

Background: Insulin stimulates glucose uptake in target tissues to promote blood glucose homeostasis, although the mechanism is not fully understood.
Results: siRNA-mediated knockdown of CLASP2 reduces insulin-stimulated glucose transport.
Conclusion: CLASP2 is a new protein involved in insulin-stimulated glucose uptake.
Significance: The identification of CLASP2 as a new player in insulin action provides an improved understanding of insulin-stimulated glucose uptake and GLUT4 trafficking.
Keywords: Glucose Transport, GLUT4, Insulin, Mass Spectrometry (MS), Phosphorylation, CLASP2
Abstract
Insulin stimulates the mobilization of glucose transporter 4 (GLUT4) storage vesicles to the plasma membrane, resulting in an influx of glucose into target tissues such as muscle and fat. We present evidence that CLIP-associating protein 2 (CLASP2), a protein previously unassociated with insulin action, is responsive to insulin stimulation. Using mass spectrometry-based protein identification combined with phosphoantibody immunoprecipitation in L6 myotubes, we detected a 4.8-fold increase of CLASP2 in the anti-phosphoserine immunoprecipitates upon insulin stimulation. Western blotting of CLASP2 immunoprecipitates with the phosphoantibody confirmed the finding that CLASP2 undergoes insulin-stimulated phosphorylation, and a number of novel phosphorylation sites were identified. Confocal imaging of L6 myotubes revealed that CLASP2 colocalizes with GLUT4 at the plasma membrane within areas of insulin-mediated cortical actin remodeling. CLASP2 is responsible for directing the distal end of microtubules to the cell cortex, and it has been shown that GLUT4 travels along microtubule tracks. In support of the concept that CLASP2 plays a role in the trafficking of GLUT4 at the cell periphery, CLASP2 knockdown by siRNA in L6 myotubes interfered with insulin-stimulated GLUT4 localization to the plasma membrane. Furthermore, siRNA mediated knockdown of CLASP2 in 3T3-L1 adipocytes inhibited insulin-stimulated glucose transport. We therefore propose a new model for CLASP2 in insulin action, where CLASP2 directs the delivery of GLUT4 to cell cortex landing zones important for insulin action.
Introduction
Insulin binding to the cell surface insulin receptor activates a highly coordinated series of signal transduction cascades, including the phosphatidylinositol 3-kinase (PI3K) pathway. Activation of PI3K by insulin results in a basolaterally localized increase in phosphatidylinositol 3,4,5-trisphosphate (PIP3)2 at the plasma membrane (PM) (1). The insulin signal then splits to activate the Akt and Rac1 pathways in parallel. Insulin stimulation of Akt releases the intracellular sequestration of GLUT4, allowing for GLUT4 cell surface translocation, PM insertion, and ultimately glucose uptake into the target cell. Insulin signaling to Rac1 within muscle cells induces cell periphery (cortical) actin reorganization and a ruffling of the cell membrane (2). The Akt and Rac1 insulin signaling pathways then converge at this point because Akt-responsive GLUT4 translocates directly to the Rac1-induced, cortical actin-reorganized membrane ruffle (3). Intracellularly, GLUT4 resides within specialized vesicles, referred to as “GLUT4 storage vesicles” or “insulin-responsive vesicles” (IRV) (4). The IRV have been shown to travel to the PM via the microtubule (MT) network, assisted by the molecular motor kinesin (KIF5B) (5), although this mechanism is not fully understood.
The microtubule-associated cytoplasmic linker protein (CLIP)-associating protein 2 (CLASP2), belongs to the plus-end tracking protein (+TIPs) family, which are proteins that localize to the distal ends of MTs and function to regulate MT dynamics. CLASP2 has an established function in cell division (6, 7) and contributes to MT stabilization at leading edges of migrating fibroblasts (8). CLASP2 can also localize to the Golgi, acting as a microtubule organizing center for Golgi-derived vesicle transport (9–11). CLASP2 attaches distal MT ends to the cell cortex (9, 12), serving to direct MT-based transport from the Golgi to the PM. Current indirect evidence supports a role for CLASP2 in insulin action. Glycogen synthase kinase 3β (GSK3β), a kinase known to be deactivated by insulin-stimulated Akt, phosphorylates CLASP2, leading to disruption of the association of CLASP2 with MTs (13, 14). CLASP2 associates with LL5β, a pleckstrin homology (PH) domain-containing protein that interacts with PI3K-generated PIP3 (9, 15). CLASP2 also associates with LL5α (alternatively known as PHLDB1) (9), a protein that undergoes insulin-stimulated translocation to the PM (16). Knockdown of LL5α in 3T3-L1 adipocytes inhibits insulin-stimulated glucose uptake (16).
We present the first direct evidence that CLASP2 responds to insulin stimulation, through insulin-stimulated phosphorylation and CLASP2 colocalization with GLUT4 at the cortical actin-reorganized PM ruffle. We present the finding that siRNA-mediated knockdown of CLASP2 in L6 myotubes inhibits insulin-stimulated GLUT4 translocation to the PM. Furthermore, siRNA-mediated knockdown of CLASP2 in 3T3-L1 adipocytes inhibits insulin-stimulated glucose transport. The fact that CLASP2, a protein known to attach MTs to the cell cortex, localizes directly to the insulin-induced membrane ruffle sheds new light onto the importance of the MT network for the trafficking of GLUT4-containing vesicles at the PM.
EXPERIMENTAL PROCEDURES
Materials
The following suppliers were used: sequencing-grade trypsin, Sigma; C18 ZipTip, Millipore; phospho-MAPK/CDK substrate (PXS*P, S*PXR/K, or PXS*PXR/K) antibody, Cell Signaling Technologies; CLASP2 antibody, Novus Biologicals; GLUT4 antibody, Abcam; Alexa Fluor 568 phalloidin, Alexa Fluor 488 donkey anti-rabbit, and Alexa Fluor 568 donkey anti-mouse, Invitrogen.
Cell Culture, Transfection, Immunoprecipitation, and Western Blot Analysis
L6 rat myoblasts were maintained in α-MEM (Invitrogen) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were subcultured by trypsinization (0.25% trypsin with ED TA) at approximately 70% confluence. Myotube differentiation was induced by placing 60–70% confluent myoblasts in α-MEM containing 2% fetal bovine serum and 1% penicillin/streptomycin for 7–8 days. Medium for all cell cultures was replaced every 24–48 h, and cultures were maintained at 37 °C and 5% CO2. The 3T3-L1 fibroblasts were grown in DMEM with 10% fetal bovine serum and differentiated into adipocytes as described (17). For cell treatment experiments, cells were starved for 4 h and then either left untreated or stimulated with the indicated time and concentration of insulin at 37 °C. Cells were lysed with lysis buffer containing 50 mm HEPES, pH 7.6, 150 mm NaCl, 1% Triton X-100, 1 mm NaF, 20 mm sodium pyrophosphate, 20 mm β-glycerol phosphate, 1 mm sodium orthovanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mm phenylmethylsulfonyl fluoride. Cell lysates were incubated on ice for 20 min followed by centrifugation (10,000 × g, 4 °C, 20 min), and the clarified supernatants were used for immunoprecipitation. For immunoprecipitation, cell lysates were incubated with specific antibodies conjugated to protein A or protein G-agarose beads overnight at 4 °C with gentle rotation. Immunoprecipitates were washed extensively with ice-cold PBS, and the proteins bound to beads were eluted by heating at 95 °C for 4 min in SDS sample loading buffer. Eluted proteins were separated by 10% SDS-PAGE and were either stained with Coomassie Blue or transferred to a nitrocellulose membrane for subsequent Western blotting.
In-gel Digestion
For the proteomics experiments, each lane of the SDS-PAGE gel was cut into 15 slices, placed in a 0.6-ml polypropylene tube, destained twice with 300 μl of 50% acetonitrile (ACN) in 40 mm NH4HCO3 and dehydrated with 100% ACN for 15 min. After removal of ACN by aspiration, the gel pieces were dried in a vacuum centrifuge at 60 °C for 30 min. Trypsin (250 ng; Sigma) in 20 μl of 40 mm NH4HCO3 was added, and the samples were maintained at 4 °C for 15 min prior to the addition of 50 μl of 40 mm NH4HCO3 containing three 10 fmol/μl peptides to serve as internal standards (bradykinin fragment 2–9, B1901, β-sheet breaker peptide, S7563, and anaphylatoxin C3a fragment, A8651; Sigma). The digestion was allowed to proceed at 37 °C overnight and was terminated by addition of 10 μl of 5% formic acid (FA). After further incubation at 37 °C for 30 min and centrifugation for 1 min, each supernatant was transferred to a clean polypropylene tube. The extraction procedure was repeated using 40 μl of 0.5% FA, and the two extracts were combined. The resulting peptide mixtures were purified by solid phase extraction (C18 ZipTip) after sample loading in 0.05% heptafluorobutyric acid:5% FA (v/v) and elution with 4 μl of 50% ACN:1% FA (v/v) and 4 μl of 80% ACN:1% FA (v/v), respectively. The eluates were combined and dried by vacuum centrifugation and 10 μl of 0.1% TFA/2% ACN (v/v) was added.
Mass Spectrometry
HPLC-ESI-MS/MSn was performed on a Thermo Finnigan (San Jose, CA) LTQ-FTICR fitted with a PicoViewTM nanospray source (New Objective, Woburn, MA). On-line HPLC was performed using a Michrom BioResources Paradigm MS4 micro two-dimensional HPLC (Alburn, CA) with a PicoFritTM column (New Objective, Woburn, MA, 75-μm inner diameter, packed with ProteoPepTM II C18 material, 300 Å); mobile phase, linear gradient of 2–27% ACN in 0.1% FA in 65 min, a hold of 5 min at 27% ACN, followed by a step to 50% ACN, hold 5 min, and then a step to 80%; flow rate, 400 nl/min.
A “top 10” data-dependent MS/MS analysis was performed (acquisition of a full scan spectrum followed by collision-induced dissociation mass spectra of the 10 most abundant ions in the survey scan). The fragment mass spectra were then searched against the human SwissProt_v 52.2 database (16,135 entries) using Mascot (Matrix Science, London, UK; version 2.1). The search variables that were used were: 10-ppm mass tolerance for precursor ion masses and 0.5 Da for product ion masses; digestion with trypsin; a maximum of two missed tryptic cleavages; variable modifications of oxidation of methionine and phosphorylation of serine, threonine, and tyrosine. Cross-correlation of Mascot search results with X! Tandem was accomplished with Scaffold (version Scaffold-01_06_19; Proteome Software, Portland, OR). Probability assessment of peptide assignments and protein identifications were made through the use of Scaffold. Only peptides with ≥95% probability were considered. Assignments of the phosphopeptides were confirmed by manual comparison of the MS/MS with the predicted fragmentation generated in silico by the MS-Product component of Protein Prospector, in addition to Ascore assignment (18) using Scaffold PTM.
Immunofluorescence and Confocal Microscopy
Myotubes were fixed with 2% paraformaldehyde, permeabilized with 2% Tween 20, and either left untreated or incubated with antibodies to CLASP2 and GLUT4 overnight with gentle rocking at 4 °C. After rinsing, the samples were incubated with either Alexa Fluor 568 phalloidin for actin and/or secondary antibodies Alexa Fluor 488 or Alexa Fluor 568 for 2 h at room temperature and then subjected to a final series of rinsing. Imaging was conducted using a Leica SP5 Multi-photon Scanning Laser Microscope housed in the W. M. Keck Bioimaging Laboratory at Arizona State University. Multiple lasers (argon, 488 nm; krypton, 561 nm; and He/Ne, 633 nm) allowed for sequential imaging including collection of differential interference contrast images of the sample. Settings for the gain and offset of the detectors were kept constant for all experiments. Using a 63× oil objective, images were scanned at 0.7-μm intervals in the z axis. The postacquisition image processing was performed using the Leica SP5 software (Leica Microsystems Inc., Bannockburn, IL) and Adobe Photoshop software (Mountain View, CA). For the quantification of the percentage of insulin-induced PM ruffles that contain GLUT4, cells were stimulated with insulin, and eight images of either negative control siRNA or CLASP2 siRNA-treated cells were obtained. Percentages of the number of ridges that contain GLUT relative to the total number of ridges within each image were calculated, and the two groups were statistically analyzed. A ridge was defined as lacking GLUT4 if there was a complete absence of GLUT4 immunofluorescence throughout the entire length of the ridge. For the -fold change of GLUT4 fluorescence per ridge calculations, cells were stimulated with insulin, and eight images of either negative control siRNA or CLASP2 siRNA-treated cells were obtained and “screen” overlaid within Adobe Photoshop, followed by analysis with NIH ImageJ software. Within the negative control image, the immediate area surrounding ridges that contain both CLASP2 and GLUT4 were highlighted using the Freehand Selections tool, followed by Measuring RGB under the PlugIns/Analyze tab. Four small background boxes near the ridge, but not overlapping, were also measured for RGB and will be highlighted using the Rectangle tool. Upon measuring the RGB, values for the mean density per pixel (Mean) and integrated density (IntDen) were calculated and displayed. The average of the background value was then subtracted from the value for the area surrounding the ridge, resulting in Corrected Integrated Density. GLUT4 immunofluorescence was then normalized by the average value of the negative control sample and then expressed as a -fold change over the negative control ± S.E.
Fluorescence Resonance Energy Transfer (FRET)
L6 myotubes were labeled with a combination of GLUT4 and CLASP2 antibodies followed by secondary antibodies Alexa Fluor 488 or 568. The myotubes were imaged on the Leica SP5 confocal microscope housed in the W. M. Keck Bioimaging facility at Arizona State University. Multiple lasers allowed for simultaneous imaging of Alexa Fluor 488 (argon (488 nm)) and Alexa Fluor 568 (krypton (568 nm)) fluorophore-labeled myotubes. The Leica SP5 FRET Analysis software was used to determine the energy efficiency by acceptor photo-bleaching (19). FRET acceptor photobleaching was conducted by double labeling the myotubes with antibodies to CLASP2 and GLUT4. The secondary antibody for GLUT4 was Alexa Fluor 568 and served as the acceptor fluorophore. The second antibody for CLASP2 was Alexa Fluor488 and served as the donor fluorophore. Myotubes were photobleached in the area of the membrane ruffle with the Kr (568 nm) laser. Photobleaching prevents energy transfer to the acceptor fluorophore which results in an increase in donor fluorophore emission.
siRNA
Rat CLASP2 smart pool siRNA was purchased from Dharmacon Research, and the targeting sequences were: AGA CAT ACG GAC TAA GAA A; GCA CGA TCC TGA CGA GAG A; GGA AAT GCC AAG ACC GAT T; and AGA AAG CAG TGT CCG GAA A. Myotubes were transfected with 15 nm siRNA on day 5 of differentiation using Lipofectamine 2000, according to the manufacturer's protocol. 72 h after transfection the cells were prepared for experimentation. For the 3T3-L1 adipocytes, mouse CLASP2 stealth RNAi duplexes were purchased from Invitrogen, and the targeting sequences were: GGA UGU GCA GCA AUC AGA UUU AUU U; CCC UAA GGU UGG AGG UCC UUC UAA A; and CCC AUG UAC CUA GAC UUA UUC CUU U. Electroporation of either the negative control siRNA duplex or the pool of 3 CLASP2 siRNA duplexes into 3T3-L1 adipocyte was performed as described (20).
3–0-[Methyl-d-1-3H]Glucose Uptake
Differentiated 3T3-L1 adipocytes seeded in 6-well plates were preincubated for 4 h in Krebs-Henseleit buffer (1.2 mm CaCl2, 4.4 mm KCl, 1.2 mm MgSO4, 1.5 mm KH2PO4, 116 mm NaCl, 29 mm NaHCO3) containing 4% bovine serum albumin and 0.5 mm cold d-glucose. In the final 15 min of incubation, appropriate wells were treated with 0.2–20 nm insulin. The buffer was then replaced with fresh prewarmed Krebs-Henseleit buffer containing 0.4–1.0 μCi/ml 3–0-[methyl-d-1-3H]glucose and 0.4–1.0 μCi/ml l-[1-14C]glucose, and were incubated for 10 s at room temperature. Glucose uptake was stopped by the addition of 0.2 mm ice-cold phloretin in PBS. The cells were then washed with ice-cold phloretin in PBS and lysed with 1% SDS, and lysates were cleared by centrifugation. The radioactivity associated with the cells was then determined by liquid scintillation of aliquots of each sample (800 μl). The uptake measurement was performed in duplicate. The protein concentration of each sample was determined by the Lowry method, and results were normalized by micrograms of total protein.
RESULTS
CLASP2 Undergoes Insulin-stimulated Phosphorylation in Rat L6 Myotubes
The rationale at the beginning of this project was that the understanding of the signaling between the insulin receptor and GLUT4 is incomplete. To address this, we performed mass spectrometry-driven proteomic profiling of phosphoproteins from either serum-starved or insulin-treated rat L6 myotubes isolated with an antibody that targets phosphoserine in a PXS*P, S*PXR/K, or PXS*PXR/K motif (Fig. 1A). Of the 406 proteins identified, CLASP2 underwent the strongest insulin-stimulated increase in abundance among proteins in the phosphoserine antibody immunoprecipitates (4.8-fold, ** p ≤ 0.01) (Table 1 and Fig. 1B). Subsequent immunoprecipitation of CLASP2 from either serum-starved or insulin-treated myotubes and Western blotting of the immunoprecipitates with the phosphoserine antibody confirmed the finding that CLASP2 undergoes insulin-stimulated phosphorylation (Fig. 1C). Western blotting of CLASP2 within L6 myotubes revealed a doublet, which may represent different isoforms of the protein (8). Mass spectrometry analysis of CLASP2 immunoprecipitates from L6 myotube lysates resulted in 68% CLASP2 sequence coverage (data not shown) and the detection of 15 CLASP2 phosphorylation sites (Table 1). Calculation of Ascore values (18) to measure the probability of correct phospho-S/T/Y assignment validated Ser-319, Ser-322, Ser-1005, and Ser-1181 as novel, previously unidentified CLASP2 phosphorylation sites (Table 2). Phosphorylation at Ser-467, Ser-469, Thr-475, Thr-477, Ser-570, and Ser-947 was also detected, although due to low Ascore values, these sites cannot be assigned unequivocally.
FIGURE 1.

CLASP2 undergoes insulin-stimulated phosphorylation. A, L6 myotubes were serum-starved and either left untreated or treated with 10 nm insulin for 15 min. The myotubes were lysed, and the whole cell lysates were immunoprecipitated with phosphoserine antibody. The immunoprecipitates were resolved by 10% SDS-PAGE and transferred to nitrocellulose membranes. The membranes containing the immunoprecipitated proteins were subjected to Western blotting for phosphoserine in a PXS*P, S*PXR/K, or PXS*PXR/K motif. B, L6 myotubes were serum-starved and either left untreated or treated with 10 nm insulin for 15 min. The myotubes were lysed, and the whole cell lysates were immunoprecipitated with phosphoserine antibody. The immunoprecipitates were resolved by 10% SDS-PAGE followed by Coomassie staining. Each lane within the SDS-PAGE gel was divided into 15 slices, ranging the span of 250 to 15 kDa, and the proteins were prepared and analyzed as described under “Experimental Procedures.” The data were then expressed as a -fold change over basal ± S.E. (error bars; n = 3; **, p < 0.01; Student's t test). C, L6 myotubes were serum-starved and either left untreated or treated with 10 nm insulin for 15 min. The myotubes were lysed, and the whole cell lysates (WCL) were immunoprecipitated (IP) with CLASP2 antibody. The immunoprecipitates were resolved by 10% SDS-PAGE and transferred to nitrocellulose membranes. The membranes containing the immunoprecipitated CLASP2 were subjected to Western blotting for phosphoserine in a PXS*P, S*PXR/K, or PXS*PXR/K motif (upper panel) or with anti-CLASP2 antibodies (lower panel). D, L6 myotubes were serum-starved and either left untreated, treated with insulin alone, or pretreated with 50 nm wortmannin (labeled W) or 50 μm PD98059 (labeled PD) for 1 h and then either left untreated or treated with 10−7 m insulin for 15 min. The myotubes were lysed, and the whole cell lysates were immunoprecipitated with CLASP2 antibody. The immunoprecipitates were resolved by 10% SDS-PAGE and transferred to nitrocellulose membranes. The membranes containing the immunoprecipitated CLASP2 were subjected to Western blotting for phosphoserine in a PXS*P, S*PXR/K, or PXS*PXR/K motif (top panel) or with anti-CLASP2 antibodies (second panel from the top).Whole cell lysates were probed for the phosphorylation of either AKT (fifth panel from the top) or MAPK (third panel from the top), or the expression of AKT (bottom panel) or MAPK (fourth panel from the top).
TABLE 1.
Identification of CLASP2 as an insulin-stimulated phosphoprotein
| Phosphoproteina | Expt. 1 |
Expt. 2 |
Expt. 3 |
|||
|---|---|---|---|---|---|---|
| Basal | Insulin | Basal | Insulin | Basal | Insulin | |
| CLASP2 SC | 8 | 56 | 6 | 46 | 14 | 35 |
| No. of IDs | 4682 | 2842 | 5276 | 3463 | 1801 | 1965 |
| NormSC | 1.71E-03 | 1.97E-02 | 1.14E-03 | 1.33E-02 | 7.77E-03 | 1.78E-02 |
a SC, spectrum counts; No. of IDs, number of assigned spectra; NormSC, normalized spectrum counts.
TABLE 2.
CLASP2 phosphorylation sites
| Start | Stop | Peptide sequencea | Phosphorylation site | Ascorec |
|---|---|---|---|---|
| 316 | 328 | SsGsVAsLPQSDR | Ser-317b | 14.04 |
| Ser-319b | 26.31 | |||
| Ser-322b | 34.3 | |||
| 316 | 344 | SSGSVASLPQSDRSSSSsQESLNRPFSSK | Ser-333 | 18.53 |
| 329 | 344 | SSSSsQESLNRPFSSK | Ser-333 | 10.11 |
| 374 | 387 | sRsDIDVNAAAGAK | Ser-374 | 9.38 |
| Ser-376 | 29.27 | |||
| 467 | 488 | sGsPGRVLtTtALSTVSSGAQR | Ser-467b | 0 |
| Ser-469b | 5.42 | |||
| Thr-475b | 0.21 | |||
| Thr-477b | 0.32 | |||
| 567 | 575 | LSSsVSAmR | Ser-570b | 0 |
| 935 | 957 | GVTEAIQNFSFRsQEDmSEPLKR | Ser-947b | 5.3 |
| 994 | 1007 | ASLLHSVPLHSsPR | Ser-1005b | 27.96 |
| 1008 | 1024 | SRDYNPYNYSDSIsPFNK | Ser-1021 | 25.7 |
| 1174 | 1192 | VLCPIIQtADYPINLAAIK | Thr-1181b | 28.73 |
a Lower case s/t represents phospho-site. Lower case m represents methionine oxidation site.
b Novel, previously unreported phosphorylation site.
c Ascore ≥20 signifies 99% certainty, 15–19 equals >90%, 3–15 is near 80% yet nonsufficient, and <3 lacks adequate information to make an assignment.
To identify the signal transduction pathway(s) responsible for insulin-stimulated phosphorylation of CLASP2 within the PXS*P, S*PXR/K, or PXS*PXR/K motifs, L6 myotubes were serum-starved and either left untreated or pretreated with 50 nm wortmannin or 50 μm PD98059 for 1 h to inhibit the insulin-stimulated activation of the PI3K and MAPK pathways, respectively. The cells were then either left untreated for basal analysis or treated with insulin. Whole cell lysates were probed for the phosphorylation of either AKT or MAPK to ensure efficient insulin stimulation and either wortmannin or PD98059 inhibition of the PI3K and MAPK pathways, respectively (Fig. 1D). The insulin-stimulated phosphorylation of CLASP2 within the proline-directed motifs was suppressed upon inhibition of the MAPK pathway and was not affected by inhibition of the PI3K pathway.
CLASP2 Colocalizes with GLUT4 at the Insulin-stimulated Membrane Ruffle in Rat L6 Myotubes
Because CLASP2 localizes to MTs, particularly at the cell cortex (9), and GLUT4 transport has been linked to traveling the MT network (21–23), we reasoned that CLASP2 cellular localization in L6 myotubes would provide insight regarding the possibility that CLASP2 is involved in GLUT4 trafficking. Cortical actin remodeling is known to serve as a focal point for insulin signaling and insulin-stimulated GLUT4 localization (3), so we chose to use confocal microscopy to test whether CLASP2 colocalizes with areas of insulin-stimulated cortical actin remodeling in L6 myotubes. Confocal imaging using rhodamine phalloidin allowed ready observation of cortical actin remodeling, similar to previous reports (3, 24) (Fig. 2, top right panel, and supplemental Fig. 1). We then took advantage of the newly advanced differential image contrasting mode of the confocal microscope to focus only on the cell surface z-plane corresponding to dorsal extension of the PM (membrane ruffle) (24) proximal to the cortical actin remodeling. We were able to capture extremely sharp images of insulin-induced membrane ruffling (Fig. 2, top left panel), whereas neither cortical actin remodeling nor membrane ruffling was observed in the serum-starved myotubes. Once an area of both cortical actin remodeling and cell surface membrane ruffling was established, we then tested CLASP2 colocalization and discovered CLASP2 along the membrane ridge, corresponding to the zone of insulin-induced actin rearrangement (Fig. 2, bottom two panels). We were not able to obtain FRET acceptor photobleaching between actin and CLASP2 (data not shown), suggesting that although these two proteins both localize to the same membrane ruffle, they are >100 Å apart and are most likely not directly interacting at the z-plane tested. We then tested for colocalization of CLASP2 with GLUT4 along the insulin-induced PM ruffle. Our strategy was to pinpoint a membrane ridge on the cell surface of an insulin-stimulated myotube using the confocal differential interference contrast mode (Fig. 3, top left panel), followed by confirmation of GLUT4 localization within the ridge (Fig. 3, top right panel). Using this approach, we discovered that CLASP2 colocalizes with GLUT4 to the cortical actin reorganized membrane ruffle (Fig. 3, bottom two panels, and supplemental Figs. 2 and 3). This colocalization was confirmed by FRET acceptor photobleaching which demonstrated up to a 19% FRET efficiency between CLASP2 and GLUT4 at the crest of the membrane ruffle (supplemental Fig. 4). These findings are strengthened by the fact that the data are all from endogenous proteins, canceling out the possibility of artifactual results due to protein overexpression issues.
FIGURE 2.

CLASP2 localizes to insulin-stimulated membrane ruffles. L6 myotubes were serum-starved and treated with 10 nm insulin for 15 min. Confocal imaging was performed as described under “Experimental Procedures.” DIC, differential interference contrast.
FIGURE 3.

CLASP2 colocalizes with GLUT4 to insulin-stimulated membrane ruffles. L6 myotubes were serum-starved and treated with 10 nm insulin for 15 min. Confocal imaging was performed as described under “Experimental Procedures.” DIC, differential interference contrast.
CLASP2 Knockdown Reduces Insulin-stimulated GLUT4 Localization to the Cortical Actin-induced Plasma Membrane Ruffle in Rat L6 Myotubes
The fact that CLASP2, a protein known to attach MTs to the cell cortex (8), localizes directly to the insulin-induced membrane ruffle supports a role for the MT network in the trafficking of GLUT4-containing vesicles at the membrane. Therefore, we chose to perform siRNA-mediated knockdown of CLASP2 protein in L6 myotubes to test the hypothesis that CLASP2 function is crucial for proper insulin-stimulated GLUT4 localization to the cortical actin induced PM ruffle. To suppress CLASP2 expression in L6 myotubes, the cells were treated with either 15 nm CLASP2-targeted siRNA or nontargeting siRNA for 72 h. We first confirmed siRNA-mediated knockdown of CLASP2 protein by Western blotting (Fig. 4A). Analysis of CLASP2 mRNA by quantitative RT-PCR revealed a 50% reduction of CLASP2 at the mRNA level (data not shown). We again focused our attention on membrane ruffles and searched for ridges that lacked CLASP2 immunofluorescence, as a result of siRNA-mediated knockdown of CLASP2 protein. Once a CLASP2-lacking membrane ruffle was found, we then discovered that ruffles that possessed reduced CLASP2 protein levels also had a decrease in the amount of insulin-stimulated GLUT4 (Fig. 4B). We quantified this phenomenon in two different ways: first, we compared the percentage of ridges observed that possessed GLUT4 in control versus CLASP2 siRNA myotubes and found that there was a significant reduction in the number of ridges that possessed GLUT4 upon CLASP2 siRNA treatment (Fig. 4C); and second, we quantified GLUT4 fluorescence intensity within the ridges that did contain GLUT4 and found that CLASP2 siRNA also significantly reduced the amount of GLUT4 per ridge (Fig. 4D).
FIGURE 4.

siRNA knockdown of CLASP2 in L6 myotubes reduces insulin-stimulated localization of GLUT4 to plasma membrane ruffles. A, siRNA knockdown of CLASP2 in L6 myotubes was performed as described under “Experimental Procedures.” B, L6 myotubes were serum-starved and either left untreated or treated with 10 nm insulin for 15 min. Confocal imaging was performed as described under “Experimental Procedures.” C, effect of siRNA knockdown of CLASP2 on the percentage of membrane ridges with GLUT4 after insulin stimulation, performed as described under “Experimental Procedures,” is quantified. D, effect of siRNA knockdown of CLASP2 on the density of GLUT4 per membrane ridge after insulin stimulation, performed as described under “Experimental Procedures,” is quantified. Error bars, S.E. *, p < 0.05.
CLASP2 Knockdown Reduces Insulin-stimulated Glucose Transport in Mouse 3T3-L1 Adipocytes
To prove that CLASP2 is vital for proper insulin action, we chose to study 3T3-L1 adipocytes because this cell model possesses an excellent capacity for insulin-stimulated glucose transport. We successfully performed siRNA-mediated knockdown of CLASP2 in 3T3-L1 adipocytes utilizing a mixture of three siRNA targeting sequences targeted toward mouse CLASP2 using a method described previously (Fig. 5A). Whereas L6 myotubes reveal a doublet when blotting for CLASP2 protein, we noticed a lack of this doublet in the 3T3-L1 adipocytes, suggesting the loss of multiple CLASP2 isoforms within adipocytes compared with myotubes, although this has yet to be proven directly. We subsequently performed 3–0-methyl-d-glucose transport assays on either negative control or CLASP2 knockdown adipocytes and found that reducing CLASP2 protein expression decreased insulin-stimulated glucose transport at all insulin doses (Fig. 5B), including the physiologically relevant dose of 0.2 nm (Fig. 5C, n = 5, * p ≤ 0.05, t test). Although significant at the 0.2 nm insulin dose, the siRNA-mediated reduction of CLASP2 protein did not completely inhibit insulin-stimulated glucose transport, which could be due to a compensatory role for the other CLASP isoform, CLASP1, or the fact that knockdown of CLASP2 was incomplete, and many cells were likely not affected by siRNA treatment. In summary, the data as a whole support an important role for CLASP2 in insulin-stimulated glucose transport.
FIGURE 5.
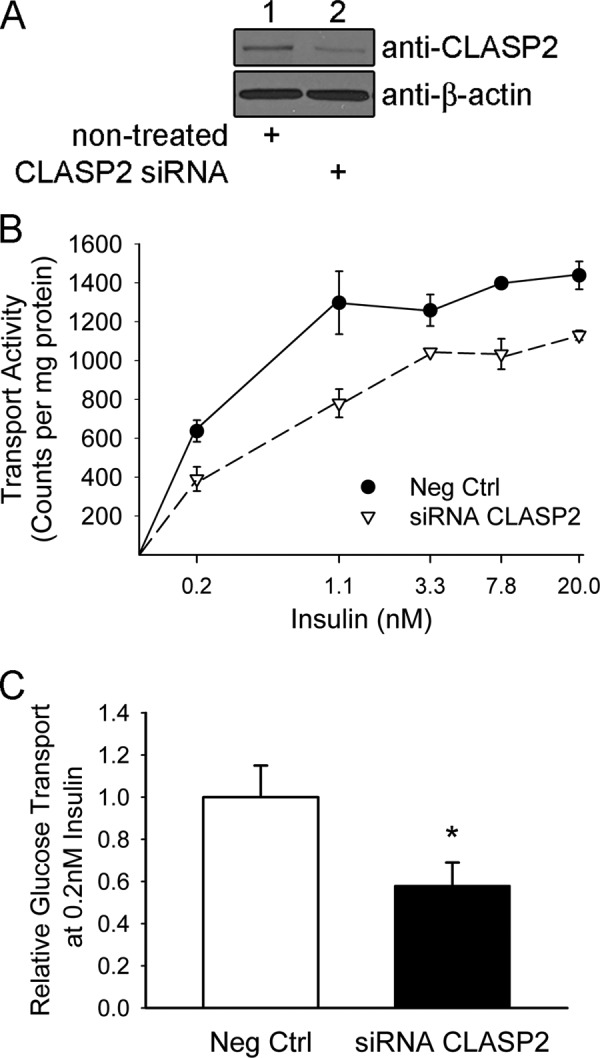
siRNA knockdown of CLASP2 in 3T3-L1 adipocytes reduces insulin-stimulated glucose transport. A, siRNA knockdown of CLASP2 in 3T3-L1 adipocytes was performed as described under “Experimental Procedures.” B, insulin-stimulated glucose transport in 3T3-L1 adipocytes was performed as described under “Experimental Procedures.” C, insulin-stimulated glucose transport in 3T3-L1 adipocytes was performed as described under “Experimental Procedures” at the physiologically relevant dose of 0.2 nm (n = 5; *, p ≤ 0.05, t test).
DISCUSSION
We present findings that the microtubule-associated protein CLASP2 plays a central part in GLUT4 trafficking. We show the first direct and known evidence that CLASP2 responds to insulin stimulation, through insulin-stimulated phosphorylation. The phosphorylation was detected with a phosphoantibody that targets phosphoserine in a PXS*P, S*PXR/K, or PXS*PXR/K motif, and this insulin-stimulated phosphorylation was blocked by inhibition of the MAPK pathway. This finding does not imply that all phosphorylation of CLASP2 is controlled by the MAPK pathway, raising the possibility that CLASP2 may undergo PI3K-controlled phosphorylation in response to insulin as well. In support of this hypothesis, it has been shown that GSK3β, a kinase known to be deactivated by insulin-stimulated Akt (25, 26), phosphorylates CLASP2, leading to disruption of the association of CLASP2 with MTs (13). In this case, insulin would suppress GSK3β phosphorylation sites within CLASP2. In fact, many of the phosphorylation sites we detected were not in classical MAPK phosphorylation motifs, and current studies are under way to explore whether insulin-induced phosphorylation of CLASP2 regulates its function with respect to GLUT4 translocation.
We also present the finding that insulin stimulates CLASP2 colocalization with GLUT4 at the cortical actin reorganized membrane ruffle. The fact that CLASP2, a protein known to attach MTs to the cell cortex, localizes directly to the insulin-induced membrane ruffle sheds new light onto the possible involvement MT network for the delivery of GLUT4-containing vesicles to the membrane. Based on our findings, we have created a working model for MT-based delivery of GLUT4 to the PM that we are currently testing, where the GLUT4 vesicle travels along the MT track to the CLASP2-directed cortical PM landing zone (Fig. 6). It is important to understand where exactly CLASP2 fits within the GLUT4 tethering/docking steps of GLUT4 vesicle transport. For instance, it is possible that CLASP2 may target the IRV to the exocyst, a stable complex of proteins located at the PM whose function is insulin-stimulated cortical immobilization of the IRV through tethering (27). Conversely, CLASP2 may be involved in the docking phase of GLUT4 trafficking, functioning together with the SNARE proteins. As an alternative to playing a direct role in the insulin-stimulated translocation of GLUT4 to the PM, MTs are also involved in endocytosis as well as trafficking toward the membrane. As a result, it is important to not discount the possibility that CLASP2 acts to inhibit the process by which GLUT4 is reinternalized, thus maintaining higher GLUT4 at the cell surface during insulin stimulation.
FIGURE 6.

Hypothetical model for CLASP2 in insulin action. The identification of the involvement of CLASP2 in insulin-stimulated glucose uptake has led to the following model. Upon insulin activation of the insulin receptor, IRS-1-mediated activation of PI-3K results in a localized increase in PIP3 at the plasma membrane. PIP3 then recruits LL5α (also known as PHLDB1), a known binding partner for CLASP2. This is followed by CLASP2 binding to LL5α, thereby directing the GLUT4 vesicle to the insulin-responsive landing zone on the cortical PM. This in turn leads to the docking and fusion of the GLUT4 vesicle with the PM, mediated by the SNARE protein complex, composed of VAMP2 within the IRV and both syntaxin 4 and SNAP23 on the PM.
Actin remodeling is central to insulin signaling activity, as WASP, Arp3, PI3K, PIP3, Akt, GLUT4, VAMP2, and additional proteins all colocalize with reorganized actin within the membrane ruffle (3, 24, 28). In addition to the importance of actin, research supports a role for the microtubule network in insulin-stimulated GLUT4 translocation. Many studies have documented that pretreatment of adipocytes with the MT-disrupting agent nocodazole inhibits insulin-stimulated glucose uptake. Total internal reflection fluorescence microscopy (29) near the cell surface of rat primary adipose cells showed GLUT4 to be highly mobile within 200 nm of the PM, moving along paths similar to MT networks, and nocodazole treatment reduces GLUT4 abundance within the total internal reflection fluorescence zone (30–32). Insulin treatment arrests GLUT4 movement near the PM, although how insulin delivers GLUT4 for contact with the PM is not known. One proposal is MT-driven delivery of the IRV to the PM at the cortical MT attachment points, using the molecular motor kinesin (KIF5B) (5, 33). Another possibility is that insulin-stimulated association of GLUT4 with α-actinin 4 (ACTN4) transfers the IRV from the MT network over to cortical actin, where the IRV then travels to the PM along actin using type I or V myosin (Myo1c and MyoVa and Vb, respectively) or nonmuscle myosin II (NM-MyoII) motors. It is important to note that there is no evidence for the switching of the IRV from MTs to cortical actin. Because the transport of GLUT4 to the PM is so important for insulin-stimulated glucose uptake, it is imperative to define the role of actin and MT cytoskeletal elements in the insulin-controlled delivery of GLUT4 to the muscle cell surface and the proteins that regulate this critical process.
This work was supported, in whole or in part, by National Institutes of Health Grant R01DK047936 (to L. J. M.).

This article contains supplemental Figs. 1–4.
- PIP3
- phosphatidylinositol 3,4,5-trisphosphate
- ACN
- acetonitrile
- CLASP2
- cytoplasmic linker protein (CLIP)-associating protein 2
- FA
- formic acid
- GLUT4
- glucose transporter 4
- GSK3β
- glycogen synthase kinase 3β
- IRV
- insulin-responsive vesicles
- MS/MS
- tandem mass spectrometry
- MT
- microtubule
- PM
- plasma membrane.
REFERENCES
- 1. Stöckli J., Fazakerley D. J., James D. E. (2011) GLUT4 exocytosis. J. Cell Sci. 124, 4147–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Raftopoulou M., Hall A. (2004) Cell migration: Rho GTPases lead the way. Dev. Biol. 265, 23–32 [DOI] [PubMed] [Google Scholar]
- 3. Khayat Z. A., Tong P., Yaworsky K., Bloch R. J., Klip A. (2000) Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J. Cell Sci. 113, 279–290 [DOI] [PubMed] [Google Scholar]
- 4. Rowland A. F., Fazakerley D. J., James D. E. (2011) Mapping insulin/GLUT4 circuitry. Traffic 12, 672–681 [DOI] [PubMed] [Google Scholar]
- 5. Semiz S., Park J. G., Nicoloro S. M., Furcinitti P., Zhang C., Chawla A., Leszyk J., Czech M. P. (2003) Conventional kinesin KIF5B mediates insulin-stimulated GLUT4 movements on microtubules. EMBO J. 22, 2387–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galjart N. (2005) CLIPs and CLASPs and cellular dynamics. Nat. Rev. Mol. Cell Biol. 6, 487–498 [DOI] [PubMed] [Google Scholar]
- 7. Maiato H., Fairley E. A., Rieder C. L., Swedlow J. R., Sunkel C. E., Earnshaw W. C. (2003) Human CLASP1 is an outer kinetochore component that regulates spindle microtubule dynamics. Cell 113, 891–904 [DOI] [PubMed] [Google Scholar]
- 8. Akhmanova A., Hoogenraad C. C., Drabek K., Stepanova T., Dortland B., Verkerk T., Vermeulen W., Burgering B. M., De Zeeuw C. I., Grosveld F., Galjart N. (2001) CLASPs are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell 104, 923–935 [DOI] [PubMed] [Google Scholar]
- 9. Lansbergen G., Grigoriev I., Mimori-Kiyosue Y., Ohtsuka T., Higa S., Kitajima I., Demmers J., Galjart N., Houtsmuller A. B., Grosveld F., Akhmanova A. (2006) CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5β. Dev. Cell 11, 21–32 [DOI] [PubMed] [Google Scholar]
- 10. Efimov A., Kharitonov A., Efimova N., Loncarek J., Miller P. M., Andreyeva N., Gleeson P., Galjart N., Maia A. R., McLeod I. X., Yates J. R., 3rd, Maiato H., Khodjakov A., Akhmanova A., Kaverina I. (2007) Asymmetric CLASP-dependent nucleation of noncentrosomal microtubules at the trans-Golgi network. Dev. Cell 12, 917–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miller P. M., Folkmann A. W., Maia A. R., Efimova N., Efimov A., Kaverina I. (2009) Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat. Cell Biol. 11, 1069–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mimori-Kiyosue Y., Grigoriev I., Lansbergen G., Sasaki H., Matsui C., Severin F., Galjart N., Grosveld F., Vorobjev I., Tsukita S., Akhmanova A. (2005) CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J. Cell Biol. 168, 141–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kumar P., Lyle K. S., Gierke S., Matov A., Danuser G., Wittmann T. (2009) GSK3β phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J. Cell Biol. 184, 895–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adachi A., Kano F., Tsuboi T., Fujita M., Maeda Y., Murata M. (2010) Golgi-associated GSK3β regulates the sorting process of post-Golgi membrane trafficking. J. Cell Sci. 123, 3215–3225 [DOI] [PubMed] [Google Scholar]
- 15. Takabayashi T., Xie M. J., Takeuchi S., Kawasaki M., Yagi H., Okamoto M., Tariqur R. M., Malik F., Kuroda K., Kubota C., Fujieda S., Nagano T., Sato M. (2010) LL5β directs the translocation of filamin A and SHIP2 to sites of phosphatidylinositol 3,4,5-triphosphate (PtdIns(3,4,5)P3) accumulation, and PtdIns(3,4,5)P3 localization is mutually modified by co-recruited SHIP2. J. Biol. Chem. 285, 16155–16165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou Q. L., Jiang Z. Y., Mabardy A. S., Del Campo C. M., Lambright D. G., Holik J., Fogarty K. E., Straubhaar J., Nicoloro S., Chawla A., Czech M. P. (2010) A novel pleckstrin homology domain-containing protein enhances insulin-stimulated Akt phosphorylation and GLUT4 translocation in adipocytes. J. Biol. Chem. 285, 27581–27589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baumann C. A., Ribon V., Kanzaki M., Thurmond D. C., Mora S., Shigematsu S., Bickel P. E., Pessin J. E., Saltiel A. R. (2000) CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature 407, 202–207 [DOI] [PubMed] [Google Scholar]
- 18. Beausoleil S. A., Villén J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 19. Zimmermann T., Rietdorf J., Girod A., Georget V., Pepperkok R. (2002) Spectral imaging and linear unmixing enables improved FRET efficiency with a novel GFP2-YFP FRET pair. FEBS Lett. 531, 245–249 [DOI] [PubMed] [Google Scholar]
- 20. Inoue M., Chiang S. H., Chang L., Chen X. W., Saltiel A. R. (2006) Compartmentalization of the exocyst complex in lipid rafts controls GLUT4 vesicle tethering. Mol. Biol. Cell 17, 2303–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang J., Imamura T., Babendure J. L., Lu J. C., Olefsky J. M. (2005) Disruption of microtubules ablates the specificity of insulin signaling to GLUT4 translocation in 3T3-L1 adipocytes. J. Biol. Chem. 280, 42300–42306 [DOI] [PubMed] [Google Scholar]
- 22. Eyster C. A., Duggins Q. S., Gorbsky G. J., Olson A. L. (2006) Microtubule network is required for insulin signaling through activation of Akt/protein kinase B: evidence that insulin stimulates vesicle docking/fusion but not intracellular mobility. J. Biol. Chem. 281, 39719–39727 [DOI] [PubMed] [Google Scholar]
- 23. Chen Y., Wang Y., Ji W., Xu P., Xu T. (2008) A pre-docking role for microtubules in insulin-stimulated glucose transporter 4 translocation. FEBS J. 275, 705–712 [DOI] [PubMed] [Google Scholar]
- 24. Patel N., Rudich A., Khayat Z. A., Garg R., Klip A. (2003) Intracellular segregation of phosphatidylinositol 3,4,5-trisphosphate by insulin-dependent actin remodeling in L6 skeletal muscle cells. Mol. Cell. Biol. 23, 4611–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang Z. Y., Zhou Q. L., Coleman K. A., Chouinard M., Boese Q., Czech M. P. (2003) Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc. Natl. Acad. Sci. U.S.A. 100, 7569–7574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Katome T., Obata T., Matsushima R., Masuyama N., Cantley L. C., Gotoh Y., Kishi K., Shiota H., Ebina Y. (2003) Use of RNA interference-mediated gene silencing and adenoviral overexpression to elucidate the roles of AKT/protein kinase B isoforms in insulin actions. J. Biol. Chem. 278, 28312–28323 [DOI] [PubMed] [Google Scholar]
- 27. Inoue M., Chang L., Hwang J., Chiang S. H., Saltiel A. R. (2003) The exocyst complex is required for targeting of GLUT4 to the plasma membrane by insulin. Nature 422, 629–633 [DOI] [PubMed] [Google Scholar]
- 28. Zaid H., Antonescu C. N., Randhawa V. K., Klip A. (2008) Insulin action on glucose transporters through molecular switches, tracks, and tethers. Biochem. J. 413, 201–215 [DOI] [PubMed] [Google Scholar]
- 29. Lizunov V. A., Matsumoto H., Zimmerberg J., Cushman S. W., Frolov V. A. (2005) Insulin stimulates the halting, tethering, and fusion of mobile GLUT4 vesicles in rat adipose cells. J. Cell Biol. 169, 481–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu Y. K., Xu K. D., Li J. Y., Feng L. Q., Lang D., Zheng X. X. (2007) Bi-directional transport of GLUT4 vesicles near the plasma membrane of primary rat adipocytes. Biochem. Biophys. Res. Commun. 359, 121–128 [DOI] [PubMed] [Google Scholar]
- 31. Chen S., Murphy J., Toth R., Campbell D. G., Morrice N. A., Mackintosh C. (2008) Complementary regulation of TBC1D1 and AS160 by growth factors, insulin, and AMPK activators. Biochem. J. 409, 449–459 [DOI] [PubMed] [Google Scholar]
- 32. Eyster C. A., Duggins Q. S., Olson A. L. (2005) Expression of constitutively active Akt/protein kinase B signals GLUT4 translocation in the absence of an intact actin cytoskeleton. J. Biol. Chem. 280, 17978–17985 [DOI] [PubMed] [Google Scholar]
- 33. Imamura T., Huang J., Usui I., Satoh H., Bever J., Olefsky J. M. (2003) Insulin-induced GLUT4 translocation involves protein kinase Cλ-mediated functional coupling between Rab4 and the motor protein kinesin. Mol. Cell. Biol. 23, 4892–4900 [DOI] [PMC free article] [PubMed] [Google Scholar]