

Abstract
We report the identification and characterization of the caz biosynthetic cluster from C. globosum and the characterization of a highly-reducing polyketide synthase (PKS) that acts in both a sequential and convergent manner with a nonreducing PKS to form the chaetomugilin and chaetoviridin azaphilones. Genetic inactivation studies verified the involvement of individual caz genes in the biosynthesis of the azaphilones. Through in vitro reconstitution, we demonstrated the in vitro synthesis of chaetoviridin A 3 from the pyrano-quinone intermediate cazisochromene 8 using the highly-reducing PKS and an acyltransferase.
Keywords: Azaphilone, filamentous fungi, acyltransferase, biosynthesis
Polyketides produced by fungi possess a diverse array of structural features and biological activities.1,2 These compounds are assembled by iterative polyketide synthases (IPKSs), which are megasynthases containing single copies of catalytic domains that are programmed to function repeatedly in different combinations.3 Additional programming complexity and structural diversity are generated when multiple IPKSs function collaboratively as illustrated in the biosynthesis of the azaphilones asperfuranone 1 and azanigerone A 2. This partnership between tandem IPKS can occur through a sequential manner in which the product of a highly-reducing PKS (HR-PKS) is transferred downstream to a nonreducing PKS (NR-PKS) for further chain elongation, as observed with 1;4 or a convergent manner, as demonstrated with 2,5 where the HR-PKS and NR-PKS function independently of one another and their individual products are combined. Understanding the functions of individual IPKSs, as well as how collaborative IPKSs are recruited to function together, are therefore important towards decoding the relationship between fungal IPKSs and their products. In this work, we discovered a single HR-PKS from Chaetomium globosum that can partner in both sequential and convergent fashions with a NR-PKS to synthesize two structurally distinct fragments that are incorporated into the antifungal chaetoviridin A 3 and cytotoxic chaetomugilin A 4 (Figure 1). This finding uncovers a previously unknown mode of collaboration among IPKSs and further demonstrates how a limited number of IPKSs can be combined to generate highly complex scaffolds.
Figure 1.
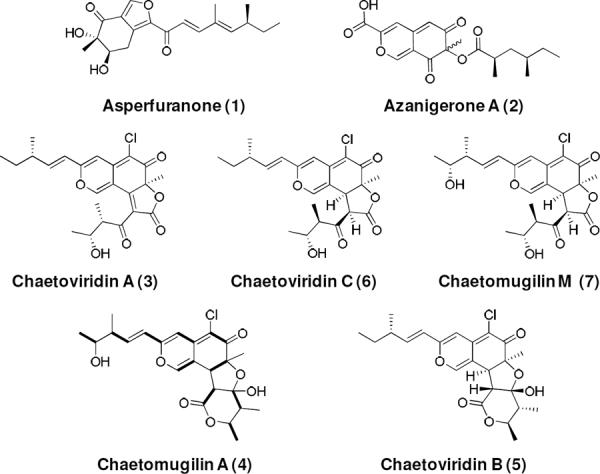
Structures of selected azaphilones and azaphilone-like molecules. The bold lines in chaetomugilin A (4) show enrichment from [1,2-13C2]acetate.
The common bicyclic pyrano-quinone scaffold observed in the azaphilones family of polyketides is biosynthesized by a NR-PKS. The reduced substituents, such as those in 1 and 2 are synthesized by a partnering HR-PKS found in each gene cluster.4,5C. globosum is a filamentous fungi that has been reported to produce a large variety of azaphilones; including the chaetoviridins 3,6 B (5)6,7 and C (6)6, as well as chaetomugilins 4,8 and M (7)9 (Figure 1). Unlike 1 and 2, the chlorinated 3, 6 and 7 contain an angular lactone ring attached to the isochromenone core, whereas 4 and 5 contain a tetracyclic isochromenone-lactol-lactone structure.7,10–12 Based on their structural similarities, we propose that 3–7 are assembled via a common biosynthetic pathway. Feeding experiments with [1,2-13C2]acetate confirmed that the entire molecule of 4 is derived from a polyketide backbone (Figures 1 and S1), which is consistent with previous isotopic experiments with structurally related azaphilones.6,13–15 Structural inspection of 3 and 4 reveals that in contrast to 1 and 2, additional IPKS machinery must be required for their biosynthesis. It can be speculated that assembly of the northern portions of 3 and 4 follows the biosynthetic logic of 1, in which a sequential collaboration of HR- and NR-PKS is required; whereas the southern portions mimic that of 2, in which the product of a different IPKS is combined with the northern portion in a convergent manner. Hence, we hypothesized that up to three IPKSs may be found in the biosynthetic gene cluster of 3–7.
Using the HR-PKS and NR-PKS sequences from the gene clusters of 1 and 2 identified in Aspergillus nidulans4 and A. niger5, respectively, the genome of C. globosum was scanned for putative azaphilone biosynthetic clusters containing two or more IPKSs. Three clusters containing a HR-PKS and a NR-PKS in close proximity to each other were identified. Additionally, each cluster contained a predicted FADH2-dependent halogenase that would be expected for introducing a chlorine atom onto the bicyclic core. Interestingly, none of these clusters contained three IPKSs as we hypothesized. Only one cluster (hereby named the caz cluster) encoded a putative acyltransferase (cazE) that would be required for transfering the convergent polyketide fragment (southern portion) to the pyranoquinone substrate (northern protion) to form 3–7. This 65-kb caz cluster was annotated to encode 16 open reading frames as shown in Figures 2A and S2, including genes encoding the HR-PKS (cazF) and NR-PKS (cazM).
Figure 2.
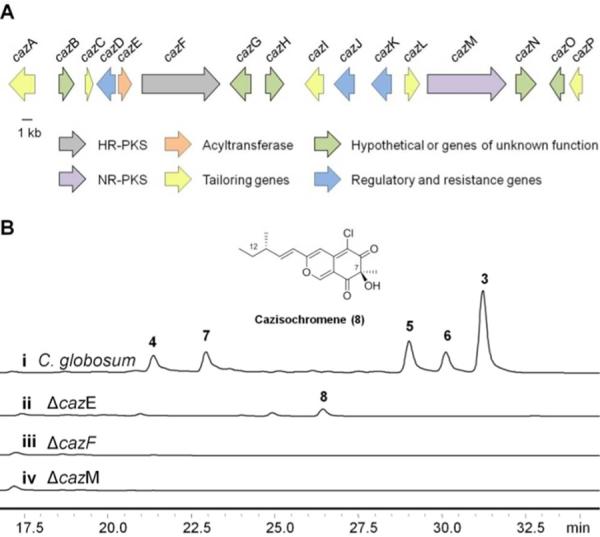
Genetic verification of the caz biosynthetic cluster in C. globosum. A) Organization of the 65 kb caz cluster. B) (i) LC-MS analysis (observed at 360 nm) of azaphilones produced by wild-type C. globosum; (ii) formation of 8 in the ΔcazE mutant; and abolishment of the production of 3–7 in the (iii) ΔcazF and (iv) ΔcazM mutants.
To confirm the involvement of the caz cluster in the biosynthesis of 3–7, gene inactivation of the NR-PKS cazM was accomplished via a fusion PCR gene targeting cassette (Figure S3).16 The ΔcazM inactivation completely abolished production of the azaphilones. Inactivation of the HR-PKS cazF also completely abolished production of 3–7 without the recovery of any pyrano-quinone intermediates, suggesting an essential role of both CazM and CazF in the synthesis of the northern portion of 3–7. This is consistent with the collaborative mode of interaction between the two IPKSs, in which the 4-methyl-hex-2-enoate 9 is the most likely product synthesized by CazF and is transferred to CazM via the Starter-unit:ACP Transacylase (SAT) domain of CazM as a starter unit for further chain elongation (Figure 3). In this model, the NR-PKS is unable to produce any polyketide product in the absence of the starter unit.17
Figure 3.

Proposed biosynthesis of the chaetoviridins and chaetomugilins from C. glbosoum. (A) Biosynthesis of 3–7 by the caz pathway. (B) Domain organization of the HR-PKS CazF and NR-PKS CazM. CazF consists of a ketosynthase (KS), malonyl-CoA:ACP acyltransferase (MAT), dehydratase (DH), methyltransferase (MT), enoyl reductase (ER), ketoreductase (KR) and acyl carrier protein (ACP). CazM consists of a starter-unit:ACP transacylase (SAT), KS, MAT, product template (PT), MT, ACP and reductive domain (R).
To further confirm this mode of interaction between the two caz IPKSs, we inactivated the acyltransferase cazE that may be involved in connecting the northern and southern portions of 3–7. We reasoned that inactivation of cazE should therefore have no effect on the sequential collaboration between CazM and CazF. Indeed, the ΔcazE mutant was unable to produce 3–7 and instead accumulated an intermediate 8 with UV absorbance consistent with that of a pyrano-quinone (Figure S4–S7). The structure of 8 was elucidated by 1D and 2D NMR spectroscopy and named cazisochromene (Figure 2B). The structure of the reduced substituent in 8 is consistent with that of 3, 5 and 6, further confirming 8 is an authentic intermediate in the caz pathway. However, the 12-hydroxyl derivative of 8 could not be detected in the culture suggesting that the hydroxyl moiety present in 4 and 7 at this position may be installed by post-PKS oxygenases from the caz cluster rather than formed from incomplete reduction by the HR-PKS CazF. This hypothesis is supported by monitoring the time-course of metabolite production and observing that the amount of 3 dramatically decreased while 4 increased over time (Figure S8).
With 8 in hand, we propose the biosynthetic pathway of 3–7 as shown in Figure 3. Following transfer of 9 from CazF to CazM, the polyketide is elongated four additional rounds and methylated once to form the heptaketide intermediate 10. While tethered to the ACP, 10 undergoes a Product Template (PT) domain-mediated regioselective cyclization via a C2–C7 aldol condensation followed by reductive release to form the benzaldehyde 11.18,19 Chlorination of the aromatic ring catalyzed by the halogenase CazI followed by a hydroxylation-catalyzed annulation at C75,20 by the predicted monooxygenase CazL would then give rise to 8. Given that the caz cluster only contains two IPKSs instead of the initially hypothesized three, either CazF or CazM must provide the additional southern triketide fragment. We propose that CazF could act as a dual functioning IPKS in that it not only supplies the reduced triketide 9 for CazM, but also a more oxidized intermediate such as 12 for CazE (Figure 3). Following CazE-catalyzed acylation, intramolecular aldol condensation of the newly formed β-keto ester yields 3. Further modification of 3 through reduction (3→6), lactone cleavage and rearrangement, and C12 oxidation furnishes products 4–7.
To test the involvement of CazF and CazE in converting 8 into 3, both enzymes were cloned and expressed from heterologous hosts for in vitro reconstitution. CazE (54 kDa) was solubly expressed as a N-terminal octahistidine-tagged protein from Escherichia coli BL21(DE3)/pJW07637.21 CazF was solubly expressed as a C-terminal hexahistidine-tagged protein from Saccharomyces cerevisiae BJ5464-NpgA/pJW07638.22, Both proteins were purified to near homogeneity with yields of 52 mg/L and 9.5 mg/L, respectively (Figures S9–S11). The minimal PKS activity of CazF was evaluated by incubating the enzyme with 2 mM malonyl-CoA. Treating the reaction mixture with 1 M NaOH (base hydrolysis) followed by LC-MS analysis of the organic extract showed production of the triacetic acid lactone 13 (Figure 4A), which is formed from the spontaneous enolization and cyclization of the unreduced triketide.17,23 Intriguingly, in the absence of base hydrolysis, significantly lower levels of 13 were detected, indicating the triketide may be fixed in an extended conformation by CazF to prevent lactonization and release. However, when CazE was added to the reaction, comparable levels of 13 to that of the base hydrolyzed reaction were produced (Figure S12). These results suggested a likely interaction between CazE and CazF, in which a conformational change in CazF after interacting with CazE may cause the unreduced triketide to be exposed to the aqueous environment and results in its subsequent lactonization and release.
Figure 4.

Reconstitution of CazE and CazF activity in vitro. (A) HPLC analysis (280 nm) of α-pyrones biosynthesized by CazF in the presence of (i) malonyl-CoA and (ii) malonyl-CoA and SAM. (B) HPLC analysis (360 nm) of chaetoviridins enzymatically synthesized in vitro when (i) CazF was incubated with 8, malonyl-CoA, NADPH and SAM; (ii) CazE was incubated with 8, malonyl-CoA, NADPH and SAM; (iii) CazE and CazF were incubated with malonyl-CoA; (iv) CazE and CazF were incubated with malonyl-CoA and SAM; (v) CazE and CazF were incubated with malonyl-CoA and NADPH; (vi) CazE and CazF were incubated with malonyl-CoA, NADPH and SAM.
We next examined the tailoring domains of CazF. The activity of the MT domain was assessed by adding 2 mM S-adenosyl-l-methionine (SAM) to CazF together with malonyl-CoA. Following incubation and base hydrolysis, a dimethylated α-pyrone 14 (Figure 4A) was isolated, which matched to that of a synthetically prepared standard (Figure S13–14).24 The formation of 14 is indicative of the correct timing of the MT domain following formation of diketide intermediates on CazF, which is required for the formation of both 9 and 12. To assay the functions of the KR, DH and ER domains, acetoacetyl-SNAC was incubated with CazF and NADPH. LC-MS analysis of the organic extract showed accumulation of 3-hydroxybutanoyl-SNAC and butyryl-SNAC, confirming that all reductive domains were functional for the purified enzyme (Figure S15).
After confirming all of the domains of CazF were catalytically active, we attempted to synthesize 3 from 8. Equimolar amounts of CazF and CazE were incubated with 8, NADPH, SAM and malonyl-CoA. LC-MS analysis of the organic extract revealed the formation of a new product; whose retention time, UV profile and isotopic mass ratio were identical to 3 (Figure 4B). When either CazF or CazE was omitted from the reaction, the formation of 3 could not be detected. Additionally, when only malonyl-CoA was added to the reaction or when NADPH was omitted, no new product formation could be detected. However, when only SAM was omitted from the reaction, a new product with a shorter retention time than 3 and loss of a methyl group by mass was detected from the in vitro assay. Based on these observations, we assigned this product to be 4'-desmethyl-chaetoviridin A (15). Therefore, the in vitro reactions unequivocally confirmed the involvement of the HR-PKS in providing the southern portion of the fragment in 3–7, as well as providing another example of an acyltransferase-mediated convergent synthesis of fungal polyketides.25 The requirement of NADPH to produce the chaetoviridins suggest that CazE may possess substrate specificity towards a triketide reduced at the δ position; while the formation of 13 in the absence of SAM indicates methylation at the γ position is not essential.
In summary, we have identified and characterized the caz biosynthetic cluster that is responsible for the production of the chaetoviridin and chaetomugilin azaphilones from C. globosum. Through in vivo gene inactivation experiments, we established that the HR-PKS CazF most likely supplies a highly reduced methylated triketide product such as 9 to the NR-PKS CazM. Since CazM cannot be solubly expressed at this point, we can not exclude the possibility that CazF transfers a polyketide product of different length to CazM as a starter unit. Additionally, through in vitro reconstitution experiments with the acyltransferase CazE, we established that CazF can also produce a more oxidized triketide product 12 that is transacylated by CazE to a pyrano-quinone intermediate 8 to afford 3. Hence, CazF serves the unprecedented dual functions in completing the biosynthesis of a polyketide product. The timing of substrate transfer from CazF, whether it is 9 or 12, must be precisely orchestrated by the two interacting acyltrasferases in the form of the freestanding CazE and the SAT domain of CazM. Our work therefore unveils a new level of programming complexity among the fungal PKSs.
Supplementary Material
ACKNOWLEDGMENT
J.M.W. is supported by a L'Oréal USA Fellowship for Women in Science. G.C. thanks the National Science Foundation for a graduate research fellowship (DGE-0707424). This work was supported by the US National Institutes of Health (1R01GM085128 and 1DP1GM106413) to Y.T; JSPS through the “Funding Program for Next Generation World-Leading Researchers” initiated by the Council for Science and Technology Policy (No. LS103), the Uehara Memorial Foundation, and by Mochida Memorial Foundation for Medical and Pharmaceutical Research to K. W. NMR instrumentation was supported by the NSF equipment grant CHE-1048804.
Footnotes
Supporting Information Available. Experimental details and NMR spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Hoffmeister D, Keller NP. Nat. Prod. Rep. 2007;24:393. doi: 10.1039/b603084j. [DOI] [PubMed] [Google Scholar]
- (2).Brase S, Encinas A, Keck J, Nising CF. Chem. Rev. 2009;109:3903. doi: 10.1021/cr050001f. [DOI] [PubMed] [Google Scholar]
- (3).Cox RJ. Org Biomol. Chem. 2007;5:2010. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]
- (4).Chiang YM, Szewczyk E, Davidson AD, Keller N, Oakley BR, Wang CC. J. Am. Chem. Soc. 2009;131:2965. doi: 10.1021/ja8088185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zabala A, Xu W, Chooi YH, Tang Y. Chem. Biol. 2012;19:1049. doi: 10.1016/j.chembiol.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Takahashi M, Koyama K, Natori S. Chem. Pharm. Bull. 1990;38:625. [Google Scholar]
- (7).Kingsland SR, Barrow RA. Aust. J. Chem. 2009;62:269. [Google Scholar]
- (8).Muroga Y, Takeshi Y, Atsushi N, Reiko T. J. Antibiot. 2008;61:615. [Google Scholar]
- (9).Muroga Y, Yamada T, Numata A, Tanaka R. Tetrahedron. 2009;65:7580. [Google Scholar]
- (10).Achard M, Beeler AB, Porco JA., Jr. ACS Comb. Sci. 2012;14:236. doi: 10.1021/co300002x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Germain AR, Bruggemeyer DM, Zhu J, Genet C, O'Brien P, Porco JA. J. Org. Chem. 2011;76:2577. doi: 10.1021/jo102448n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Musso L, Dallavalle S, Merlini L, Bava A, Nasini G, Penco S, Giannini G, Giommarelli C, De Cesare A, Zuco V, Vesci L, Pisano C, Castorina M, Milazzo F, Cervoni ML, Dal Piaz F, De Tommasi N, Zunino F. Bioorg. Med. Chem. 2010;18:6031. doi: 10.1016/j.bmc.2010.06.068. [DOI] [PubMed] [Google Scholar]
- (13).Ogihara J, Kato J, Oishi K, Fujimoto Y. J. Biosci. Bioeng. 2000;90:678. doi: 10.1263/jbb.90.678. [DOI] [PubMed] [Google Scholar]
- (14).Hajjaj H, Klaebe A, Loret MO, Goma G, Blanc PJ, Francois J. Appl. Environ. Microbiol. 1999;65:311. doi: 10.1128/aem.65.1.311-314.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hajjaj H, Klaebe A, Goma G, Blanc PJ, Barbier E, Francois J. Appl. Environ. Microbiol. 2000;66:1120. doi: 10.1128/aem.66.3.1120-1125.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Szewczyk E, Nayak T, Oakley CE, Edgerton H, Xiong Y, Taheri-Talesh N, Osmani SA, Oakley BR. Nat. Protoc. 2006;1:3111. doi: 10.1038/nprot.2006.405. [DOI] [PubMed] [Google Scholar]
- (17).Zhou H, Qiao K, Gao Z, Meehan MJ, Li JW, Zhao X, Dorrestein PC, Vederas JC, Tang Y. J. Am. Chem. Soc. 2010;132:4530. doi: 10.1021/ja100060k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Crawford JM, Thomas PM, Scheerer JR, Vagstad AL, Kelleher NL, Townsend CA. Science. 2008;20:243. doi: 10.1126/science.1154711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bailey AM, Cox RJ, Harley K, Lazarus CM, Simpson TJ, Skellam E. Chem. Commun. (Camb) 2007:4053. doi: 10.1039/b708614h. [DOI] [PubMed] [Google Scholar]
- (20).Davison J, al Fahad A, Cai M, Song Z, Yehia SY, Lazarus CM, Bailey AM, Simpson TJ, Cox RJ. Proc. Natl. Acad. Sci. U. S. A. 2012;109:7642. doi: 10.1073/pnas.1201469109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jez JM, Ferrer JL, Bowman ME, Dixon RA, Noel JP. Biochemistry. 2000;39:890. doi: 10.1021/bi991489f. [DOI] [PubMed] [Google Scholar]
- (22).Lee KK, Da Silva NA, Kealey JT. Anal. Biochem. 2009;394:75. doi: 10.1016/j.ab.2009.07.010. [DOI] [PubMed] [Google Scholar]
- (23).Ma SM, Li JW, Choi JW, Zhou H, Lee KK, Moorthie VA, Xie X, Kealey JT, Da Silva NA, Vederas JC, Tang Y. Science. 2009;326:589. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yokoe H, Mitsuhashi C, Matsuoka Y, Yoshimura T, Yoshida M, Shishido K. J. Am. Chem. Soc. 2011;133:8854. doi: 10.1021/ja202874d. [DOI] [PubMed] [Google Scholar]
- (25).Xie X, Meehan MJ, Xu W, Dorrestein PC, Tang Y. J. Am. Chem. Soc. 2009;131:8388. doi: 10.1021/ja903203g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.