butyl group is akin to the isopropyl side chain of the P4 residue Leu-6 and is also located in the S4 pocket (Fig. 3, B and C).
FIGURE 3.

Binding of peptide CP5-46-A to NS3-4A compared with the natural substrate and small molecule inhibitors. A, binding of CP5-46-A (green) compared with the natural substrate 4A5B (orange, PDB code 3M5M). NS3 protease domain is shown in blue with modeled surface and side chains of active site residues in orange. B, binding of CP5-46-A (green) compared with small molecule inhibitor ITMN-191 (blue, PDB code 3M5L). C, binding of CP5-46-A (green) compared with small molecule inhibitor boceprevir (yellow, PDB code 2OC8). The side chains binding to the protease subsites are indicated (P1′–P6). Structures were aligned based on NS3/4 backbone residues.
Tight Coordination Is Correlated with a Novel Tyrosine Finger Motif
The C-terminal section of CP5-46-A is located at the protease prime (S′) site. Residues 13–17 fold back, creating a finger-like structure (aa sequence PGYDP). Interestingly, the central motif that binds to the bottom of the pocket (PGY) was also found in peptide K6-10 from the initial panning with competitive elution (Table 1). No similarly structured tyrosine finger with a PGY(D,Y)(P,A) pattern was found in the PDB using the PDBeMotif server. The sequences observed with a similar pattern do not show such an extended finger-like structure and sequester the tyrosine (supplemental Fig. S8). This tyrosine finger covers a pocket located next to the NS4A peptide and is mainly hydrogen-bonded with NS3 aa Ser-7, Ser-37, Arg-109, and several water molecules (Fig. 1D). The tyrosine finger is presumably responsible for the tight binding of IPs. This is supported by lower local atomic B-factors that match those of surrounding protease residues compared with the rest of the peptide (Fig. 2C). The tyrosine finger extends the binding of the IP at the S′ site of the protease to a region probably not probed by natural substrates. Additionally, His-19 of CP5-46-A forms an intramolecular salt bridge with Asp-11, further stabilizing the tyrosine finger and the IP.
Increasing the Potency of CP5-46-A by Structure-based Modifications
Based on the NS3-4A/CP5-46-A complex, a prognosis was made for several amino acid substitutions that might improve binding of IPs. Amino acids that may enhance binding to the S1′ and S4 subsites of the protease were replaced. Leu-10 at position P1′ was modified to Glu or Gln trying to facilitate hydrogen bond interactions with N3/4A residue Gln-51 and His-57 by occupying residual space in S1′. Additionally substitutions with Gln, Glu, or Asp of peptide residue Leu-6 at position P4 were generated. However, unfortunately none of those assayed peptides showed any inhibitory effect. We interpret this result as an indication that the original peptides are already highly accommodated and that alterations lead to disruption of steric geometry of the interaction (supplemental Table S3).
Finally, alterations were considered for the constant N-terminal part of the peptide, i.e. that region which had not been subjected to randomization in the gene library, largely influenced by considerations similar to those undertaken in early work of others, based on homology to natural peptide substrate. Comparison of CP5-46-A with bound substrate 4A4B (PDB code 3M5M) revealed that peptide backbone atoms of Gly-4 and Arg-5 coordinate akin to P5-Glu and P6-Asp of 4A4B (Fig. 2A). Positions P5 and P6 are known to be important for 4A4B binding mainly due to electrostatic interactions with basic amino acids in particular P6-Asp with protease Lys-165 (30, 31). Hence, we introduced a G4D and R5E mutation to mimic this interaction. This led to a 6-fold increase in inhibitory potency of CP5-46-A (from IC50 = 124 nm to 23 nm (Table 1)). In the longer and more potent CP5-46 (IC50, 11.3 nm) a 10-fold improvement of IC50 value (1.1 nm) and Ki (∼530 pm; Table 1 and supplemental Fig. S3) was observed.
Crystals of NS3-4A in complex with the optimized peptide CP5-46A-4D5E were obtained under similar conditions and diffracted to 2.2 Å in the same space group (supplemental Table S2). A superposition based on the NS3-4A residues shows that both peptides bind in a similar way (Fig. 2C and supplemental Fig. S5); the double substitution with acidic amino acids (G4D,R5E) did not alter overall coordination. However, the N- as well as C-terminal amino acids of CP5-46A-4D5E (residues 4–6 and 19–21) have higher atomic B-factors compared with CP5-46-A (Fig. 2C). This indicates a more loose and flexible binding, also reflected by the weak electron density observed for those residues (supplemental Fig. S5, B and C). Notably, here again the central tyrosine finger shows the lowest mobility, which is in the same range as that for CP5-46A and the NS3 protease. As predicted the position of Glu-5 is similar to P5-Glu of the substrate 4A4B (Fig. 3A). The ionic interactions between the inhibitor Asp-4 and protease Lys-165 have apparently not formed. Instead, the side chain of Asp-4 points in the opposite direction (supplemental Fig. S5D).
Inhibitory Peptide Shows Negligible Susceptibility to Known NS3-4A Resistance Mutations
A persistent problem of viral inhibitors under development is the rapid emergence of resistant variants due to the low viral RNA polymerase fidelity and the high viral replication rate (32). Hence, peptidomimetic monotherapy rapidly leads to selection of resistant protease variants (12). Several mutations of NS3 that cause resistance against current peptidomimetic protease inhibitors are in close proximity to the bound peptide CP5-46-A. For instance, A156V was reported to have a great impact on the activity of linear α-ketoamide inhibitors as well as the macrocyclic inhibitors (12). It was thus of primary interest to investigate the effect of A156V mutation on CP5-46-4D5E binding. The site-directed NS3 mutation A156V was generated and validated to have WT activity (supplemental Table S4). The optimized inhibitory peptide CP5-46-4D4E, assayed with this NS3-4A variant, has an IC50 value of 1.65 nm. This is in the same range (IC50 = 1.14 nm) as tested with the WT protease (Table 2A). Compared with other NS3-4A inhibitors the peptide shows negligible susceptibility to the A156V resistance mutation. CP5-46A-4D5E was tested in a replicon cell culture model with replicons carrying either the A156V or R155K NS3-4A resistance mutations. The R155K mutation is known to resist all known linear α-ketoamide or macrocyclic inhibitors (12). CP5-46A-4D5E showed negligible susceptibility to these NS3 mutations with less than a 2-fold increase in their IC50 values (Table 2B).
TABLE 2.
Effect of NS3 mutations on inhibitory potency of peptide CP5-46–4D5E
A, NS3-4A A156V mutant. Fold change in the IC50 for the mutant NS3-4A protease compared with that for the wild-type (WT) protease. Measurements were carried out in duplicate, the curves were fitted using GraphPad Prism with a nonlinear regression model, and the S.D. is shown. B, effect of mutants R155K and A156V on peptide CP5-46A-4D5E in the cell culture replicon assay. Fold change in the IC50 for the mutant NS3-4A protease compared with that for the WT protease. Measurements were carried out in triplicate and mean values are shown.
| A. Protease | IC50 | Change |
|---|---|---|
| nm | -Fold | |
| NS3-4A WT | 1.14 ± 0.047 | |
| NS3-4A A156V | 1.65 ± 0.248 | 1.5 |
| B. Peptide | IC50 |
Change |
||||
|---|---|---|---|---|---|---|
| WT | R155K | A156V | WT | R155K | A156V | |
| μm | -Fold | |||||
| CP5-46A-4D5E-Ant | 36.4 | 49.3 | 72.5 | 1.0 | 1.4 | 2.0 |
| Ant-CP5-46A-4D5E | 48.4 | 56.7 | 51.4 | 1.0 | 1.2 | 1.1 |
CP5-46 Intracellular Delivery via Cell-penetrating Peptides (CPPs) Shows in Vivo Inhibition
Given that NS3-4A is an intracellular target, inhibitors need to cross the plasma membrane to reach their site of action. The intracellular delivery of optimized IPs was attempted using fusions with CPPs, short peptides that can traverse the cell membrane of eukaryotic cells (33). For peptide delivery, several CPPs were tested (34): HIV-Tat peptide (35), the Antennapedia peptide (36), the nona-arg peptide (37), and the PTD-5 peptide (supplemental Table S5) (38). The Antennapedia peptide was found to be best suited for intracellular delivery. The IC50 value for inhibition of viral replication was determined with 48 and 36 μm for the N- and C-terminal fusion of the Antennapedia peptide to CP5-46A-4D5E, respectively (Table 2B). This is the first time that inhibition of viral replication was achieved with large peptide inhibitors that are usually unable to cross the cell membrane without a carrier.
DISCUSSION
A combinatorial biology approach was used to generate novel peptide lead sequences for an inhibitor of HCV NS3-4A. These were optimized leading to a 20-fold improvement in binding and inhibitory efficiency with a final Ki of 530 pm. This high affinity achieved for a peptide based ligand is quite remarkable because optimization of “professional ligands” such as antibodies, through directed evolution, may yield only subnanomolar affinities via the creation of well adapted, deep (often hydrophobic) troughs or grooves to bind extended peptides (39). Even under optimal conditions, it is difficult to obtain high affinity peptide ligands due to their higher flexibility and chain entropy compared with more rigid artificial compounds especially with a shallow pocket as target (39, 40).
The key features apparently critical for the subnanomolar affinity of the optimized peptide are as follows: (i) half of the solvent accessible surface of the peptide is buried within the interface with NS3 (supplemental Table S6), anchoring the inhibitor in the shallow NS3 binding pocket (Fig. 1A). Although it is tightly coordinated, the peptide just “caps” the active site and thus is not processed. Notably, the peptide neither geometrically intervenes with catalytic triad geometry nor causes major structural rearrangements within NS3 but may diminish the function of the oxyanion hole; (ii) the N-terminal anti-parallel β-sheet (Fig. 1B) further stabilizes the peptide-protease interactions. The peptide affinity in this region was enhanced 6-fold by a double substitution mimicking substrate binding of 4A4B, although the proposed ionic interaction of substrate Asp-6 with protease Lys-165 (30, 31) did not form in the co-complex as expected (supplemental Fig. S5D). (iii) The C-terminal part of the peptide, which could not be co-crystallized, seems to synergistically increase the inhibitory effect, presumably by additional interactions with the protease surface and/or stabilizing secondary interactions (41). (iv) Finally, an important feature contributing to the high affinity gain is the tyrosine finger motif. It extends the peptide coordination to a pocket remote from the catalytic triad, thus functioning as a C-terminal anchor for the peptide. This is supported by the fact that atomic B-factors of the tyrosine finger match those of surrounding protease residues, whereas those for remaining peptide residues are significantly higher, which is particularly seen for the final and best inhibitory peptide CP5-46-4D4E (Fig. 2C). We note that the low B-factors for the tyrosine finger are not caused by crystal contacts. In addition an intramolecular salt bridge stabilizes the finger and restrains it. There is a close coordination of the tyrosine finger with side chains (Fig. 2D) involving tyrosine ring in the environment of the hydrophobic region of Glu-9, hydrogen bonds (supplemental Table S6) to Ser-7 and Arg-109 involving three coordinated water molecules. In view of this tight coordination and the absence of further tyrosine variants being selected during the library screening, we have not attempted to vary residues within the finger structure, concentrating instead on sequence variation in adjacent sequences that might enhance binding affinity.
Although CP5-46-A is refractory to resistance mutations compared with peptidomimetic inhibitors, it is surprising that they occupy similar NS3 substrate pockets (Fig. 3, B and C). The functional groups are similarly positioned, e.g. analogous atom types bind to the NS3 S1 and S3 subsites (30). Analysis with PDBePISA (42) shows that the N-terminal half of CP5-46-A occupies a surface area (391 Å2) in the same range as those covered by peptidomimetic inhibitors (301–386 Å2) (supplemental Table S7). Differences in properties thus correlate with extended binding of the peptide inhibitor to regions not occupied by small molecular weight inhibitors.
Why Is Inhibitory Capacity Unaffected by Mutations Conferring Resistance to Peptidomimetic PIs?
Usually resistance mutations occur at positions that have no severe influence on substrate binding but weaken inhibitor binding (30). For the new drugs telaprevir and boceprevir, various viral resistance mutations have been observed in vitro and in vivo, thus precluding their use in monotherapy (12, 13). Notably, residues Arg-155 and Ala-156 interact with both inhibitors where they extend most extensively beyond the “substrate envelope” (30). Mutations at these positions confer resistance against nearly all reported drugs in the literature (12, 13). In contrast CP5-46-4D5E shows only negligible susceptibility in vitro to the A156V mutation (1.5-fold increase of IC50 value). Similar results were achieved with A156V or R155K NS3 mutants in the cell culture model. Surface models show that both mutations are not likely to affect binding of CP5-46-4D5E (supplemental Fig. S6). Hence, the optimized peptides, compared with small molecule inhibitors, are far less susceptible for such crucial resistance mutations. Notably, our in silico modeling of CP-46-A on the NS3 surface of HCV genotypes 1 through 6 (supplemental Fig. S7) suggests that the peptide inhibitor may have a higher barrier for viral resistance development. It is not unlikely that resistance mutations can also occur against the peptide inhibitors. However, because of the alternative binding mechanism and because of their larger interactive surface, we predict that single HCV mutations should less frequently result in resistance. Clearly, additional cell culture studies are warranted to define possible viral resistance mechanisms against these peptide inhibitors and to explore viral cross-resistance toward other PIs.
Intracellular Delivery of Peptide Inhibitors Was Demonstrated
Although we have shown inhibition of HCV in a cellular model with fusions to a CPP, the inhibitory concentration is far higher (36 μm) than that required in vitro (23 nm). Several CPP-conjugated drugs are in clinical development with promising results (43). Because fusion to the Antennapedia peptide had no influence on in vitro inhibition (Table 1), the reduced in vivo inhibition presumably results from inefficient cell delivery and/or from low stability of the peptides in cell culture. It is a sobering fact that there are no general rules governing the efficacy of CPPs for cell penetration (44, 45, 46). Nevertheless, this was the first time viral HCV replication was impeded with large peptide inhibitors that are unable to cross the cell membrane on their own.
The Tyrosine Finger Presents an Alternative Antiviral Scaffold
We show that the cosmix-plexing approach facilitates rapid development of novel peptide variants capable of inhibiting mutants resistant to small molecular weight inhibitors because there remains an unmet need for rescue therapy of patients with HCV resistance to currently developed PIs. In conclusion, we note that by searching new structural diversity, unique compounds were selected that highlight novel structures for the development of drug derivatives with high efficacy as HCV NS3-4A protease inhibitors. In particular, we suggest that the tyrosine finger can be used as a promising scaffold for further HCV drug development because it is particularly important in this interaction and binds at an alternative site.
Acknowledgments
We thank Prof. Ralf Bartenschlager (University of Heidelberg) for providing the HCV replicon I389/NS3–3′UTR (AJ242654); Dr. Werner Tegge (HZI) for chemical peptide synthesis and critical discussions, and Juliane Lindner (HZI) for technical assistance testing peptides display valences.

This article contains supplemental Methods, Tables S1–S7, Figs. S1–S8, and additional references.
- HCV
- hepatitis C virus
- IP
- inhibitory peptide
- aa
- amino acid(s)
- PI
- protease inhibitor
- NS
- nonstructural.
REFERENCES
- 1. Shepard C. W., Finelli L., Alter M. J. (2005) Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5, 558–567 [DOI] [PubMed] [Google Scholar]
- 2. de Bruijne J., Weegink C. J., Jansen P. L., Reesink H. W. (2009) New developments in the antiviral treatment of hepatitis C. Vox Sang. 97, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartenschlager R., Frese M., Pietschmann T. (2004) Novel insights into hepatitis C virus replication and persistence. Adv. Virus Res. 63, 71–180 [DOI] [PubMed] [Google Scholar]
- 4. Bartenschlager R., Lohmann V. (2000) Replication of the hepatitis C virus. Baillieres Best Pract. Res. Clin. Gastroenterol. 14, 241–254 [DOI] [PubMed] [Google Scholar]
- 5. Lin C., Thomson J. A., Rice C. M. (1995) A central region in the hepatitis C virus NS4A protein allows formation of an active NS3-NS4A serine proteinase complex in vivo and in vitro. J. Virol. 69, 4373–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim J. L., Morgenstern K. A., Lin C., Fox T., Dwyer M. D., Landro J. A., Chambers S. P., Markland W., Lepre C. A., O'Malley E. T., Harbeson S. L., Rice C. M., Murcko M. A., Caron P. R., Thomson J. A. (1996) Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 87, 343–355 [DOI] [PubMed] [Google Scholar]
- 7. Tomei L., Failla C., Vitale R. L., Bianchi E., De Francesco R. (1996) A central hydrophobic domain of the hepatitis C virus NS4A protein is necessary and sufficient for the activation of the NS3 protease. J. Gen. Virol. 77, 1065–1070 [DOI] [PubMed] [Google Scholar]
- 8. Landro J. A., Raybuck S. A., Luong Y. P., O'Malley E. T., Harbeson S. L., Morgenstern K. A., Rao G., Livingston D. J. (1997) Mechanistic role of an NS4A peptide cofactor with the truncated NS3 protease of hepatitis C virus: elucidation of the NS4A stimulatory effect via kinetic analysis and inhibitor mapping. Biochemistry 36, 9340–9348 [DOI] [PubMed] [Google Scholar]
- 9. Manns M. P., McHutchison J. G., Gordon S. C., Rustgi V. K., Shiffman M., Reindollar R., Goodman Z. D., Koury K., Ling M., Albrecht J. K. (2001) Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomized trial. Lancet 358, 958–965 [DOI] [PubMed] [Google Scholar]
- 10. Fried M. W., Shiffman M. L., Reddy K. R., Smith C., Marinos G., Gonçales F. L., Jr., Häussinger D., Diago M., Carosi G., Dhumeaux D., Craxi A., Lin A., Hoffman J., Yu J. (2002) Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347, 975–982 [DOI] [PubMed] [Google Scholar]
- 11. Jensen D. M. (2011) A new era of hepatitis C therapy begins. N. Engl. J. Med. 364, 1272–1274 [DOI] [PubMed] [Google Scholar]
- 12. Halfon P., Locarnini S. (2011) Hepatitis C virus resistance to protease inhibitors. J. Hepatol. 55, 192–206 [DOI] [PubMed] [Google Scholar]
- 13. Sarrazin C., Zeuzem S. (2010) Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138, 447–462 [DOI] [PubMed] [Google Scholar]
- 14. He Y., King M. S., Kempf D. J., Lu L., Lim H. B., Krishnan P., Kati W., Middleton T., Molla A. (2008) Relative replication capacity and selective advantage profiles of protease inhibitor-resistant hepatitis C virus (HCV) NS3 protease mutants in the HCV genotype 1b replicon system. Antimicrob. Agents Chemother. 52, 1101–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gentile I., Carleo M. A., Borgia F., Castaldo G., Borgia G. (2010) The efficacy and safety of telaprevir–a new protease inhibitor against hepatitis C virus. Expert Opin. Investig. Drugs 19, 151–159 [DOI] [PubMed] [Google Scholar]
- 16. Sato A. K., Viswanathan M., Kent R. B., Wood C. R. (2006) Therapeutic peptides: technological advances driving peptides into development. Curr. Opin. Biotechnol. 17, 638–642 [DOI] [PubMed] [Google Scholar]
- 17. Collins J., Horn N., Wadenbäck J., Szardenings M. (2001) Cosmix-plexing: a novel recombinatorial approach for evolutionary selection from combinatorial libraries. J. Biotechnol. 74, 317–338 [DOI] [PubMed] [Google Scholar]
- 18. Taliani M., Bianchi E., Narjes F., Fossatelli M., Urbani A., Steinkühler C., De Francesco R., Pessi A. (1996) A continuous assay of hepatitis C virus protease based on resonance energy transfer depsipeptide substrates. Anal. Biochem. 240, 60–67 [DOI] [PubMed] [Google Scholar]
- 19. Misialek S., Rajagopalan R., Stevens S. K., Beigelman L., Seiwert S. D., Kossen K. (2009) Optimization of the multiple enzymatic activities of the hepatitis C virus NS3 protein. Anal. Biochem. 394, 138–140 [DOI] [PubMed] [Google Scholar]
- 20. Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 21. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 22. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adams P. D., Gopal K., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Pai R. K., Read R. J., Romo T. D., Sacchettini J. C., Sauter N. K., Storoni L. C., Terwilliger T. C. (2004) Recent developments in the PHENIX software for automated crystallographic structure determination. J. Synchrotron Radiat. 11, 53–55 [DOI] [PubMed] [Google Scholar]
- 25. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 28. Dimasi N., Pasquo A., Martin F., Di Marco S., Steinkühler C., Cortese R., Sollazzo M. (1998) Engineering, characterization and phage display of hepatitis C virus NS3 protease and NS4A cofactor peptide as a single-chain protein. Protein Eng. 11, 1257–1265 [DOI] [PubMed] [Google Scholar]
- 29. Urbani A., Bianchi E., Narjes F., Tramontano A., De Francesco R., Steinkühler C., Pessi A. (1997) Substrate specificity of the hepatitis C virus serine protease NS3. J. Biol. Chem. 272, 9204–9209 [DOI] [PubMed] [Google Scholar]
- 30. Romano K. P., Ali A., Royer W. E., Schiffer C. A. (2010) Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc. Natl. Acad. Sci. U.S.A. 107, 20986–20991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steinkühler C., Biasiol G., Brunetti M., Urbani A., Koch U., Cortese R., Pessi A., De Francesco R. (1998) Product inhibition of the hepatitis C virus NS3 protease. Biochemistry 37, 8899–8905 [DOI] [PubMed] [Google Scholar]
- 32. Gaudieri S., Rauch A., Pfafferott K., Barnes E., Cheng W., McCaughan G., Shackel N., Jeffrey G. P., Mollison L., Baker R., Furrer H., Günthard H. F., Freitas E., Humphreys I., Klenerman P., Mallal S., James I., Roberts S., Nolan D., Lucas M. (2009) Hepatitis C virus drug resistance and immune-driven adaptations: relevance to new antiviral therapy. Hepatology 49, 1069–1082 [DOI] [PubMed] [Google Scholar]
- 33. Schwarze S. R., Hruska K. A., Dowdy S. F. (2000) Protein transduction: unrestricted delivery into all cells? Trends Cell Biol. 10, 290–295 [DOI] [PubMed] [Google Scholar]
- 34. Lohmann V., Hoffmann S., Herian U., Penin F., Bartenschlager R. (2003) Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77, 3007–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Green M., Loewenstein P. M. (1988) Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 55, 1179–1188 [DOI] [PubMed] [Google Scholar]
- 36. Joliot A., Pernelle C., Deagostini-Bazin H., Prochiantz A. (1991) Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 88, 1864–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wender P. A., Mitchell D. J., Pattabiraman K., Pelkey E. T., Steinman L., Rothbard J. B. (2000) The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc. Natl. Acad. Sci. U.S.A. 97, 13003–13008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tilstra J., Rehman K. K., Hennon T., Plevy S. E., Clemens P., Robbins P. D. (2007) Protein transduction: identification, characterization, and optimization. Biochem. Soc. Trans. 35, 811–815 [DOI] [PubMed] [Google Scholar]
- 39. Zahnd C., Spinelli S., Luginbühl B., Amstutz P., Cambillau C., Plückthun A. (2004) Directed in vitro evolution and crystallographic analysis of a peptide-binding single chain antibody fragment (scFv) with low picomolar affinity. J. Biol. Chem. 279, 18870–18877 [DOI] [PubMed] [Google Scholar]
- 40. Njoroge F. G., Chen K. X., Shih N. Y., Piwinski J. J. (2008) Challenges in modern drug discovery: a case study of boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection. Acc Chem. Res. 41, 50–59 [DOI] [PubMed] [Google Scholar]
- 41. Fersht A. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, W. H. Freeman and Company, New York [Google Scholar]
- 42. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 43. Johnson R. M., Harrison S. D., Maclean D. (2011) Therapeutic applications of cell-penetrating peptides. Methods Mol. Biol. 683, 535–551 [DOI] [PubMed] [Google Scholar]
- 44. van den Berg A., Dowdy S. F. (2011) Protein transduction domain delivery of therapeutic macromolecules. Curr. Opin. Biotechnol. 22, 888–893 [DOI] [PubMed] [Google Scholar]
- 45. Duchardt F., Fotin-Mleczek M., Schwarz H., Fischer R., Brock R. (2007) A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 8, 848–866 [DOI] [PubMed] [Google Scholar]
- 46. Patel L. N., Zaro J. L., Shen W. C. (2007) Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm. Res. 24, 1977–1992 [DOI] [PubMed] [Google Scholar]
- 47. Ingallinella P., Altamura S., Bianchi E., Taliani M., Ingenito R., Cortese R., De Francesco R., Steinkühler C., Pessi A. (1998) Potent peptide inhibitors of human hepatitis C virus NS3 protease are obtained by optimizing the cleavage products. Biochemistry 37, 8906–8914 [DOI] [PubMed] [Google Scholar]