

Abstract
Fluorescent liposomal nanovesicles (liposomes) are commonly used for lipid research and/or signal enhancement. However, the problem of self-quenching with conventional fluorescent liposomes limits their applications because these liposomes must be lysed to detect the fluorescent signals. Here, we developed a nonquenched fluorescent (NQF)1 liposome by optimizing the proportion of sulforhodamine B (SRB) encapsulant and lissamine rhodamine B-dipalmitoyl phosphatidylethanol (LRB-DPPE) on a liposomal surface for signal amplification. Our study showed that 0.3% of LRB-DPPE with 200 μm of SRB provided the maximal fluorescent signal without the need to lyse the liposomes. We also observed that the NQF liposomes largely eliminated self-quenching effects and produced greatly enhanced signals than SRB-only liposomes by 5.3-fold. To show their application in proteomics research, we constructed NQF liposomes that contained phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) and profiled its protein interactome using a yeast proteome microarray. Our profiling led to the identification of 162 PI(3,5)P2-specific binding proteins (PI(3,5)P2-BPs). We not only recovered many proteins that possessed known PI(3,5)P2-binding domains, but we also found two unknown Pfam domains (Pfam-B_8509 and Pfam-B_10446) that were enriched in our dataset. The validation of many newly discovered PI(3,5)P2-BPs was performed using a bead-based affinity assay. Further bioinformatics analyses revealed that the functional roles of 22 PI(3,5)P2-BPs were similar to those associated with PI(3,5)P2, including vesicle-mediated transport, GTPase, cytoskeleton, and kinase. Among the 162 PI(3,5)P2-BPs, we found a novel motif, HRDIKP[ES]NJLL that showed statistical significance. A docking simulation showed that PI(3,5)P2 interacted primarily with lysine or arginine side chains of the newly identified PI(3,5)P2-binding kinases. Our study showed that this new tool would greatly benefit profiling lipid–protein interactions in high-throughput studies.
Cell viability and physiological functions are maintained through a complex biomolecular interaction network. One of the key components in the regulatory system includes lipid–protein interactions that mediate various cell responses and metabolisms. Increasing evidence shows that such interactions have profound influences on cell polarization, the cell cycle, and other cellular processes. To date, in vitro characterizations of lipid interactions with other biomolecules are often conducted using artificial membrane models, such as liposomal nanovesicles, to mimic biological membranes. Liposomal nanovesicles, termed liposomes, are spherical vesicles that are surrounded by phospholipid bilayers in which the lipid of interest can be incorporated. An important benefit of liposomes is the ease in which a large number of fluorescent molecules can be encapsulated so that the liposome binding signals can be greatly enhanced for detection (1–4). Therefore, liposomes have become a practical and popular tool for use as a model membrane or fluorophore-loaded vehicle to study signal amplification (1–4) and/or lipid research (5–9).
In general, liposomes are capable of encapsulating hundreds of millions of fluorescent dye molecules, thereby providing greatly enhanced signals (1–4). However, high concentrations of fluorophores often lead to self-quenching, and as a result, the fluorescent signals cannot be detected without first lysing the liposomes (1–4). This issue has limited their applications for real-time detection and high-density chip assays. To solve this problem, we developed a novel non-quenched fluorescent (NQF) liposome with the capability of signal amplification. During the fabrication procedure, we used sulforhodamine B (SRB) as an encapsulant and incorporated lissamine rhodamine B-dipalmitoyl phosphatidylethanol (LRB-DPPE) within the liposomal bilayer.
Profiling phosphatidylinositide-protein interactions is of particular interest because these lipids have been implicated in a wide variety of cell functions, including cell signaling, actin cytoskeletal reorganization, exocytosis, and intracellular trafficking (10–14). Among the phosphatidylinositides, phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) is one of the most important mediators of signal transduction (15, 16). Intensive studies over the past decade have shown that PI(3,5)P2 is involved in protein sorting into multivesicular bodies (MVBs), membrane recycling/turnover, and the vacuole acidification (17–19). Like other phosphatidylinositides, PI(3,5)P2 may regulate downstream pathways through the binding of the myo-inositol head group to proteins containing phosphoinositide-binding domains (20, 21). Thus far, a handful of modular phosphoinositide-binding domains have been identified, including C2 (Protein Kinase C homology 2) (22), a WD-40 motif (tryptophan-aspartic acid repeats) that folds as β-propellers (23), ARRB1 (β-arrestin 1), and a number of actin regulatory domains (e.g. the gelsolin/villin family, cofilin, and profilin) (20).
To globally profile PI(3,5)P2-binding proteins as the foundation for a better understanding of the biology of PI(3,5)P2, we employed the newly developed PI(3,5)P2-NQF liposomes to probe the Saccharomyces cerevisiae proteome microarray. We not only recovered many proteins that contained known PI(3,5)P2-binding domains, but we also validated many newly discovered PI(3,5)P2-binding proteins using a bead-based affinity assay. Representing both a signal and an analyte carrier, the NQF liposomes should provide a new research model for studying lipid–protein interactions in the future.
MATERIALS AND METHODS
Optimization of SRB Concentration
The SRB fluorophore (Sigma-Aldrich Co.) was dissolved in Tris-buffered saline (0.02 m Tris, 0.2 m NaCl, pH 7.5) and was full wavelength-scanned by a spectrophotometer (U-2000, Hitachi, Yokohama, Japan). The optimal emission wavelength was determined by fluorescence spectroscopy. In a 96-well black plate, fluorescent signals from various SRB concentrations (0, 50, 100, 125, 150, 175, 200, 225, 250, 300, 350, and 400 μm) were detected by a fluorometer.
Optimization of LRB-DPPE Proportion on Liposomal Surface
Different molar ratios of LRB-DPPE were examined to obtain the optimum proportion for incorporation into the liposomal bilayer. Lipid components containing 9.6 μmol of cholesterol, 10.2–10.4 μmol of dipalmitoyl phophatidylcholine (DPPC), and 0–0.2 μmol of LRB-DPPE (lipids above were purchased from Avanti Polar Lipids, Alabaster, AL) were dissolved in 1 ml chloroform/methanol (volume ratio = 5:1). The lipid mixture was sonicated homogeneously and hydrated with 0.3 ml 200 μm SRB encapsulant (optimized concentration) at 50 °C. The organic solvent was subsequently removed using a vacuum rotary evaporator (Eyela N-1000, Japan) in a 50 °C water bath. An additional 0.3 ml encapsulant solution was added for further hydration, and the raw product was then placed in a 45 °C water bath to rotate for 30 min. The formed liposomes were extruded through a polycarbonate syringe filter (Avanti Polar Lipids, Alabaster, AL) and size excluded by a Sephadex G-25 column (Pharmacia AB Biotechnology Uppsala, Sweden). The concentration of each fabricated liposome batch was determined using the Bartlett assay (24). To determine the optimal proportion of LRB-DPPE, the fluorescence intensities of the fabricated liposomes (5 nmol total lipid/μl) with the varied proportions of LRB-DPPE were measured by a fluorometer.
Fabrication of PI(3,5)P2 and Phosphatidylinositol (PI) NQF Liposomes
Lipid mixtures including 9 μmol cholesterol (45%), 1 μmol dipalmitoyl phosphatidylglycerol (DPPG) (5%), 9.86 μmol DPPC (49.3%), 0.06 μmol LRB-DPPE (0.3%), and 0.08 μmol PI(3,5)P2 or PI (0.4%) (Echelon Biosciences, Salt Lake City, UT) were dissolved in a 1 ml chloroform/methanol mixture. The organic solvent was completely removed with nitrogen to yield a thin lipid film. The formed lipid layer was then hydrated with 0.3 ml 200 μm SRB encapsulant and incubated for 45 min at 60 °C. The liposome suspension was then incubated for 30 min to allow the spherical vesicles to further mature and fully hydrate. Finally, the fabricated NQF liposomes were extruded through a 50 nm-diameter polycarbonate syringe filter and purified using Zeba desalt spin columns (Thermo, Rockford, IL).
Proteome Chip Assays
Yeast proteome microarrays that were composed of ∼5,800 N-terminal GST-fusion proteins were constructed on nitrocellulose-coated glass slides (FAST slides, Whatman), as previously reported (9). After blocking with 1% BSA in TBS, the yeast proteome chips were probed with 110 μl NQF liposomes containing PI(3,5)P2, PI, or structural lipids only. The binding reactions were performed on microarrays in the dark at room temperature for 80 min. To remove the unbound liposomes, chips were gently washed with osmolarity-adjusted TBS and air dried. The slides were then scanned with a GenePix 4000B array scanner (Axon Instruments, Union City, CA). Binding signals were acquired and analyzed with the GenePix Pro 6.0 software. Each liposome-binding experiment was conducted in duplicate.
Bead-based Affinity Assays
Each protein for the binding assays with the PI(3,5)P2-containing liposomes was freshly purified. The protein purification protocol was modified from the high-throughput yeast protein purification procedure developed by Zhu et al. (9). The bead-based assay in a 96-well filter plate format was modified from the earlier reports (25–27). Yeast colonies on agar plates were inoculated in a 2.2 ml 96-deep well plate containing 1 ml URA-/glucose liquid media. When the OD600 reached 4.0 at 30 °C with vigorous shaking, 5 μl the cultures was transferred to 12-channel reagent reservoirs that contained 6 ml of URA-/raffinose media. After the culture reached an OD600 of 0.6–1.0, 316 μl of a 40% galactose solution was added to each well (final concentration of 2%) and incubated at 30 °C for 4 h with moderate shaking to induce protein expression. The cells were harvested by centrifugation, transferred to a 96-deep well plate and subjected to a snap freeze at −80 °C. The frozen culture was mixed with 200 μl zirconia beads (0.7 mm, Biospec Product, Germany) and 400 μl lysis buffer containing fresh protease inhibitors (9). After thawing the culture, the deep well plate was sealed by a cap mat and vortexed at 4 °C for 30 min. An additional 400 μl lysis buffer was added to each well, and the debris was spun down at 4000 rpm for 15 min. The supernatants were transferred to another 96-deep well plate and mixed with 100 μl pre-washed glutathione-Sepharose beads (GE Healthcare, Sweden). To capture the GST-tagged PI(3,5)P2-BPs, the plate was incubated at 4 °C for 80 min with gentle shaking. The glutathione bead-supernatant mixtures were transferred and applied to the membrane of a 96-well filter plate (Nunc, Rochester, NY). The beads were washed with 400 μl wash buffer I and wash buffer II (9) three times. After removing the buffer by centrifuging at 800 g for 1 min, 50 μl the quenched PI(3,5)P2 fluorescent liposomes encapsulating 150 mm of SRB were added into each well and incubated with moderate shaking at 4 °C for 1 h. Unbound liposomes were removed by gently washing ten times with 200 μl osmolarity-adjusted TBS. After the complete removal of the washing solution, 60 μl 30 mm n-octyl-β-d-glucopyranoside (n-OG) was added to each well and incubated with vigorous shaking at 4 °C for 5 min to lyse the bound liposomes. The eluates were collected in a 96-well black plate (Nunc). The fluorescent signals were measured at 590 nm with excitation at 540 nm using a Synergy 2 plate reader (Biotek, Winooski, VT). To monitor the quality of the purification, proteins were eluted and separated by SDS-PAGE, followed by Coomassie blue staining.
Databases
Potential protein–protein interactions among the PI(3,5)P2-BPs were processed by the STRING database (http://string-db.org/; version 9.0) (28). Human orthologs of the identified proteins were examined using the KEGG Sequence Similarity DataBase (http://www.genome.jp/kegg/ssdb/) and compared with previously reported phosphoinositide binding profiles in LIM1215 colon carcinoma cell lines (5, 6). Protein domain architectures and their biological functions were retrieved from the Pfam database (http://pfam.sanger.ac.uk/), UniProt Knowledgebase (http://www.uniprot.org/uniprot/), and the Saccharomyces Genome Database (http://www.yeastgenome.org).
Protein Domain Enrichment and Motif Search by MEME
Domain annotations of the yeast proteome were retrieved from Pfam (29). A total of 5,594 yeast proteins were covered by the Pfam-defined domains. Of the 162 identified PI(3,5)P2-BPs, 144 proteins possessed Pfam domains and were further analyzed in the R environment. A total of 135 of the phosphoinositide-interacting proteins reported by Zhu et al. (9) contained Pfam-defined domains and were subjected to the same analysis. The enrichment of protein families and domains was determined by Fisher's exact test with a p value ≤ 0.05. Additionally, 22 identified PI(3,5)P2-BPs that had overlapping functions with PI(3,5)P2 (Table II) were analyzed by MEME (version 4.6.1) (30) using the following parameters: (1) occurrences of a single motif were set to zero or one per sequence; (2) the minimum number of sites was 8; (3) the minimum width of each motif was 10 and the maximum width was 20. Multiple sequence alignment was conducted by ClustalX2 (version 2.0.12) (31).
Table II. Identified PI(3,5)P2-specific binding proteins that had overlapping functions with PI(3,5)P2.
| Systematic name | Protein name | Functional annotation | |
|---|---|---|---|
| Vesicle-mediated transport | Ydl099w | Bug1p | ER-Golgi vesicle mediated transport |
| Ydl072c | Yet3p | ER-Golgi vesicle mediated transport | |
| Ydr100w | Tvp15p | Vesicle-mediated transport | |
| Ylr447c | Vma6p | Vacuolar acidification; vacuolar transport | |
| Ydr065w | Rrg1p | Vacuolar acidification | |
| Ykr001c | Vps1p | Vacuolar protein sorting; actin cytoskeleton organization; GTPase activity | |
| GTPases and associated regulators | Yor134w | Bag7p | Rho GTPase activator activity |
| Ydl240w | Lrg1p | Rho GTPase activator activity | |
| Yhr168w | Mtg2p | GTPase | |
| Ygl161c | Yip5p | Rab GTPase binding | |
| Cytoskeleton regulation | Ygl106w | Mlc1p | Myosin motor activity |
| Ygl075c | Mps2p | Structural constituent of cytoskeleton; spindle pole body | |
| Ydr356w | Spc110p | Structural constituent of cytoskeleton; spindle pole body | |
| Ygl061c | Duo1p | Structural constituent of cytoskeleton; spindle pole body | |
| Yal020c | Ats1p | Tubulin; microtubule cytoskeleton organization | |
| Ylr370c | Arc18p | Arp2/3 complex; actin binding | |
| Kinases | Ynr031c | Ssk2p | Serine/threonine protein kinase |
| Ygr040w | Kss1p | Serine/threonine protein kinase | |
| Ygr052w | Fmp48p | Serine/threonine protein kinase | |
| Yar019c | Cdc15p | Serine/threonine protein kinase | |
| Ygl179c | Tos3p | Serine/threonine protein kinase | |
| Ybr135w | Cks1p | Protein kinase activator |
Docking Simulation
Protein structures were retrieved from the Protein Data Bank (PDB) (32) or modeled by the SwissModel (33). Some of the modeled protein structures were retrieved from the Protein Model Portal (http://www.proteinmodelportal.org/) (34). To assess the quality of the structure modeling, QMEAN Z-scores (35, 36) were determined, and if the Z-score > −4, the model was applied to the docking simulation. If the Z-score < −4, the structure was remodeled by the I-tasser (37) and ModWeb (38). The PI(3,5)P2 structure was retrieved from the PubChem Compound Database (39) (Compound ID: 643961) and modified by the OpenBabel (version 2.3.0) (http://openbabel.sourceforge.net/) (40) to simulate its protonation state under physiological conditions. The molecular docking was conducted by the SwissDock (41) and was then estimated by DSX scoring (42).
RESULTS
Construction of a Novel NQF Liposome
To eliminate the self-quenching problem of the SRB encapsulant, we first attempted to obviate solvent relaxation. Solvent relaxation is a physical property of fluorophores that fluctuates with the surrounding environment, causing changes in the absorption and emission spectra. Thus, the SRB encapsulant dissolved in 0.02 m TBS was full wavelength-scanned to determine the optimal excitation wavelength. The maximal absorbance of SRB was determined to be 565 nm, and the corresponding emission wavelength was determined to be 585 nm. The fluorescence of serial diluted SRB in TBS was examined to determine the optimal concentration that showed a minimum quenching effect. As shown in Fig. 1A, the dose-response curve displayed a proportional increase in SRB fluorescence that stalled when the concentration reached 100 μm and gained maximal fluorescence intensity at 200 μm. Higher concentrations of SRB caused the fluorescent signal to decrease as a result of self-quenching.
Fig. 1.

Construction of NQF liposomes to eliminate the problem of self-quenching. A, Optimization of SRB concentration. The fluorescence maximum of SRB in 0.02 m TBS was revealed at 200 μm. B, Optimization of LRB-DPPE proportion. Liposomal nanovesicles containing 200 μm SRB and various proportions (0–1%) of LRB-DPPE were fabricated. The amount of liposomes per batch was adjusted to the same concentration by using the Bartlett assay (24), and the fluorescence intensities (AU) were measured by a fluorometer. Liposomes that contained 200 μm SRB and 0.3% LRB-DPPE produced the maximal fluorescent signal, which was higher than the liposomes that contained only 200 μm SRB without LRB-DPPE by 5.3-fold. C, Structure representation of PI(3,5)P2-containing NQF liposomes. The concentration-optimized SRB (200 μm) was encapsulated in liposomes containing 0.3% LRB-DPPE, 0.4% PI(3,5)P2, and 99.3% structural lipids.
We next modified the liposomal surface to further enhance the fluorescent signals. We opted for LRB-DPPE because its fluorescent spectrum is close to that of SRB. Different fluorescent liposomes containing various proportions (0%–1%) of LRB-DPPE and 200 μm SRB (optimized concentration) were prepared. These liposomes were adjusted to the same concentration (5 nmol of total lipid per microliter) according to the Bartlett assay (24), and the fluorescence intensities were quantitatively measured (Fig. 1B). The maximal fluorescence intensity was obtained when 0.3% of LRB-DPPE and 200 μm SRB were used to prepare the fluorescent liposomes. Interestingly, the liposomes that contained 0.3% of LRB-DPPE and 200 μm SRB generated a higher signal than the liposomes containing only 200 μm SRB without the LRB-DPPE by 5.3-fold (Fig. 1B). This result indicated that the LRB-DPPE on the membrane surface contributed the most to the fluorescent signals.
Global profiling of PI(3,5)P2-protein Interactions Using Developed NQF Liposomes
To show the superiority and analytical capabilities of the NQF liposomes, we first fabricated a conventional PI(3,5)P2-containing fluorescent liposome and probed the liposomes to the yeast proteome microarray. However, the spot signals from the conventional liposomes would seriously overlap and blur the chip image (supplementary Information S1), resulting in an unanalyzable chip data. This assay showed the limitation of conventional fluorescent liposomes on high-density chip assays. Alternatively, we constructed PI(3,5)P2-containing NQF liposomes to determine PI(3,5)P2-protein interactions using yeast proteome chips (9). The NQF liposomes were constructed by loading the SRB encapsulant onto a lipid bilayer that contained 0.3% LRB-DPPE, 0.4% PI(3,5)P2, and 99.3% structural lipids (i.e., 45% cholesterol, 49.3% DPPC, and 5% DPPG) (Fig. 1C). Two types of liposomes were also constructed as controls: one containing 0.4% PI as a replacement for the PI(3,5)P2 and the other containing no phosphoinositides at all. These liposomes were separately incubated on the yeast proteome chips to identify their binding proteins (Fig. 2A). After the unbound liposomes were removed by gentle washes in osmolarity-adjusted TBS, the slides were dried and scanned with a microarray scanner to acquire the binding signals (Fig. 2B).
Fig. 2.
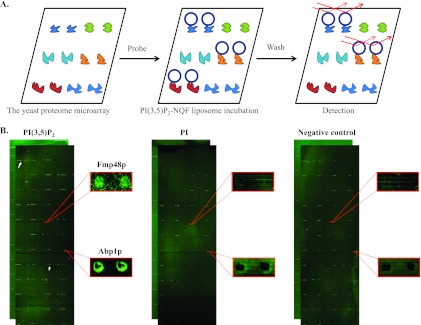
A schematic representation of PI(3,5)P2 binding assays using yeast proteome chips. A, A batch of PI(3,5)P2-NQF liposome was fabricated and incubated on the yeast proteome chips, followed by a wash step to remove excess unbound liposomes. The chips were then scanned and further analyzed by a comparison with control liposomes. B, Yeast proteome chips probed with PI(3,5)P2 (left panel), PI (middle panel) and control (right panel) NQF liposomes. The binding signals of Fmp48p were only observed in the PI(3,5)P2 probing results, and thus was identified to be PI(3,5)P2-specific. However, Abp1p was shown in both PI(3,5)P2 and PI experiments, which was then categorized to bind both PI(3,5)P2 and PI.
To perform a quantitative analysis of the binding signals, we generated the signal intensity for each spot as the value of median foreground minus the median background signals. To identify positive proteins that were bound by individual liposomes, a histogram of the signal intensity of every protein spot in the microarray was plotted to determine the standard deviation (S.D.) value. Because we observed a wide range of the normal distribution curve, which indicated that the S.D. value of the dataset was quite high, we first used a cut-off of ≥ mean + 1 S.D. to identify positive liposome-binding proteins (hits). To further strengthen our ability to identify more relevant hits, we applied additional criteria to improve the fidelity of the results: (1) both duplicate protein spots were required to show significant signal intensity to be considered a true hit; and (2) a hit was required to be reproducible in the repeated chip binding assays. The PI(3,5)P2-specific binding proteins (PI(3,5)P2-BPs) were identified by removing the hits that were also identified in the PI- and structural lipid-containing liposomes (supplementary Information S2). Under such stringent criteria, a total of 162 proteins were identified (supplementary Information S2). Similarly, 12 additional proteins were shown to interact with both PI(3,5)P2- and PI-containing liposomes.
Validation of PI(3,5)P2-binding Proteins by Bead-based Affinity Assays
To validate the PI(3,5)P2-protein interactions that were identified with the NQF liposomes on the yeast proteome chips, we employed a bead-based affinity assay using conventional quenched liposomes in a 96-filter plate format (Fig. 3A). Briefly, 29 random proteins were expressed and purified as GST fusions from yeast (9). The purified proteins that were captured on the glutathione (GSH) beads were loaded into a 96-filter plate and incubated with quenched PI(3,5)P2 fluorescent liposomes. After incubation, the unbound liposomes were removed, and the retained liposomes were detected after lysis of the liposome to release the fluorescent signals. Each protein sample here was conducted in triplicate. To normalize the assay results, the binding signals of the proteins were divided by the signal of the GSH beads without protein to generate the fold enrichment. Of the 29 random proteins, 7 of them were not detectable on an SDS-PAGE gel stained with Coomassie brilliant blue (data not shown). Of the 22 proteins that were visualized on the SDS-PAGE, 14 (64%) showed statistically significant enrichment in the bead-based affinity assays (Fig. 3B and supplementary Information S3; p value < 0.05; n = 3), indicating that our dataset was of high fidelity.
Fig. 3.
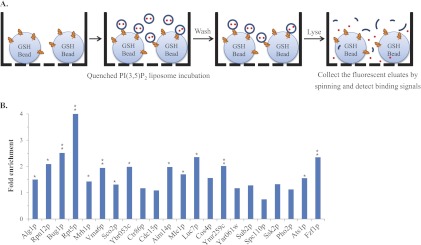
The bead-based affinity assays confirmed the PI(3,5)P2-protein interactions revealed by the NQF liposomes. A, A schematic representation of the bead-based affinity assay. The proteins of interest were purified by the GST-glutathione (GSH) bead affinity purification procedure (9). Prior to elution, fabricated PI(3,5)P2 fluorescent liposomes were added into each well of a 96-filter plate and allowed to interact with the purified target proteins. After the complete removal of unbound liposomes, the bound liposomes were lysed with 30 mm n-OG, and the fluorescent eluates were collected in a 96-well black plate to detect each binding signal. B, Validation of identified PI(3,5)P2-protein interactions. We chose 29 PI(3,5)P2-BPs to conduct the bead-based affinity assays, and each protein sample was conducted in triplicate. For normalization, the binding signals of the proteins were divided by the signal of the glutathione beads without proteins to generate the fold enrichment. Among the 29 proteins, seven were not detectable on an SDS-PAGE gel stained with Coomassie brilliant blue (data not shown). Therefore, 14 (64%) of the 22 proteins showed more than a 1.3-fold enrichment with statistical significance (* p < 0.05, ** p < 0.005; n = 3).
PI(3,5)P2- and PI-binding Proteins
The function and domain annotations of the 12 proteins that interacted with both PI(3,5)P2 and PI are summarized in Table I. Among them, Sro77p, Pkc1p, and Abp1p have previously been reported to possess phosphoinositide-binding domains. Sro77p contains 14 repeated WD-40 motifs, which have been reported to interact specifically with PI(3,5)P2 (23). Pkc1p and Abp1p contain a C2 and cofilin domain, respectively, both of which are also known to interact with phosphoinositides (20, 22). Although Abp1p was scored positive in both PI(3,5)P2- and PI-NQF liposome probing assays, its signal intensity from the PI(3,5)P2-NQF liposomes was much higher than that from the PI-NQF liposomes, indicating that Abp1p may be able to differentiate PI(3,5)P2 from PI (Fig. 2B). Interestingly, Abp1p has been reported to be involved in the activation of the Arp2/3 complex, a key mediator of actin cytoskeleton organization and cell morphology (43, 44). Because a reported PI(3,5)P2 effector, Ent3p, is necessary for normal actin cytoskeleton organization (45), it is plausible that the interaction between Abp1p and PI(3,5)P2 might serve as an alternative mechanism for initiating cytoskeleton remodeling. Furthermore, other newly identified proteins are known to be associated with known cellular processes involving PI(3,5)P2, such as vacuolar transport, lipid turnover, and cytoskeletal regulation. For example, Erv14p is required for endoplasmic reticulum to Golgi transport (46), whereas Atg15p participates in the breakdown of MVB vesicles and membrane recycling (47), indicating potential connections with PI(3,5)P2.
Table I. Identified proteins that interacted specifically with both PI(3,5)P2 and PI.
| Systematic name | Protein name | Functional annotation | Phosphoinositide recognition domain |
|---|---|---|---|
| Ydl068w | Ydl068w | Function unknown | |
| Ygl054c | Erv14p | ER-Golgi vesicle mediated transport | |
| Ybl106c | Sro77p | Golgi-plasma membrane transport | WD-40 repeats |
| Ybl105c | Pkc1p | Serine/threonine protein kinase | C2 |
| Ybr211c | Ame1p | Attachment of spindle microtubules to kinetochore | |
| Ycr068w | Atg15p | MVB disassembly; vacuolar protein processing | |
| Ydr005c | Maf1p | Transcription factor | |
| Cmd1p | Cmd1p | Calcium ion binding | |
| Ygr082w | Tom20p | Protein import into mitochondrial matrix | |
| Yer087w | Aim10p | Prolyl-tRNA aminoacylation | |
| Ycr088w | Abp1p | Actin cytoskeleton regulation | Cofilin |
| Yel075c | Yel075c | Function unknown |
PI(3,5)P2-specific Binding Proteins
In a previous report by Zhu et al. (9), biotinylated liposomes containing five different phosphatidylinositides were probed to the same yeast proteome microarrays, resulting in 150 phosphoinositide-binding proteins. However, these assays did not include PI(3,5)P2. Given the important role of PI(3,5)P2 in cells, we decided to compare the two datasets as an evaluation of binding specificity. Although five proteins (i.e., Ssb1p, Tos3p, Pau15p, Fmp48p, and Kss1p) that were identified in this study were also reported to interact with PI3P, PI4P, and PI(4,5)P2 (9), we did not observe substantial overlapping hits, indicating that the newly discovered PI(3,5)P2-binding proteins were quite specific. In Zhu's study, ∼39% of the phosphoinositide-binding proteins were, or were predicted to be, membrane associated (9). In comparison, we found that 44 (27%) of the 162 PI(3,5)P2-specific binding proteins were known to be associated with membranes, such as the plasma membrane (2%), ER membrane (4%), Golgi apparatus membrane (1%), and mitochondrial membrane (3%), to name a few (supplementary Information 2). We also identified one GPI anchor protein, similar to the results from the previous screening (9). Other membrane-associated PI(3,5)P2-BPs included Rfs1p, which is involved in membrane rafts. Membrane rafts are lipid microdomains that are enriched in signaling molecules such as sphingolipids and phosphoinositides (48). Interestingly, the STRING analysis revealed that 15 identified PI(3,5)P2-BPs were co-expressed or interacted (edges ≤ 3) with Rfs1p (Fig. 4A). These included three proteins (Yet3p, Yip5p, and Tvp15p) in vesicle-mediated transport and one (Arc18p) in actin regulation. Additionally, an identified PI(3,5)P2-specific binding γ subunit of the trimeric G protein (Gγ), Gpg1p, has been reported to be co-expressed with Rfs1p (49). Gγ is well known to regulate the G protein-coupled receptor (GPCR) by GPI modification and target membrane rafts to trigger its downstream pathways. In our identified list, we also found another membrane protein, Ste3p, which belongs to GPCR. It is therefore conceivable that PI(3,5)P2 might mediate cell responses via GPCR signaling.
Fig. 4.

Bioinformatic analyses of PI(3,5)P2-specific binding proteins. A, Protein–protein interactions among the 162 PI(3,5)P2-BPs revealed by the STRING analysis (version 9.0) (28). The identified proteins that co-expressed or had experimental evidence to interact with Rfs1p (edges ≤ 3) are shown. B, Statistical analysis of enriched Pfam domains. Protein families or domains that were significantly enriched in the set of PI(3,5)P2-specific binding proteins (left) and in the previously reported phosphoinositide-interacting partners (9) (right) are shown (p < 0.05). The histogram indicates the frequency of each enriched domain, i.e., the number of identified proteins classified in the Pfam domain was divided by the number of total proteins in the Pfam domain. C, Functional classification. According to corresponding biological functions, the 162 identified PI(3,5)P2-BPs and 150 phosphoinositide-interacting proteins reported by Zhu et al. (9) were classified into nine groups. Biological processes and functional annotations were retrieved from the UniProt Knowledgebase and Saccharomyces Genome Database.
For overall characterization of these proteins, we first conducted a Pfam domain analysis to discover enriched domains from the identified PI(3,5)P2-BPs and those in Zhu's report (9). A total of ten significantly overrepresented Pfam-defined domains were found in our study (Fig. 4B; left), whereas six were observed in other phosphoinositides from Zhu's study (9) (Fig. 4B; right) (p < 0.05, Fisher's exact test). Interestingly, we discovered that an unknown Pfam domain, Pfam-B_8509, was enriched in both datasets, including two overlapping hits, Tos3p and Fmp48p. In our study, another unknown Pfam domain (Pfam-B_10446) and three membrane-associated domains (LMBR1, Complex1_LYR, and cytochrome b5-like (Cyt-b5) domain) showed significant enrichment. In addition, we also found a Cenp-F_leu_zip (leucine-rich repeats of Cenp-F/LEK1) family that was enriched in the identified PI(3,5)P2-BPs. The Cenp-F family is involved in microtubule binding and is responsible for the kinetochore function in mitosis (50). These findings appeared to indicate that PI(3,5)P2 and other phosphatidylinositides might participate in their currently known functions via the binding of the enriched Pfam domains.
To obtain a deeper insight into the role of PI(3,5)P2 in lipid-mediated cell regulation, the 162 identified PI(3,5)P2-BPs were manually classified into nine groups according to their own biological functions (Fig. 4C; left panel). The functional classification was also compared with that of the other phosphoinositide-binding proteins from the previous report (9) (Fig. 4C; right panel). The distribution of each group was broadly similar between the two datasets, whereas kinases accounted for a higher percentage (14.1%) in the previous screening of other phosphatidylinositides (9). For PI(3,5)P2-binding proteins, most of them were evenly distributed (2.5%–6.8%) throughout the nine groups. In our profiling, 22 (13.6%) PI(3,5)P2-BPs in the vesicle-mediated transport, GTPase, cytoskeleton, and kinase group were thought to have overlapping functions with PI(3,5)P2 (Table II). Herein, we concentrated on the interpretation of these proteins in sections Vesicle-mediated Transport, GTPases and Associated Regulators, Cytoskeleton Regulation, and Kinases.
Vesicle-mediated Transport
Vacuole protein sorting, such as the MVB-sorting pathway, is a crucial mechanism for directing proteins toward vacuoles and transporting them into appropriate positions. Some intracellular biological processes, such as vacuolar acidification, are known to assist in this vesicle-mediated transport (51, 52). PI(3,5)P2 has been reported to be involved in vacuolar acidification and protein sorting into MVBs through the regulation of a variety of proteins (17–19). Remarkably, six (3.7%) newly identified PI(3,5)P2-BPs were responsible for vesicle-mediated transport (Bug1p, Yet3p, and Tvp15p), vacuolar acidification (Vma6p, Rrg1p), and vacuolar protein sorting (Vps1p) (Table II). These results implied that PI(3,5)P2-coordinated cellular transportation may be conferred by the direct interactions of PI(3,5)P2 with these proteins.
GTPases and Associated Regulators
In our PI(3,5)P2-binding protein list, we also found two Rho GTPase-activating proteins (RhoGAP), Bag7p and Lrg1p (Table II). It is intriguing that both Bag7p and Lrg1p are able to activate Rho1p, a key regulator of controlling cell polarization (53). Current evidence shows that Rho1p regulates MPK1-signaling pathway through the binding of protein kinase C 1 (PKC1) to control actin cytoskeleton organization, indicating a possible cascade with which PI(3,5)P2 may be involved (54–56). Other GTPase (Mtg2p) and the GTPase regulator (Yip5p) also showed PI(3,5)P2 binding affinities (Table II).
Cytoskeleton Regulation
A large number of cytoskeletal and actin-related proteins have been reported to interact with PI(3,5)P2 (5). Similarly, six (3.7%) identified PI(3,5)P2-BPs are known to be involved in the cytoskeleton system (Table II). These included three structural proteins of the cytoskeleton (Mps2p, Spc110p, and Duo1p), one protein implicated in microtubule organization (Ats1p) (57), and one protein involved in the mediation of actin polymerization/nucleation (Arc18p) (58). It is likely that PI(3,5)P2 regulated yeast cytoskeleton organization via these interactions.
Kinases
Phosphorylation by kinase activity is a common way to regulate the complex cellular signal transduction. Through the combination of NQF liposomes and the yeast proteome microarray, five kinases, and one kinase activator were identified as PI(3,5)P2-BPs in this study (Table II). The identified Ssk2p, a MAPKKK involved in the yeast HOG pathway, has been shown to mediate an osmo-sensing MAP kinase cascade and to promote actin cytoskeleton recovery following osmotic stress (59–61). It is worth noting that the rapid accumulation of PI(3,5)P2 in yeast occurs when sustaining hyperosmotic stress via activation of the PI3P 5-OH kinase (15). Therefore, PI(3,5)P2 accumulation that is triggered by osmotic stress might provide clues in stress response and actin cytoskeleton organization, presumably through the binding with Ssk2p. Other identified kinases include Kss1p, which is involved in the control of filamentous growth (62). Overall, these results provided potential downstream effectors of PI(3,5)P2 and showed the utility of NQF liposomes.
Motif Search and Docking Analysis
To assess whether novel consensus sequences existed among the newly identified 22 PI(3,5)P2-BPs (Table II), we performed a MEME motif search using their primary amino acid sequences (30). The result showed that 8 of the 22 PI(3,5)P2-BPs shared a consensus sequence, [YS][JA]H[SA][AL]G[IV][IP]HRDIKP[ES]NJLL, in which J represents leucine or isoleucine (E-value = 3.8E-7) (Fig. 5A). To assess whether the motif also showed significance in the set of the 162 PI(3,5)P2-BPs, we used the more conserved part, HRDIKP[ES]NJLL, to map our dataset against the yeast proteome. The result showed that this conserved motif was statistically significant among the 162 PI(3,5)P2-BPs (p value < 0.005). This motif was located within the RhoGAP domain of Lrg1p, the Bap31 domain of Yet3p, the dynamin central region of Vps1p, and the annotated catalytic sites in the PKc-like kinase domains (PSSM-ID: 200772) of several protein kinases (i.e. Tos3p, Fmp48p, Ssk2p, Kss1p, and Cdc15p). Thus far, quite a few kinases have been shown to physically interact with PI(3,5)P2 or to be involved with its metabolism using a variety of high-throughput approaches (5–7, 9). One of the most important kinases in yeast is Fab1p, which phosphorylates PI3P to produce PI(3,5)P2. To further explore the potential significance of this motif, especially in proteins with kinase activity, we scanned the amino acid sequence of Fab1p with the conserved motif. Surprisingly, we discovered a similar motif located in the region (position 800–1500 aa) that interacts with the PI(3,5)P2 regulatory complex (Fig. 5A). This interaction affects the level of PI(3,5)P2 and vacuole morphology (63). Our finding implied that this conserved motif in the 8 PI(3,5)P2-BPs might also interact with the PI(3,5)P2 regulatory complex to affect the level of PI(3,5)P2.
Fig. 5.

Motif search and docking simulation. A, consensus motif identified by MEME (version 4.6.1) (30). Eight of 22 PI(3,5)P2-BPs that had overlapping functions with PI(3,5)P2 contained a consensus sequence, [YS][JA]H[SA][AL]G[IV][IP]HRDIKP[ES]NJLL, where J represents leucine or isoleucine (E-value = 3.8E-7). The circled part showed statistical significance among the 162 PI(3,5)P2-BPs with a p value < 0.005 (Fisher's exact test). The conserved sequences were aligned against Fab1p by ClustalX2 (version 2.0.12) (31) using default settings. Numbers in each parenthesis indicate the start site of the motif in each amino acid sequence. B, The identified PI(3,5)P2-binding kinases were processed by the SwissDock (41) to simulate the molecular interactions. The ribbon diagram shows the secondary structures of the five serine/threonine protein kinases and one kinase activator. The positively charged residues near the PI(3,5)P2 head group docking site are highlighted. The surface representation of each model is also shown in the same orientation (blue, positively charged; red, negatively charged).
Although we identified a conserved motif in the newly identified PI(3,5)P2-BPs, this motif still might not be the binding site of PI(3,5)P2 because it is located at the catalytic site of the protein kinase domain (PSSM-ID: 200772), which is responsible for transferring the γ phosphate of ATP to the substrate proteins. If PI(3,5)P2 were bound to the identified motif, it might inhibit the kinase activity as a competitor. However, this hypothesis is quite unlikely because the resulting recruitment of protein kinases to the membrane microdomain via PI(3,5)P2 would block kinase activity rather than activate downstream signaling. Therefore, we also used docking simulations to identify potential PI(3,5)P2-binding sites of these kinases. Given the fact that PI(3,5)P2 binds to its interacting proteins via the inositol head group, we removed its hydrophobic tail and only used the deprotonated head group to dock against the five protein kinases and one kinase activator (Table II) (21, 64). Meanwhile, we chose seven reported PI(3,5)P2-binding proteins as positive controls (Table III) (65–70). The DSX score system was applied for the assessment of docking significance, and the QMEAN Z-scores were also determined to control the quality of each protein structure created from homology modeling. The potential docking pockets with their DSX scores are summarized in Table III. Other information such as the protein structure sources, the amino acid fragments that were covered by the protein models, the QMEAN Z-scores, and the PDB codes of the modeled protein structures are summarized in the supplementary Information S4. To our surprise, all of the kinases received a score close to those of the 7 positive controls, suggesting that the identified PI(3,5)P2-binding kinases were of high confidence. None of the identified docking pockets in any kinase shared homology with the known PIP-binding motifs. Although we did not recover any conserved binding domain from among the kinases, our docking model indicated that the PI(3,5)P2-kinase interactions would predominantly be achieved through the binding of lysine and arginine side chains to the phosphate groups on the PI(3,5)P2 inositol head (Fig. 5B). This phenomenon is similar to the reported docking of other phosphatidylinositides to kinases (64). Consistently, the electrostatic interactions between PI(3,5)P2 and positively charged residues play a major role in PI(3,5)P2-protein interactions.
Table III. Molecular docking of PI(3,5)P2 to the identified PI(3,5)P2-binding kinases.
| Ordered locus name | Protein name | DSXa |
|---|---|---|
| Ybr135w | Cks1p | –79.674 |
| Ygl105wb | Arc1p | –64.801 |
| Ykr003wb | Osh6p | –59.202 |
| Yjr125cb | Ent3p | –56.153 |
| Ygl179c | Tos3p | –54.999 |
| Ypl232wb | Sso1p | –54.343 |
| Ydr153cb | Ent5p | –53.411 |
| Ygr052w | Fmp48p | –52.078 |
| Ybr260cb | Rgd1p | –51.512 |
| Yhr193cb | Egd2p | –51.453 |
| Ynr031c | Ssk2p | –50.359 |
| Ygr040w | Kss1p | –50.186 |
| Yar019c | Cdc15p | –47.694 |
a Lower the DSX showed, higher the docking success obtained.
b Reported PI(3,5)P2-binding proteins were employed as positive controls.
DISCUSSION
Many signal transduction and metabolic pathways are coordinated by lipid–protein interactions. It is therefore important to develop appropriate membrane models to monitor the molecular interactions in vitro. Liposomes are one of the most common tools used in signal amplification and lipid research. Nevertheless, conventional liposomes for signal amplification that are composed of a large number of fluorescent molecules usually result in self-quenching problems and cannot be detected without the lysis of the liposomes, thus causing limited applications. In this study, we successfully developed an NQF liposome by optimizing the proportion of SRB encapsulant and LRB-DPPE on the liposomal surface for signal amplification. Note that when 0.3% LRB-DPPE was incorporated in the liposomal bilayer, it provided higher signals than the 200 μm SRB-encapsulated liposomes without LRB-DPPE by ∼5.3-fold. The capability of the NQF liposomes to enhance the signal intensity without lysis should extend its application in biosensing technologies and investigations of lipid interactions.
To show the usefulness of the NQF liposomes, we incorporated PI(3,5)P2 into the phospholipid bilayer to fabricate the PI(3,5)P2-NQF liposome, which was then applied to probe yeast proteome microarrays for identifying PI(3,5)P2-binding proteins. Herein, a total of 162 PI(3,5)P2-BPs and 12 proteins that targeted both PI(3,5)P2 and PI were identified. Three proteins that bound to both PI(3,5)P2 and PI in this study possessed previously reported phosphoinositide-binding domains (20, 22, 23). Additionally, the domain annotations of the PI(3,5)P2-BPs were also examined in this study. We found that three PI(3,5)P2-BPs (Met30p, Rtt10p, and Dug2p) contained the known PI(3,5)P2-specific targeting motif, WD-40, and one (Rim8p) contained an arrestin structure that binds to phosphoinositides (20) (supplementary Information S2). To further investigate the correlation between PI(3,5)P2 and the identified proteins, an analysis of human homologs was conducted using the KEGG Sequence Similarity DataBase to compare with the known PI(3,5)P2-binding proteins in humans. Catimel et al. (5, 6) conducted a large-scale screening of phosphoinositide-binding proteins from human colonic carcinoma cell lines using affinity assays and LC/MS/MS. Consistent with this report, six human homologs of identified PI(3,5)P2-BPs were found to overlap with the previously reported human phosphoinositide-interacting proteins (5, 6) (supplementary Information S2). All of these findings indicated positive results in this proteome chip-based assay and strongly supported the feasibility of the NQF liposome as a model for studying lipid–protein interactions.
To date, a total of 8 proteins have been experimentally verified to be “PI(3,5)P2 binding” in yeast according to the Saccharomyces Genome Database; however, none of the 8 proteins overlapped with our dataset (162 PI(3,5)P2-BPs). To address this issue, we conducted an assay by probing the DyLightTM549-conjugated anti-GST antibody on the yeast proteome microarray to determine whether the 8 proteins were covered by the chip. It turned out that only 3 of the 8 proteins (Egd2p, Rgd1p, and Tup1p) were available on the chip. All 3 proteins showed moderate PI(3,5)P2-binding signals on the yeast proteome microarrays, but the signals were lower than the cut-off that we set. According to our literature searches, Egd2p has been reported to have a stronger binding affinity to PI3P and only have moderate binding affinity to PI(3,5)P2 (67). Additionally, it has been reported that the N-terminal region of Rgd1p is required for interaction with PI(3,5)P2 (69). Given that our proteins in the microarray contained GST-fusions at the N terminus, it is thus likely that some potential PI(3,5)P2-binding proteins like Rgd1p were not identified in this study. Tup1p is the most recently identified PI(3,5)P2-binding protein (71), and thus there is a lack of information about the binding sites or interaction kinetics between Tup1p and PI(3,5)P2.
From a systematic point of view, we categorized the 162 PI(3,5)P2-BPs into nine groups, and, according to this profiling, the biological functions of 22 identified PI(3,5)P2-BPs were highly relevant to the functional role of PI(3,5)P2, including protein sorting, vacuolar acidification and cytoskeleton organization. The functionally uncharacterized proteins and the proteins with scattered functions were classified into “Others” (Fig. 4C). A total of 30 (18.5%) identified PI(3,5)P2-BPs were annotated to be “uncharacterized” according to the Saccharomyces genome database (72). One possible explanation for the identification of such percentage of functionally uncharacterized proteins is that until now the yeast proteome has not been systematically screened for binding property to PI(3,5)P2 because of its poor solubility and a lack of detection reagents. In addition, membrane proteins are notoriously difficult to work with. Using the newly developed detection method, coupled with the proteome microarray technology, we were able to screen for PI(3,5)P2-interactors on a proteomic scale in an unbiased fashion. That may partially explain this phenomenon. Similarly, many uncharacterized yeast proteins were also identified in a similar screen that was performed by Zhu et al. in 2001 (9). Among the functionally characterized PI(3,5)P2-BPs, interestingly, some proteins contained more than one function related to PI(3,5)P2. Vps1p, for example, is a vacuolar-sorting protein that is also important for protein retention and actin cytoskeleton organization (73), revealing a strong connection with PI(3,5)P2. Another example is Mlc1p, a cytoskeleton protein that is also required for vesicle targeting and secretion (74). Additionally, several newly identified PI(3,5)P2-binding GTPases have also been implicated in cytoskeleton regulation (Bag7p, Lrg1p) and vesicle-mediated transport (Yip5p) (75).
We also examined the functional enrichment of the PI(3,5)P2-BPs within biological networks. GO categories that were statistically overrepresented (p value ≤ 0.001) in the yeast protein interacting network were illustrated by GO trees (GO level ≥ 5) (supplementary Information S5). Generally, predominant functional classes included the protein phosphorylation (yellow), cytoskeleton organization (green), protein sorting (red), macromolecular complex assembly (blue), and other functions (gray). Among them, interestingly, the TOR signaling cascade (GO: 0031929) was enriched in the identified 162 PI(3,5)P2-BPs. In yeast, the TOR complex has been implicated to be an important regulator of cytoskeleton (76). In mammals, mTOR is a member of the PI3/PI4-kinase family that is mediated by the Akt pathway, and it has been reported to modulate the actin cytoskeleton (77). The overrepresentation of the TOR signaling cascade in our network analysis indicated a potential pathway associated with PI(3,5)P2.
We performed a docking simulation to show the identified PI(3,5)P2-kinase interactions (Fig. 5B). Our results showed that the PI(3,5)P2 head group was successfully docked into the five kinases and one kinase activator compared with the currently known PI(3,5)P2-binding proteins. According to the simulation, these interactions were achieved through the binding of lysine and arginine side chains to the phosphate groups on PI(3,5)P2. It is well known that certain factors can affect the accuracy of the docking simulations, including the input protein structure (78), the docking algorithm (79), and the scoring function that was applied to score the docking poses (80). In this study, the main limitation to the accuracy of the docking simulations was the input protein structures. To retrieve a more accurate receptor-ligand binding pose, the input structures should contain an actual docking site that was in the binding conformation. This could have compromised the results of the docking simulation.
Zhu et al. (9) performed a global profiling of PI3P, PI4P, PI(3,4)P2, PI(4,5)P2, and PI(3,4,5)P3 using biotinylated liposomes in yeast proteome microarrays. Alternatively, we used NQF liposomes to straightforwardly detect signals from microarrays rather than through the binding of a biotin-streptavidin dye. Because Zhu et al. did not include PI(3,5)P2, we employed a similar approach to globally characterize binding proteins of PI(3,5)P2 using the newly developed NQF liposomes, which resulted in the identification of a large number of novel PI(3,5)P2-binding proteins. We then implemented a bead-based affinity assay to validate the identified PI(3,5)P2-protein interactions. As presented, we successfully showed the interactions and further confirmed the feasibility of the NQF liposomes by application of conventional quenched liposomes. A bioinformatics analysis of the results obtained in this study indicated that PI(3,5)P2 may be involved in many additional cellular functions that are yet to be fully understood. It is expected that our study will serve as a stepping stone to fully elucidate the biological significance of PI(3,5)P2 and the relevant downstream signaling cascades. In conclusion, the newly developed NQF liposomes provide an excellent membrane model for investigations into lipid–protein interactions with the capability of signal amplification in the future.
Supplementary Material
Acknowledgments
We thank the Department of Food Science at National Taiwan Ocean University for providing the instruments and Yu-Chieh Wang for engaging the discussion and providing helpful suggestions.
Footnotes
* This research was supported in part by the National Science Council, Taiwan (NSC99-2627-M-008-003, NSC100-2320-B-008-001, and NSC100-2627-M-008-003 to C-S Chen), Veterans General Hospitals and University System of Taiwan (VGHUST99-G4-1 to C-S Chen), Cathy General Hospital, Taiwan (99CGH-NCU-A3 and 100CGH-NCU-A3 to C-S Chen), Landseed Hospital, Taiwan (NCU-LSH-100-A-012), the Ministry of Education, Taiwan, the State Key Development Program for Basic Research of China (2010CB529205 to C-S Tao), the Program for New Century Excellent Talents in University (NCET-09-551 to C-S Tao), the Shanghai “Phosphor” Science Foundation (10QA1403800 to C-S Tao), the National Natural Science Foundation (31000388 to C-S Tao), SRF for ROCS, S.E. (C-S Tao), and the NIH (R01 GM076102 and U54 RR020839 to H. Zhu).
 This article contains supplemental Information S1 to S5.
This article contains supplemental Information S1 to S5.
1 The abbreviations used are:
- NQF
- nonquenched fluorescence
- SRB
- sulforhodamine B
- LRB-DPPE
- lissamine rhodamine B-dipalmitoyl phosphatidylethanol
- PI(3,5)P2
- phosphatidylinositol 3,5 bisphosphate
- MVB
- multivesicular bodies.
REFERENCES
- 1. Annie Ho J. A., Wu L. C., Chang L. H., Hwang K. C., Reuben Hwu J. R. (2009) Liposome-based immunoaffinity chromatographic assay for the quantitation of immunoglobulin E in human serum. J. Chromatogr. B 878, 172–176 [DOI] [PubMed] [Google Scholar]
- 2. Chen C. S., Baeumner A. J., Durst R. A. (2005) Protein G-liposomal nanovesicles as universal reagents for immunoassays. Talanta 67, 205–211 [DOI] [PubMed] [Google Scholar]
- 3. Edwards K. A., Wang Y., Baeumner A. J. (2010) Aptamer sandwich assays: human α-thrombin detection using liposome enhancement. Anal Bioanal Chem. 398, 2645–2654 [DOI] [PubMed] [Google Scholar]
- 4. Ho J. A., Wu L. C., Huang M. R., Lin Y. J., Baeumner A. J., Durst R. A. (2007) Application of ganglioside-sensitized liposomes in a flow injection immunoanalytical system for the determination of cholera toxin. Anal. Chem. 79, 246–250 [DOI] [PubMed] [Google Scholar]
- 5. Catimel B., Schieber C., Condron M., Patsiouras H., Connolly L., Catimel J., Nice E. C., Burgess A. W., Holmes A. B. (2008) The PI(3,5)P2 and PI(4,5)P2 interactomes. J. Proteome Res. 7, 5295–5313 [DOI] [PubMed] [Google Scholar]
- 6. Catimel B., Yin M. X., Schieber C., Condron M., Patsiouras H., Catimel J., Robinson D. E., Wong L. S., Nice E. C., Holmes A. B., Burgess A. W. (2009) PI(3,4,5)P3 Interactome. J. Proteome Res. 8, 3712–3726 [DOI] [PubMed] [Google Scholar]
- 7. Gallego O., Betts M. J., Gvozdenovic-Jeremic J., Maeda K., Matetzki C., Aguilar-Gurrieri C., Beltran-Alvarez P., Bonn S., Fernandez-Tornero C., Jensen L. J., Kuhn M., Trott J., Rybin V., Muller C. W., Bork P., Kaksonen M., Russell R. B., Gavin A. C. (2010) A systematic screen for protein–lipid interactions in Saccharomyces cerevisiae. Mol. Syst. Biol. 6, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peetla C., Stine A., Labhasetwar V. (2009) Biophysical interactions with model lipid membranes: applications in drug discovery and drug delivery. Mol. Pharm. 6, 1264–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu H., Bilgin M., Bangham R., Hall D., Casamayor A., Bertone P., Lan N., Jansen R., Bidlingmaier S., Houfek T., Mitchell T., Miller P., Dean R. A., Gerstein M., Snyder M. (2001) Global analysis of protein activities using proteome chips. Science 293, 2101–2105 [DOI] [PubMed] [Google Scholar]
- 10. Michell R. H., Heath V. L., Lemmon M. A., Dove S. K. (2006) Phosphatidylinositol 3,5-bisphosphate: metabolism and cellular functions. Trends Biochem. Sci 31, 52–63 [DOI] [PubMed] [Google Scholar]
- 11. Martin T. F. (1998) Phosphoinositide lipids as signaling molecules: common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu. Rev. Cell Dev. Biol. 14, 231–264 [DOI] [PubMed] [Google Scholar]
- 12. De Camilli P., Emr S. D., McPherson P. S., Novick P. (1996) Phosphoinositides as regulators in membrane traffic. Science 271, 1533–1539 [DOI] [PubMed] [Google Scholar]
- 13. Di Paolo G., De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657 [DOI] [PubMed] [Google Scholar]
- 14. Strahl T., Thorner J. (2007) Synthesis and function of membrane phosphoinositides in budding yeast, Saccharomyces cerevisiae. Biochim. Biophys. Acta 1771, 353–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dove S. K., Cooke F. T., Douglas M. R., Sayers L. G., Parker P. J., Michell R. H. (1997) Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature 390, 187–192 [DOI] [PubMed] [Google Scholar]
- 16. Whiteford C. C., Brearley C. A., Ulug E. T. (1997) Phosphatidylinositol 3,5-bisphosphate defines a novel PI 3-kinase pathway in resting mouse fibroblasts. Biochem. J. 323, 597–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Odorizzi G., Babst M., Emr S. D. (1998) Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell 95, 847–858 [DOI] [PubMed] [Google Scholar]
- 18. Bonangelino C. J., Catlett N. L., Weisman L. S. (1997) Vac7p, a novel vacuolar protein, is required for normal vacuole inheritance and morphology. Mol. Cell. Biol. 17, 6847–6858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dove S. K., McEwen R. K., Mayes A., Hughes D. C., Beggs J. D., Michell R. H. (2002) Vac14 controls PtdIns(3,5)P(2) synthesis and Fab1-dependent protein trafficking to the multivesicular body. Curr. Biol. 12, 885–893 [DOI] [PubMed] [Google Scholar]
- 20. Balla T. (2005) Inositol–lipid binding motifs: signal integrators through protein–lipid and protein–protein interactions. J. Cell Sci. 118, 2093–2104 [DOI] [PubMed] [Google Scholar]
- 21. Lemmon M. A. (2003) Phosphoinositide recognition domains. Traffic 4, 201–213 [DOI] [PubMed] [Google Scholar]
- 22. Mehrotra B., Myszka D. G., Prestwich G. D. (2000) Binding kinetics and ligand specificity for the interactions of the C2B domain of synaptogmin II with inositol polyphosphates and phosphoinositides. Biochemistry 39, 9679–9686 [DOI] [PubMed] [Google Scholar]
- 23. Dove S. K., Piper R. C., McEwen R. K., Yu J. W., King M. C., Hughes D. C., Thuring J., Holmes A. B., Cooke F. T., Michell R. H., Parker P. J., Lemmon M. A. (2004) Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. EMBO J. 23, 1922–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bartlett G. R. (1959) Phosphorus assay in column chromatography. J. Biol. Chem. 234, 466–468 [PubMed] [Google Scholar]
- 25. Davidson W. S., Ghering A. B., Beish L., Tubb M. R., Hui D. Y., Pearson K. (2006) The biotin-capture lipid affinity assay: a rapid method for determining lipid binding parameters for apolipoproteins. J. Lipid Res. 47, 440–449 [DOI] [PubMed] [Google Scholar]
- 26. Scholler N., Crawford M., Sato A., Drescher C. W., O'Briant K. C., Kiviat N., Anderson G. L., Urban N. (2006) Bead-based ELISA for validation of ovarian cancer early detection markers. Clin. Cancer Res. 12, 2117–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schwenk J. M., Lindberg J., Sundberg M., Uhlén M., Nilsson P. (2007) Determination of binding specificities in highly multiplexed bead-based assays for antibody proteomics. Mol. Cell. Proteomics 6, 125–132 [DOI] [PubMed] [Google Scholar]
- 28. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., von Mering C. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Finn R. D., Mistry J., Tate J., Coggill P., Heger A., Pollington J. E., Gavin O. L., Gunasekaran P., Ceric G., Forslund K., Holm L., Sonnhammer E. L., Eddy S. R., Bateman A. (2010) The Pfam protein families database. Nucleic Acids Res. 38, D211–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bailey T. L., Elkan C. (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Int. Conf. Intelligent Syst. Mol. Biol. 2, 28–36 [PubMed] [Google Scholar]
- 31. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 32. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 34. Arnold K., Kiefer F., Kopp J., Battey J. N., Podvinec M., Westbrook J. D., Berman H. M., Bordoli L., Schwede T. (2009) The protein model portal. J. Struct. Funct. Genomics 10, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Benkert P., Biasini M., Schwede T. (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benkert P., Tosatto S. C., Schomburg D. (2008) QMEAN: A comprehensive scoring function for model quality assessment. Proteins 71, 261–277 [DOI] [PubMed] [Google Scholar]
- 37. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eswar N., John B., Mirkovic N., Fiser A., Ilyin V. A., Pieper U., Stuart A. C., Marti-Renom M. A., Madhusudhan M. S., Yerkovich B., Sali A. (2003) Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 31, 3375–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Y., Xiao J., Suzek T. O., Zhang J., Wang J., Bryant S. H. (2009) PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 37, W623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guha R., Howard M. T., Hutchison G. R., Murray-Rust P., Rzepa H., Steinbeck C., Wegner J., Willighagen E. L. (2006) The Blue Obelisk-interoperability in chemical informatics. J. Chem. Inf. Model. 46, 991–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grosdidier A., Zoete V., Michielin O. (2011) SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 39, W270–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Neudert G., Klebe G. (2011) DSX: a knowledge-based scoring function for the assessment of receptor-ligand complexes. J. Chem. Inf. Model. 51, 2731–2745 [DOI] [PubMed] [Google Scholar]
- 43. Drubin D. G., Mulholland J., Zhu Z. M., Botstein D. (1990) Homology of a yeast actin-binding protein to signal transduction proteins and myosin-I. Nature 343, 288–290 [DOI] [PubMed] [Google Scholar]
- 44. Goode B. L., Rodal A. A., Barnes G., Drubin D. G. (2001) Activation of the Arp2/3 complex by the actin filament binding protein Abp1p. J. Cell Biol. 153, 627–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Friant S., Pécheur E. I., Eugster A., Michel F., Lefkir Y., Nourrisson D., Letourneur F. (2003) Ent3p Is a PtdIns(3,5)P2 effector required for protein sorting to the multivesicular body. Dev. Cell 5, 499–511 [DOI] [PubMed] [Google Scholar]
- 46. Powers J., Barlowe C. (2002) Erv14p directs a transmembrane secretory protein into COPII-coated transport vesicles. Mol. Biol. Cell 13, 880–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Epple U. D., Eskelinen E. L., Thumm M. (2003) Intravacuolar membrane lysis in Saccharomyces cerevisiae. Does vacuolar targeting of Cvt17/Aut5p affect its function? J. Biol. Chem. 278, 7810–7821 [DOI] [PubMed] [Google Scholar]
- 48. Pike L. J. (2003) Lipid rafts: bringing order to chaos. J. Lipid Res. 44, 655–667 [DOI] [PubMed] [Google Scholar]
- 49. Uetz P., Giot L., Cagney G., Mansfield T. A., Judson R. S., Knight J. R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Qureshi-Emili A., Li Y., Godwin B., Conover D., Kalbfleisch T., Vijayadamodar G., Yang M., Johnston M., Fields S., Rothberg J. M. (2000) A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 403, 623–627 [DOI] [PubMed] [Google Scholar]
- 50. Liao H., Winkfein R. J., Mack G., Rattner J. B., Yen T. J. (1995) CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J. Cell Biol. 130, 507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mellman I., Fuchs R., Helenius A. (1986) Acidification of the endocytic and exocytic pathways. Annu. Rev. Biochem. 55, 663–700 [DOI] [PubMed] [Google Scholar]
- 52. Yamashiro C. T., Kane P. M., Wolczyk D. F., Preston R. A., Stevens T. H. (1990) Role of vacuolar acidification in protein sorting and zymogen activation: a genetic analysis of the yeast vacuolar proton-translocating ATPase. Mol. Cell. Biol. 10, 3737–3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Roumanie O., Weinachter C., Larrieu I., Crouzet M., Doignon F. (2001) Functional characterization of the Bag7, Lrg1 and Rgd2 RhoGAP proteins from Saccharomyces cerevisiae. FEBS Lett. 506, 149–156 [DOI] [PubMed] [Google Scholar]
- 54. Kamada Y., Qadota H., Python C. P., Anraku Y., Ohya Y., Levin D. E. (1996) Activation of yeast protein kinase C by Rho1 GTPase. J. Biol. Chem. 271, 9193–9196 [DOI] [PubMed] [Google Scholar]
- 55. Nonaka H., Tanaka K., Hirano H., Fujiwara T., Kohno H., Umikawa M., Mino A., Takai Y. (1995) A downstream target of RHO1 small GTP-binding protein is PKC1, a homolog of protein kinase C, which leads to activation of the MAP kinase cascade in Saccharomyces cerevisiae. EMBO J. 14, 5931–5938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schmidt A., Schmelzle T., Hall M. N. (2002) The RHO1-GAPs SAC7, BEM2 and BAG7 control distinct RHO1 functions in Saccharomyces cerevisiae. Mol. Microbiol. 45, 1433–1441 [DOI] [PubMed] [Google Scholar]
- 57. Kirkpatrick D., Solomon F. (1994) Overexpression of yeast homologs of the mammalian checkpoint gene RCC1 suppresses the class of α-tubulin mutations that arrest with excess microtubules. Genetics 137, 381–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Winter D., Podtelejnikov A. V., Mann M., Li R. (1997) The complex containing actin-related proteins Arp2 and Arp3 is required for the motility and integrity of yeast actin patches. Curr. Biol. 7, 519–529 [DOI] [PubMed] [Google Scholar]
- 59. Yuzyuk T., Foehr M., Amberg D. C. (2002) The MEK kinase Ssk2p promotes actin cytoskeleton recovery after osmotic stress. Mol. Biol. Cell 13, 2869–2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tatebayashi K., Takekawa M., Saito H. (2003) A docking site determining specificity of Pbs2 MAPKK for Ssk2/Ssk22 MAPKKKs in the yeast HOG pathway. EMBO J. 22, 3624–3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Maeda T., Wurgler-Murphy S. M., Saito H. (1994) A two-component system that regulates an osmosensing MAP kinase cascade in yeast. Nature 369, 242–245 [DOI] [PubMed] [Google Scholar]
- 62. Cook J. G., Bardwell L., Thorner J. (1997) Inhibitory and activating functions for MAPK Kss1 in the S. cerevisiae filamentous-growth signalling pathway. Nature 390, 85–88 [DOI] [PubMed] [Google Scholar]
- 63. Botelho R. J., Efe J. A., Teis D., Emr S. D. (2008) Assembly of a Fab1 phosphoinositide kinase signaling complex requires the Fig 4 phosphoinositide phosphatase. Mol. Biol. Cell 19, 4273–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Moravcevic K., Mendrola J. M., Schmitz K. R., Wang Y. H., Slochower D., Janmey P. A., Lemmon M. A. (2010) Kinase associated-1 domains drive MARK/PAR1 kinases to membrane targets by binding acidic phospholipids. Cell 143, 966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chidambaram S., Mullers N., Wiederhold K., Haucke V., von Mollard G. F. (2004) Specific interaction between SNAREs and epsin N-terminal homology (ENTH) domains of epsin-related proteins in trans-Golgi network to endosome transport. J. Biol. Chem. 279, 4175–4179 [DOI] [PubMed] [Google Scholar]
- 66. Eugster A., Pecheur E. I., Michel F., Winsor B., Letourneur F., Friant S. (2004) Ent5p is required with Ent3p and Vps27p for ubiquitin-dependent protein sorting into the multivesicular body. Mol. Biol. Cell 15, 3031–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fernandez-Murray J. P., McMaster C. R. (2006) Identification of novel phospholipid binding proteins in Saccharomyces cerevisiae. FEBS Lett. 580, 82–86 [DOI] [PubMed] [Google Scholar]
- 68. Mendonsa R., Engebrecht J. (2009) Phosphatidylinositol-4,5-bisphosphate and phospholipase D-generated phosphatidic acid specify SNARE-mediated vesicle fusion for prospore membrane formation. Eukaryotic Cell 8, 1094–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Prouzet-Mauléon V., Lefebvre F., Thoraval D., Crouzet M., Doignon F. (2008) Phosphoinositides affect both the cellular distribution and activity of the F-BAR-containing RhoGAP Rgd1p in yeast. J. Biol. Chem. 283, 33249–33257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang P., Duan W., Munn A. L., Yang H. (2005) Molecular characterization of Osh6p, an oxysterol binding protein homolog in the yeast Saccharomyces cerevisiae. FEBS J. 272, 4703–4715 [DOI] [PubMed] [Google Scholar]
- 71. Han B. K., Emr S. D. (2011) Phosphoinositide [PI(3,5)P2] lipid-dependent regulation of the general transcriptional regulator Tup1. Genes Dev. 25, 984–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cherry J. M., Hong E. L., Amundsen C., Balakrishnan R., Binkley G., Chan E. T., Christie K. R., Costanzo M. C., Dwight S. S., Engel S. R., Fisk D. G., Hirschman J. E., Hitz B. C., Karra K., Krieger C. J., Miyasato S. R., Nash R. S., Park J., Skrzypek M. S., Simison M., Weng S., Wong E. D. (2012) Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 40, D700–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wilsbach K., Payne G. S. (1993) Vps1p, a member of the dynamin GTPase family, is necessary for Golgi membrane protein retention in Saccharomyces cerevisiae. EMBO J. 12, 3049–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wagner W., Bielli P., Wacha S., Ragnini-Wilson A. (2002) Mlc1p promotes septum closure during cytokinesis via the IQ motifs of the vesicle motor Myo2p. EMBO J. 21, 6397–6408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Inadome H., Noda Y., Kamimura Y., Adachi H., Yoda K. (2007) Tvp38, Tvp23, Tvp18 and Tvp15: novel membrane proteins in the Tlg2-containing Golgi/endosome compartments of Saccharomyces cerevisiae. Exp. Cell Res. 313, 688–697 [DOI] [PubMed] [Google Scholar]
- 76. Aronova S., Wedaman K., Anderson S., Yates J., 3rd, Powers T. (2007) Probing the membrane environment of the TOR kinases reveals functional interactions between TORC1, actin, and membrane trafficking in Saccharomyces cerevisiae. Mol. Biol. Cell. 18, 2779–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kim D. H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 [DOI] [PubMed] [Google Scholar]
- 78. Fan H., Irwin J. J., Webb B. M., Klebe G., Shoichet B. K., Sali A. (2009) Molecular docking screens using comparative models of proteins. J. Chem. Inf. Modeling 49, 2512–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sperandio O., Miteva M. A., Delfaud F., Villoutreix B. O. (2006) Receptor-based computational screening of compound databases: the main docking-scoring engines. Curr. Protein Peptide Sci. 7, 369–393 [DOI] [PubMed] [Google Scholar]
- 80. Coupez B., Lewis R. A. (2006) Docking and scoring–theoretically easy, practically impossible? Curr. Med. Chem. 13, 2995–3003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.