

Abstract
Lipoprotein lipase (LPL) is secreted into the interstitial spaces by adipocytes and myocytes but then must be transported to the capillary lumen by GPIHBP1, a glycosylphosphatidylinositol-anchored protein of capillary endothelial cells. The mechanism by which GPIHBP1 and LPL move across endothelial cells remains unclear. We asked whether the transport of GPIHBP1 and LPL across endothelial cells was uni- or bidirectional. We also asked whether GPIHBP1 and LPL are transported across cells in vesicles and whether this transport process requires caveolin-1. The movement of GPIHBP1 and LPL across cultured endothelial cells was bidirectional. Also, GPIHBP1 moved bidirectionally across capillary endothelial cells in live mice. The transport of LPL across endothelial cells was inhibited by dynasore and genistein, consistent with a vesicular transport process. Also, transmission electron microscopy (EM) and dual-axis EM tomography revealed GPIHBP1 and LPL in invaginations of the plasma membrane and in vesicles. The movement of GPIHBP1 across capillary endothelial cells was efficient in the absence of caveolin-1, and there was no defect in the internalization of LPL by caveolin-1-deficient endothelial cells in culture. Our studies show that GPIHBP1 and LPL move bidirectionally across endothelial cells in vesicles and that transport is efficient even when caveolin-1 is absent.
Keywords: transcytosis, triglycerides, caveolae, chylomicrons, lipid metabolism
The lipolytic processing of triglyceride-rich lipoproteins (e.g., chylomicrons, very low density lipoproteins) by lipoprotein lipase (LPL) is the central event in plasma triglyceride metabolism and plays a crucial role in the delivery of lipid nutrients to parenchymal cells (e.g., adipocytes, myocytes) (1–4). LPL is synthesized by parenchymal cells and secreted into the interstitial spaces, but it needs to reach the capillary lumen in order to hydrolyze the triglycerides in plasma lipoproteins. Recent studies showed that GPIHBP1, a glycosylphosphatidylinositol-anchored protein of capillary endothelial cells, binds LPL avidly (5) and is required for the transport of LPL to the capillary lumen (6). In the absence of GPIHBP1, LPL accumulates in the interstitial spaces around parenchymal cells and is unable to process triglyceride-rich lipoproteins in the bloodstream, resulting in markedly elevated plasma triglyceride levels and interfering with the delivery of lipid nutrients to parenchymal cells (5–7).
Although GPIHBP1 is essential for the delivery of LPL to the luminal face of capillaries, the cellular mechanisms for moving GPIHBP1 and LPL across endothelial cells are poorly defined. It is unclear whether GPIHBP1 and LPL move unidirectionally from the basolateral face of endothelial cells to LPL's site of action along the capillary lumen or whether “backwards trafficking” also occurs (i.e., movement of GPIHBP1 and LPL from the lumen to the basolateral face of cells). A second issue is whether GPIHBP1 and LPL are transported across cells in vesicles [as has been reported to be the case for albumin and its binding partner gp60 (8–10)] and, if so, whether this transport is dependent on caveolin-1, a caveolar protein. Here, we addressed these issues with a combination of in vitro studies in cultured cells and in vivo studies in wild-type and genetically modified mice.
MATERIALS AND METHODS
Gpihbp1−/− mice have been described previously (5). Cav1−/− mice (11) were obtained from the Jackson Laboratory. Mice were fed a chow diet and housed in a barrier facility with a 12 h light-dark cycle. All studies were approved by UCLA's Animal Research Committee.
Rat heart microvessel endothelial cells (RHMVEC) were purchased from VEC Technologies. Lung endothelial cells derived from Cav1+/+ and Cav1−/− mice (12) were a gift from Dr. Carlos Fernández-Hernando (Yale University). Endothelial cells were grown in MCDB-131 complete medium (VEC Technologies). Lentiviruses encoding S-protein-tagged versions of GPIHBP1 and CD59 (13) were transduced into endothelial cells as described (6).
Human LPL was concentrated from the medium of a Chinese hamster ovary (CHO) cell line stably expressing V5-tagged human LPL (14). LPL transport assays were performed as described (6). Briefly, RHMVECs were grown on transwell filters (polyethylene terephthalate, 1.1-cm2 filtration area, 1 μm pore size; Millipore) that had been coated with 50 μg/ml fibronectin (BD Biosciences). Cells were grown until they formed tight monolayers and did not leak medium over a 24 h period. V5-tagged human LPL (hLPL) was added to either the apical (upper) or basolateral (lower) medium, and PBS was added to the other chamber. Rabbit immunoglobulin was added into the medium along with LPL, which allowed us to detect leakage of medium from one chamber to the other. In some experiments, cells were treated with genistein (Sigma), dynasore (Sigma), or vehicle (DMSO) alone. After 1–2 h at 37°C, LPL on the cell surface was released with heparin (100 U/ml; APP Pharmaceuticals). Apical and basolateral samples were dot-blotted onto a nitrocellulose membrane. LPL was detected with a mouse monoclonal antibody (5D2) against LPL (15) (a gift from Dr. John Brunzell, University of Washington) at a 1:200 dilution. Secondary antibody dilutions were 1:500 for an IRDye800-labeled donkey anti-mouse IgG and an IRDye680-labeled donkey anti-rabbit IgG (both from LI-COR Biosciences). All blots were scanned and quantified with the Odyssey infrared system (LI-COR).
To measure transport of GPIHBP1 across endothelial cells in vivo, Alexa555- (Invitrogen) or CF568- (Biotium) labeled antibody 11A12 (a rat monoclonal antibody against mouse GPIHBP1) (14) was injected into the brown adipose tissue (BAT) of wild-type, Gpihbp1−/−, or Cav1−/− mice. After 20–120 min, the mice were euthanized, perfused with PBS, and fixed with 4% paraformaldehyde (PFA). In some experiments, mice were injected intravenously with 40 µg of CF568-labeled antibody 11A12. After waiting for 15 or 120 min, the mice were perfused with PBS and fixed in situ with 4% PFA. BAT was then embedded in OCT and processed for immunohistochemistry.
Immunohistochemistry was performed on 10 µm thick frozen sections on glass slides. Sections were fixed with 4% PFA, permeablized twice with 0.2% Triton X-100 in PBS for 30 min, and then incubated in blocking buffer containing 10% donkey serum and 0.2% Triton X-100 in PBS for 30 min at room temperature. The sections were incubated with either a hamster antibody against CD31 (1:100; Millipore), a goat antibody against mouse LPL (1:100) (16), a goat antibody against podocalyxin (1:200; R and D systems), or a rabbit antibody against caveolin-1 (1:500) in blocking buffer containing 0.2% Triton X-100 at 4°C overnight. Slides were incubated with secondary antibodies, an Alexa488-labeled anti-goat IgG (1:500; Invitrogen) or a Dylight 488-labeled anti-hamster IgG (1:200; Jackson ImmunoResearch), at room temperature for 1 h. Slides were also stained with DAPI to identify nuclei. Confocal fluorescence microscopy was performed with a Zeiss LSM700 laser-scanning microscope. Fluorescence signals were quantified with ImageJ software (17). A line was drawn over the luminal or basolateral membrane, and the integrated density was measured with the “Measure” command. Integrated density was divided by area and corrected for background. Background was calculated by measuring the integrated density of a line drawn between the basolateral and luminal membranes. For each condition, fluorescence signals in 6–10 capillary cross-sections were recorded.
Electron microscopy (EM) studies of cultured cells and mouse tissues were performed with monoclonal antibody 11A12 conjugated to 6 nm gold beads (Electron Microscopy Supplies). For cell culture studies, the antibody was added to the apical surface of cells and incubated for 2 h at 37°C. In other experiments, the apical face of endothelial cells was incubated with V5-tagged human LPL for 1 h at 37°C, washed, and then incubated for 1 h at 37°C with an anti-V5 tag-specific monoclonal antibody conjugated to 6 nm gold beads. For all experiments, cells were fixed in 2.5% glutaraldehyde in 100 mM cacodylate buffer (pH 7.4). To detect GPIHBP1 transport in mouse tissues, gold-conjugated 11A12 was injected directly into the quadriceps of wild-type or Gpihbp1−/− mice. After 2 h, mice were perfused with PBS and fixed with 2.5% glutaraldehyde in 100 mM cacodylate buffer (pH 7.4).
Fixed cells and tissues were incubated with 2.5% glutaraldehyde and 2 mM MgCl2 in 100 mM cacodylate buffer (pH 7.4) overnight. Samples were then treated with 1% osmium tetroxide in 100 mM cacodylate buffer for 1 h, washed in distilled water four times (10 min/wash), and then treated with 1–2% aqueous uranyl acetate overnight at 4°C in the dark. Samples were then washed and sequentially dehydrated with increasing concentrations of acetone (20, 30, 50, 70, 90, and 100%) for 30 min each, followed by three additional treatments with 100% acetone for 20 min each. Samples were then infiltrated with increasing concentrations of epon or Spurr's resin (25% for 1 h, 50% for 1 h, 75% for 1 h, 100% for 1 h, 100% overnight at room temperature), and then incubated overnight at 70°C in a resin mold. Sections of 50–90 nm were cut on a Leica ultramicrotome with a diamond knife.
In some studies, samples were incubated in a solution containing 2% glutaraldehyde and 0.5% tannic acid in 0.1 M PBS (pH 7.4) for 2 h at room temperature, then washed five times in 0.1 M PBS buffer, and postfixed in 1% osmium tetroxide in PBS. The samples were then washed four times in sodium acetate buffer (pH 5.5) and block-stained in 0.5% uranyl acetate in 0.1 M Na acetate buffer for 12 h at 4°C. Samples were dehydrated in graded ethanol (50%, 75%, 95%, 100%, 100%, 100%) for 10 min each, passed through propylene oxide, and infiltrated in mixtures of Epon 812 and propylene oxide (1:1 and then 2:1 for 2 h each). Samples were then infiltrated in pure Epon 812 overnight, embedded in pure Epon 812, and cured for 48 h at 60°C. Sections of 60 nm were cut on an ultramicrotome (RMC MTX) with a diamond knife. The sections were deposited on single-hole grids coated with Formvar and carbon and double-stained in aqueous solutions of 8% uranyl acetate for 25 min at 60°C and lead citrate for 3 min at room temperature.
For routine EM, samples were examined with a FEI Tecnai 12 electron microscope operating at 80 kV or a 100CX JEOL electron microscope. For EM tomography, 250 nm thick sections were collected on formvar-coated copper slot grids. Following staining, 10 nm colloidal gold particles were applied to both surfaces of the grid to serve as fiducial markers for subsequent image analysis. Dual-axis tilt series (–65° to +65° at 1° intervals) were obtained with a computerized tilt stage in a Tecnai 12 electron microscope operating at 120 kV. Tomographic reconstruction and modeling was performed with the IMOD software package (18).
LPL internalization was measured by incubating endothelial cells with V5-tagged human LPL for 2 h at 37°C, or by incubating cells with LPL for 1 h at 4°C and then at 37°C for the indicated times. For some experiments, DMSO (0.2%), genistein (100 μM), or heparin (500 U/ml) was added with the LPL. For time-course experiments, endothelial cells were washed with PBS and then incubated with V5-tagged human LPL for 1 h at 4°C and then at 37°C for 0, 15, or 30 min. For all experiments, cells were then treated with sulfo-NHS-SS-biotin (Thermo Scientific) in PBS (two 15 min cycles at 4°C) to biotinylate proteins on the cell surface. After quenching the biotinylation reaction with 100 mM glycine (two 15 min cycles at 4°C), cell extracts were collected in radioimmune precipitation buffer (PBS containing 1% Triton X-100, 0.5% deoxycholate, and 0.02% SDS) with Complete Mini EDTA-free protease inhibitors (Roche Applied Science). An aliquot of the cell extracts was saved to measure total LPL. Biotinylated proteins were removed with NeutrAvidin agarose beads (Thermo Scientific) overnight at 4°C. After centrifugation, extracts were transferred to new tubes and treated with fresh NeutrAvidin beads twice (3 h at 4°C) to remove residual biotinylated proteins. Whole-cell extracts and the internalized proteins (the supernatant fluid after removal of biotinylated proteins) were subjected to Western blotting with antibodies against the V5 tag with a monoclonal antibody against the V5 tag (1:200, Invitrogen) and actin (1:1000). Band intensities were quantified with an Odyssey infrared scanner.
For siRNA knockdown studies, RHMVECs were transfected with S-tagged GPIHBP1 (5) and either control siRNA (Invitrogen #46-2001) or an siRNA against Cav1 (Invitrogen #CAV1RSS352112). Transfected cells were grown on transwell filters for 48 h, and LPL transport was quantified as described earlier.
To measure LPL mass in pre- and postheparin plasma, blood samples from wild-type and Cav1−/− mice were collected under anesthesia from the retro-orbital sinus before and 5 min after an intravenous injection of 50 U heparin sodium in 150 μl of normal saline. Plasma samples were preincubated in 1.2 M guanidium HCl, 0.8 M NaCl, 1% BSA, 0.05% Tween 20, 10 mM sodium phosphate (pH 7.4) for 1 h at 4°C. The samples were then diluted to 0.24 M guanidium-HCl prior to addition to microtiter wells coated with 1 μg/ml anti-mouse LPL antibodies (1 μg/well) (19). The samples were incubated overnight at 4°C, and the microtiter wells were then washed with an automated plate washer 6 times with PBS containing 0.05% Tween 20. The wells were then incubated with biotinylated goat anti-mouse LPL immunoglobulins overnight at 4°C. The microtiter wells were then washed 9 times and subsequently incubated with streptavidin-coupled horseradish peroxidase for 2 h at 37°C. Nonspecifically bound streptavidin-horseradish peroxidase was removed by washing 12 times with phosphate-buffered saline containing 0.05% Tween 20 before color development with o-phenylenediamine dihydrochloride and reading at OD490. A standard curve ranging from 0.02 to 2.0 ng of recombinant mouse LPL was included on each plate.
RESULTS
To investigate whether LPL moves bidirectionally across endothelial cells, GPIHBP1-expressing rat heart microvessel endothelial cells (RHMVEC) were seeded on transwell filters and grown until they formed tight monolayers. LPL was added to the basolateral medium of GPIHBP1-expressing endothelial cells, and the LPL that reached the apical face of cells was released by heparin and quantified (Fig. 1A). As expected, LPL was transported across GPIHBP1-expressing RHMVECs, and the level of LPL transport was greater than in control cells expressing CD59, a GPI-anchored protein that lacks the capacity to bind LPL (Fig. 1A). GPIHBP1-expressing RHMVECs also transported LPL in the reverse direction; when LPL was added to the apical face of cells, the LPL was transported to the basolateral face of cells (Fig. 1B). The amount of LPL transport in either direction was similar.
Fig. 1.

Bidirectional movement of LPL and GPIHBP1 across endothelial cells. (A, B) Transport of LPL after 1 h from the basolateral chamber to the apical membrane (A) or from the apical chamber to the basolateral membrane (B). Transport assays were performed on confluent monolayers of GPIHBP1-expressing endothelial cells grown on transwell filters. Bar graphs show the fold change (mean ± SEM of triplicate samples) in LPL transport across cells in GPIHBP1-expressing cells (GPIHBP1) compared with CD59-expressing cells (Control). (C) Transport of Alexa555-labeled antibody against GPIHBP1 (11A12) from the basolateral to the luminal face of capillary endothelial cells in BAT after 20 or 120 min. Sections were stained with an antibody against CD31 (green) to identify endothelial cells and DAPI (blue) to identify nuclei. The luminal face of endothelial cells is marked by arrowheads. (D) Transport of CF568-labeled antibody 11A12 (red) from the luminal to the basolateral face of capillary endothelial cells in BAT after 15 or 120 min. The basolateral face of endothelial cells is marked by arrows.
We next examined whether GPIHBP1 moves bidirectionally across capillary endothelial cells in BAT of live mice. Normally, GPIHBP1 is located along both the basolateral and apical faces of capillary endothelial cells (6); this localization can be easily documented by photographing cross-sections of capillaries containing endothelial cell nuclei (supplementary Fig. I). In contrast to GPIHBP1, another endothelial cell protein, podocalyxin, is found only on the luminal face of capillaries (supplementary Fig. I). To assess basolateral-to-luminal movement of GPIHBP1, we injected a fluorescently labeled antibody against GPIHBP1 (11A12) into the interstitium of BAT of wild-type and Gpihbp1−/− mice. After the injection, the mice (which were awake, ambulatory, and euthermic) were returned to their cages. After 20 or 120 min, the mice were euthanized and perfusion-fixed, and sections of BAT were stained with a fluorescently labeled antibody against CD31 (to visualize endothelial cells) and DAPI (to visualize cell nuclei). At the 20 min time point, antibody 11A12 was bound to endothelial cells of wild-type mice (Fig. 1C, supplementary Fig. I), but little could be found at the luminal face of cells (77 ± 3% on the basolateral surface versus 23 ± 3% on the luminal surface; Fig. 1C, arrowhead). After 2 h, however, substantial amounts of antibody 11A12 had been transported to the lumen (51 ± 4% basolateral versus 49 ± 4% luminal; Fig. 1C, supplementary Fig. I). In anesthetized mice, when the body temperature inadvertently fell to 25°C, antibody 11A12 became associated with endothelial cells but did not reach the capillary lumen, even at the 2 h time point (supplementary Fig. II). As expected, antibody 11A12 did not bind to capillaries in Gpihbp1−/− mice (supplementary Fig. I).
Movement of GPIHBP1 from the luminal-to-basolateral face of endothelial cells was assessed by injecting fluorescently labeled antibody 11A12 intravenously and then examining cross-sections of capillaries in BAT. After 15 min, antibody 11A12 was bound to the luminal face of capillaries in wild-type mice (91 ± 3% luminal versus 9 ± 3% basolateral; Fig. 1D, supplementary Fig. I) but not in Gpihbp1−/− mice (supplementary Fig. I). After 2 h, substantial amounts of antibody 11A12 in wild-type mice had been transported to the basolateral face of capillaries (58 ± 2% luminal versus 42 ± 2% basolateral; Fig. 1D, supplementary Fig. I).
We suspected that GPIHBP1 and LPL were likely carried across cells in vesicles and that transport across cells might be inhibited by the dynamin inhibitor dynasore (20). [Dynamin is required for many vesicle fission events (21).] Indeed, dynasore reduced LPL transport across cells in a dose-dependent fashion (Fig. 2A). Genistein, a tyrosine kinase inhibitor that is thought to interfere with caveolar-dependent endocytosis (22, 23), also inhibited LPL transport (Fig. 2B).
Fig. 2.
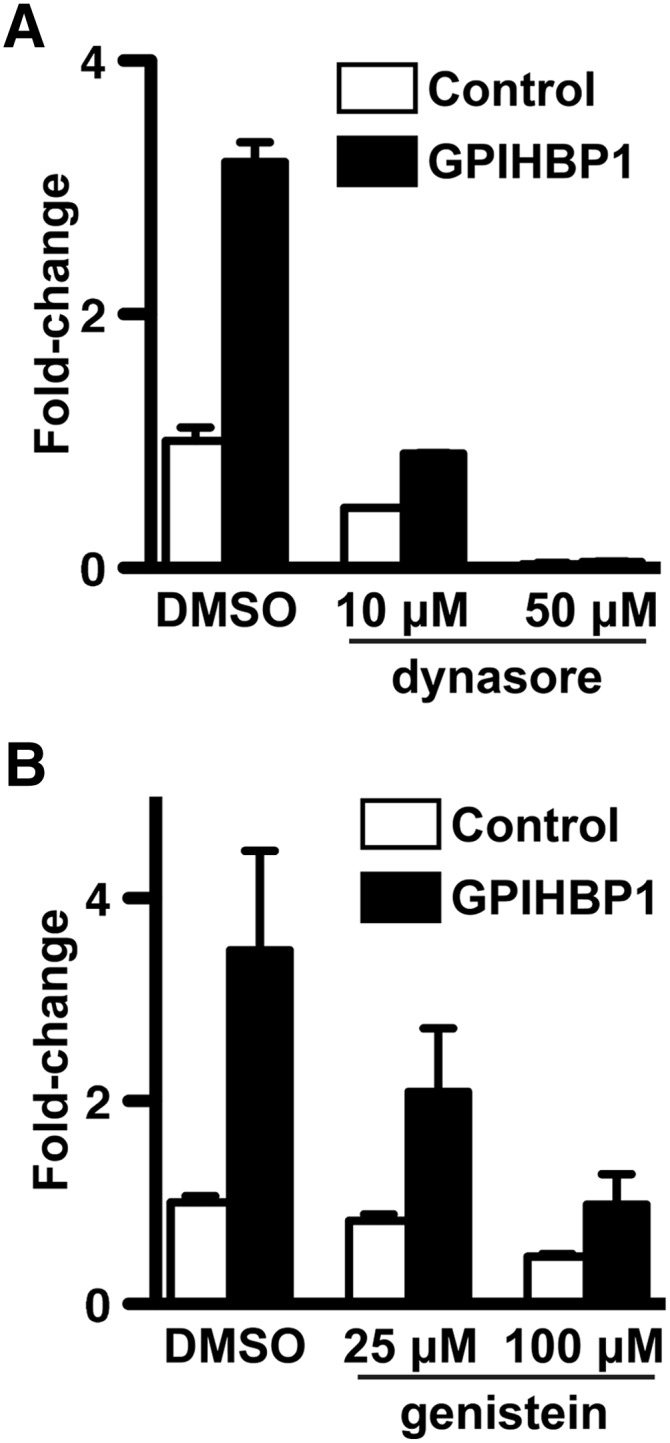
Inhibition of LPL transport across GPIHBP1-expressing endothelial cells grown on transwell filters by dynasore (A) and genistein (B). Bar graphs show the fold change (normalized to vehicle-treated CD59-expressing control cells; mean ± SEM;) in the presence of inhibitors or vehicle (DMSO) alone. Both dynasore (P = 0.0003; duplicate samples for each dose) and genistein (P = 0.0009; two sets of experiments each done in duplicate) significantly reduced transport as judged by one-way ANOVA.
To further investigate the possibility of vesicular trafficking, we examined GPIHBP1 localization by immunogold EM. We cultured endothelial cells on transwell filters until they formed tight monolayers, and gold-conjugated antibody 11A12 was added to the apical face of cells. After 2 h at 37°C, the cells were fixed and processed for EM. We found gold beads clustered in ∼60–100 nm invaginations at both the apical and basolateral faces of cells (Fig. 3A): 64.1% of gold particles were within invaginations or internalized vesicles; 22.4% were at the necks of invaginations; and 13.5% were on the plasma membrane but not associated with invaginations. No gold-conjugated antibody 11A12 was found on endothelial cells that expressed CD59 rather than GPIHBP1 (supplementary Fig. III).
Fig. 3.

Electron micrographs showing GPIHBP1 and LPL on plasmalemmal invaginations. (A, B) Localization of gold-labeled antibodies against GPIHBP1 (A) or V5-LPL (B) after their addition to the apical face of GPIHBP1-expressing endothelial cells grown on filters. Gold particles are indicated by white arrows. (C, D) Localization of gold-conjugated antibody 11A12 in endothelial cells 2 h after injection of the antibody into the quadriceps of a wild-type mouse (C) or a Gpihbp1−/− mouse (D). Scale bar, 100 nm.
The distribution of LPL in endothelial cells resembled that of GPIHBP1. When GPIHBP1-expressing endothelial cells were incubated with V5-tagged human LPL followed by gold-conjugated anti-V5 IgG, gold beads were observed in apical and basolateral plasma membrane invaginations and in what appeared to be intracellular vesicles (Fig. 3B). No gold beads were observed on cells expressing CD59 (supplementary Fig. III).
We also examined GPIHBP1 localization in vivo. Gold-conjugated antibody 11A12 was injected into the quadriceps of Gpihbp1+/+ and Gpihbp1−/− mice. After 2 h, muscle biopsies were prepared for EM. Gold particles were observed in plasma membrane invaginations of endothelial cells and in what appeared to be intracellular vesicles in wild-type mice (Fig. 3C) but not in Gpihbp1−/− mice (Fig. 3D).
To determine whether GPIHBP1 could be found in bona fide vesicles that had lost connections to the plasma membrane, we examined cultured endothelial cells with dual-axis electron tomography. These studies showed that gold beads could be found in plasma membrane invaginations and in multilobed vesicular structures; some gold beads were found in vesicles that had lost connections to the plasma membrane (Fig. 4, supplementary Videos I–VI).
Fig. 4.

Dual-axis electron tomography on GPIHBP1-expressing endothelial cells grown on transwell filters and incubated with gold-conjugated antibody 11A12. Left column: electron micrographs showing structures containing gold beads. 3-D modeling was carried out by drawing contours around membranes of each section of the tomogram (middle column) and then reconstructing the contours to generate the 3-D model (right column). Tomography revealed gold particles in plasma membrane invaginations and vesicles near the apical surface (A), in multi-lobed invaginations and vesicular structures (B), and in vesicles near the basolateral surface (C). Insets show higher magnification images of the boxed areas. Modeling of one vesicle in (C) revealed that it had no connections with the plasma membrane (see also supplementary Videos I–VI). Red, plasma membrane and uncoated vesicles; green, clathrin-coated vesicle; yellow, gold particles; light blue, edge of transwell filter. Scale bar, 200 nm.
Caveolae are flask-shaped plasma membrane invaginations (50–100 nm in diameter) that are abundant in many cells types, particularly endothelial cells (24), and resemble the types of plasma membrane invaginations where we found GPIHBP1 and LPL. Caveolin-1 is thought to be a key protein in the formation of caveolae (11, 25). We investigated whether caveolin-1 had an important role in GPIHBP1-mediated LPL transport. We reasoned that an important role for caveolin-1 in LPL transport was possible but perhaps unlikely, given that caveolin-1-deficient mice have only slightly elevated triglyceride levels (whereas GPIHBP1-deficient mice have very severe hypertriglyceridemia) (5, 26). In our initial experiments, we tested whether siRNA knockdown of caveolin-1 in RHMVECs would affect LPL transport. We observed no significant difference in LPL transport between GPIHBP1-expressing RHMVECs that had been transfected with a Cav1 siRNA and those transfected with a control siRNA (Fig. 5A). However, because we were able to achieve only a ∼50% knockdown of caveolin-1 expression, as judged by Western blotting (Fig. 5B), we next attempted to quantify LPL transport with endothelial cells isolated from the lungs of wild-type and Cav1−/− mice. Unfortunately, these experiments proved to be impossible because Cav1−/− endothelial cells do not form tight monolayers on transwell grids. As an alternative, we measured LPL internalization. We reasoned that vesicular transport depends on internalization and that internalization might be a reasonable surrogate for transport. We found that LPL internalization, like LPL transport, is temperature sensitive and inhibited by genistein (supplementary Fig. IV). When LPL internalization was quantified, we found that the amount of LPL internalized by Gpihbp1-expressing Cav1−/− endothelial cells was similar to the amount of LPL internalized by Gpihbp1-expressing wild-type endothelial cells (Fig. 5C). When Gpihbp1-expressing Cav1−/− endothelial cells were incubated with V5-tagged human LPL followed by gold-conjugated anti-V5 IgG, gold beads were observed in what appeared to be intracellular vesicles, despite the relative paucity of invaginations (consistent with caveolin-1 deficiency) (Fig. 5D).
Fig. 5.

Effects of caveolin-1 deficiency on LPL movement in endothelial cells. (A) Transport of LPL across GPIHBP1- and mock-transfected RHMVECs that had also been transfected with control or Cav1 siRNA. Bar graphs are composites of two independent experiments done in triplicate and show the fold change (mean ± SEM) in LPL transport compared with cells transfected only with control siRNA. (B) Caveolin-1 protein levels (mean ± SEM) in control and Cav1 siRNA-transfected cells as judged by quantitative Western blotting (control set to 100%). (C) LPL internalization by GPIHBP1-expressing mouse lung endothelial cells derived from wild-type or Cav1−/− mice after incubation with V5-tagged LPL for 1 h at 4°C and then for 0, 15, or 30 min at 37°C. Chart shows percentage of bound LPL that was internalized (triplicate samples normalized to actin; mean ± SEM) with time 0 set at 0% internalization. Curves were not significantly different as judged by two-way ANOVA (P = 0.6759). (D) Localization of gold-labeled antibodies against V5-LPL after their addition to the apical face of GPIHBP1-expressing Cav1−/− endothelial cells grown on filters. Scale bar, 100 nm.
A previous report found that the LPL activity released into the plasma after an injection of heparin was normal in Cav1−/− mice (26). Consistent with that report, we found that pre- and postheparin plasma LPL mass levels were normal in Cav1−/− mice (Fig. 6A). We also examined GPIHBP1 transport and LPL localization in endothelial cells from wild-type and Cav1−/− mice. As expected, the tissues of Cav1−/− mice lacked caveolin-1, as judged by immunohistochemistry (Fig. 6B), and capillary endothelial cells in these mice had far fewer invaginations (supplementary Fig. V). When fluorescently labeled 11A12 was injected into the interstitium of BAT, the level of fluorescence at the capillary lumen after 2 h was not perceptibly different in capillaries of Cav1+/+ and Cav1−/− mice (44 ± 4% versus 41 ± 2%; Fig. 6C). Also, when we performed immunohistochemical studies on sections of BAT with an LPL-specific antibody, the relative amounts of LPL at the basolateral and luminal faces of endothelial cells were similar in capillaries of wild-type and Cav1−/− mice (47 ± 4% luminal versus 49 ± 2%, respectively; Fig. 6D).
Fig. 6.

Effects of caveolin-1 deficiency on GPIHBP1 and LPL transport across endothelial cells. (A) Pre-and postheparin plasma LPL mass levels in wild-type and Cav1−/− mice (n = 3/genotype). (B) Immunohistochemical detection of caveolin-1 (green) in BAT of Cav1+/+ and Cav1−/− mice. (C) Transport of CF568-labeled antibody 11A12 from the interstitial spaces to the capillary lumen in BAT of wild-type (Cav1+/+) and Cav1−/− mice after 2 h. The luminal face of endothelial cells is marked by arrowheads. (D) Immunohistochemical detection of LPL along the luminal face of endothelial cells in BAT of Cav1+/+ and Cav1−/− mice.
CD36 is thought to be important in moving the fatty acid products of lipolysis across endothelial cells and into parenchymal cells (27). Like GPIHBP1, CD36 (a lipid raft protein) does not require caveolin 1 for internalization (28). We hypothesized that CD36 and GPIHBP1 might colocalize on the surface of cells. Indeed, GPIHBP1 and CD36 colocalize quite well on the surface of GPIHBP1-expressing cultured endothelial cells (supplementary Fig. VI).
DISCUSSION
Previous studies showed that GPIHBP1 is required for moving LPL to the capillary lumen (6), but the cellular mechanisms underlying the transport have remained unclear. In the current studies, we showed by EM that GPIHBP1 and LPL are located in invaginations along the plasma membrane and in cytoplasmic vesicles, strongly suggesting that GPIHBP1 and LPL traverse endothelial cells in transcytotic vesicles. The inhibition of LPL transport by dynasore (an inhibitor of dynamin) and genistein further supports a vesicular transport mechanism. We found no evidence that LPL transport depends on caveolin-1. Preheparin plasma LPL levels, which are low in GPIHBP1-deficient mice (16), were normal in Cav1-deficient mice. By EM, LPL added to Cav1-deficient endothelial cells was found in both plasma membrane invaginations and in vesicles. Moreover, GPIHBP1-mediated LPL internalization is preserved in Cav1-deficient endothelial cells, and LPL reaches the capillary lumen normally in Cav1-deficient mice.
Given that LPL's site of action is at the apical face of endothelial cells, we initially suspected that the movement of LPL to the capillary lumen would be unidirectional (from the basolateral to the apical face of endothelial cells). However, we found that LPL and GPIHBP1 move bidirectionally across endothelial cells. After reflection, we realized that bidirectional trafficking could make physiologic sense. Some LPL is released into the circulation during the lipolytic processing of triglyceride-rich lipoproteins (29); thus, bidirectional movement would make it possible for GPIHBP1 to return to the basolateral face of cells and carry more LPL to the lumen. Recent studies by Olafsen and Fong strongly suggested that GPIHBP1 is a long-lived molecule, remaining in capillary endothelial cells for hours or days (30), presumably moving back and forth across cells continuously. Conceivably, continuous bidirectional transport could lead to an accumulation of LPL on the luminal face of capillaries. However, this would require that GPIHBP1 “hand off” LPL to another protein within the capillary lumen. Given that the amounts of LPL on the luminal and basolateral faces of capillaries appear to be equal (6), the more likely explanation is that the LPL within the lumen is bound to GPIHBP1.
It is possible that bidirectional movement of LPL plays a role in the regulation of LPL activity. For example, transporting luminal LPL to the subendothelial spaces could facilitate inhibition of LPL activity by ANGPTL4. ANGPTL4 is present in the subendothelial spaces (31–33), and this may be an important site for its regulation of LPL activity (34). ANGPTL4 is also present in the plasma, but plasma ANGPTL4 levels do not correlate with triglyceride levels in humans (35, 36), and a recent study showed that plasma lipoproteins can interfere with the inhibition of LPL by ANGPTL4 (34).
GPIHBP1 and related transport machinery could conceivably be important for the uptake of triglyceride-rich lipoproteins. Bartelt et al. (27) recently reported that, during cold stress and active lipolysis, intact lipoproteins move across capillary endothelial cells in BAT. We would not be surprised if GPIHBP1 ultimately proves to be relevant to that process.
Whether GPIHBP1 is relevant to the transport of other proteins, aside from LPL, is unknown. Other lipase family members, for example, hepatic lipase and endothelial lipase, lack the ability to bind avidly to GPIHBP1 (37). However, it is quite possible that GPIHBP1 could play a role in binding other proteins and shuttling them across endothelial cells. This possibility deserves further scrutiny.
Cav1 deficiency has been reported to reduce the number of invaginations and vesicles in endothelial cells (11, 25), a finding that we have verified. However, we did not detect a defect in GPIHBP1 or LPL movement across Cav1−/− endothelial cells. Our observations indicate that caveolin-1 is either unimportant or minimally important in the movement of GPIHBP1 and LPL across endothelial cells. Caveolar-independent endocytosis of GPI-anchored proteins has been described previously [reviewed by Howes, Mayor, and Parton (38)]. For example, interleukin 2 receptor endocytosis is clathrin- and caveolin-independent but dynamin-dependent (39). It seems likely that such a mechanism is operative in the transport of GPIHBP1 and LPL across endothelial cells.
The current studies have filled in important blanks regarding cellular mechanisms of GPIHBP1 and LPL transport across endothelial cells, but our understanding of the cast of molecules involved in the transport process remains incomplete. And not surprisingly, our understanding of hypertriglyceridemia is also incomplete. Although some cases of hypertriglyceridemia are caused by LPL and GPIHBP1 mutations (4, 40), the pathogenesis of most cases of hypertriglyceridemia in lipid clinics is mysterious. A more complete understanding of the molecular machinery for transporting LPL to the capillary lumen should yield an improved understanding of hypertriglyceridemia in humans.
Supplementary Material
Acknowledgments
The authors thank UCLA's Brain Research Institute for assistance with EM.
Footnotes
Abbreviations:
- ANGPTL4
- angiopoietin-like 4
- BAT
- brown adipose tissue
- DAPI
- 4′,6-diamidino-2-phenylindole
- EM
- electron microscopy
- GPIHBP1
- glycosylphosphatidylinositol-anchored high density lipoprotein binding protein 1
- PFA
- paraformaldehyde
- RHMVEC
- rat heart microvessel endothelial cell
This work was supported by Scientist Development Awards from the American Heart Association (B.S.J.D. and A.P.B.); a postdoctoral fellowship award from the American Heart Association (C.G.); and National Institutes of Health Grants HL-094732 (A.P.B.), HL-86683 (L.G.F.), HL-089781 (L.G.F.), HL-089781 (S.G.Y.), HL-090553 (S.G.Y.), and HL-087228 (S.G.Y.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. Lentiviruses were prepared by the UCLA Vector Core facility, which is supported by JCCC/P30 CA-016042 and CURE/P30 DK-041301.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of methods, six figures, and six videos.
REFERENCES
- 1.Havel R. J. 2010. Triglyceride-rich lipoproteins and plasma lipid transport. Arterioscler. Thromb. Vasc. Biol. 30: 9–19 [DOI] [PubMed] [Google Scholar]
- 2.Havel R. J., Kane J. P.2001. Introduction: structure and metabolism of plasma lipoproteins. In The Metabolic and Molecular Bases of Inherited Disease. C. R. Scriver, A. L. Beaudet, W. S. Sly et al., editors. McGraw-Hill, New York. 2705–2716.
- 3.Olivecrona T., Olivecrona G.2009. The ins and outs of adipose tissue. In Cellular Lipid Metabolism. C. Ehnholm, editor. Springer Berlin Heidelberg. 315–369.
- 4.Wang H., Eckel R. H. 2009. Lipoprotein lipase: from gene to obesity. Am. J. Physiol. Endocrinol. Metab. 297: E271–E288 [DOI] [PubMed] [Google Scholar]
- 5.Beigneux A. P., Davies B. S., Gin P., Weinstein M. M., Farber E., Qiao X., Peale F., Bunting S., Walzem R. L., Wong J. S., et al. 2007. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 5: 279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies B. S., Beigneux A. P., Barnes R. H., 2nd, Tu Y., Gin P., Weinstein M. M., Nobumori C., Nyren R., Goldberg I., Olivecrona G., et al. 2010. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 12: 42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinstein M. M., Goulbourne C. N., Davies B. S., Tu Y., Barnes R. H., 2nd, Watkins S. M., Davis R., Reue K., Tontonoz P., Beigneux A. P., et al. 2012. Reciprocal metabolic perturbations in the adipose tissue and liver of GPIHBP1-deficient mice. Arterioscler. Thromb. Vasc. Biol. 32: 230–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minshall R. D., Tiruppathi C., Vogel S. M., Niles W. D., Gilchrist A., Hamm H. E., Malik A. B. 2000. Endothelial cell-surface gp60 activates vesicle formation and trafficking via G(i)-coupled Src kinase signaling pathway. J. Cell Biol. 150: 1057–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tiruppathi C., Song W., Bergenfeldt M., Sass P., Malik A. B. 1997. Gp60 activation mediates albumin transcytosis in endothelial cells by tyrosine kinase-dependent pathway. J. Biol. Chem. 272: 25968–25975 [DOI] [PubMed] [Google Scholar]
- 10.Vogel S. M., Minshall R. D., Pilipovic M., Tiruppathi C., Malik A. B. 2001. Albumin uptake and transcytosis in endothelial cells in vivo induced by albumin-binding protein. Am. J. Physiol. Lung Cell. Mol. Physiol. 281: L1512–L1522 [DOI] [PubMed] [Google Scholar]
- 11.Razani B., Engelman J. A., Wang X. B., Schubert W., Zhang X. L., Marks C. B., Macaluso F., Russell R. G., Li M., Pestell R. G., et al. 2001. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 276: 38121–38138 [DOI] [PubMed] [Google Scholar]
- 12.Fernández-Hernando C., Yu J., Suarez Y., Rahner C., Davalos A., Lasuncion M. A., Sessa W. C. 2009. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab. 10: 48–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gin P., Yin L., Davies B. S., Weinstein M. M., Ryan R. O., Bensadoun A., Fong L. G., Young S. G., Beigneux A. P. 2008. The acidic domain of GPIHBP1 is important for the binding of lipoprotein lipase and chylomicrons. J. Biol. Chem. 283: 29554–29562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beigneux A. P., Gin P., Davies B. S., Weinstein M. M., Bensadoun A., Fong L. G., Young S. G. 2009. Highly conserved cysteines within the Ly6 domain of GPIHBP1 are crucial for the binding of lipoprotein lipase. J. Biol. Chem. 284: 30240–30247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang S. F., Reich B., Brunzell J. D., Will H. 1998. Detailed characterization of the binding site of the lipoprotein lipase-specific monoclonal antibody 5D2. J. Lipid Res. 39: 2350–2359 [PubMed] [Google Scholar]
- 16.Weinstein M. M., Yin L., Beigneux A. P., Davies B. S., Gin P., Estrada K., Melford K., Bishop J. R., Esko J. D., Dallinga-Thie G. M., et al. 2008. Abnormal patterns of lipoprotein lipase release into the plasma in GPIHBP1-deficient mice. J. Biol. Chem. 283: 34511–34518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider C. A., Rasband W. S., Eliceiri K. W. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mastronarde D. N. 1997. Dual-axis tomography: an approach with alignment methods that preserve resolution. J. Struct. Biol. 120: 343–352 [DOI] [PubMed] [Google Scholar]
- 19.Page S., Judson A., Melford K., Bensadoun A. 2006. Interaction of lipoprotein lipase and receptor-associated protein. J. Biol. Chem. 281: 13931–13938 [DOI] [PubMed] [Google Scholar]
- 20.Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., Kirchhausen T. 2006. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell. 10: 839–850 [DOI] [PubMed] [Google Scholar]
- 21.Mayor S., Pagano R. E. 2007. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 8: 603–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y., Norkin L. C. 1999. Extracellular simian virus 40 transmits a signal that promotes virus enclosure within caveolae. Exp. Cell Res. 246: 83–90 [DOI] [PubMed] [Google Scholar]
- 23.Pelkmans L., Kartenbeck J., Helenius A. 2001. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 3: 473–483 [DOI] [PubMed] [Google Scholar]
- 24.Bastiani M., Parton R. G. 2010. Caveolae at a glance. J. Cell Sci. 123: 3831–3836 [DOI] [PubMed] [Google Scholar]
- 25.Drab M., Verkade P., Elger M., Kasper M., Lohn M., Lauterbach B., Menne J., Lindschau C., Mende F., Luft F. C., et al. 2001. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 293: 2449–2452 [DOI] [PubMed] [Google Scholar]
- 26.Razani B., Combs T. P., Wang X. B., Frank P. G., Park D. S., Russell R. G., Li M., Tang B., Jelicks L. A., Scherer P. E., et al. 2002. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J. Biol. Chem. 277: 8635–8647 [DOI] [PubMed] [Google Scholar]
- 27.Bartelt A., Bruns O. T., Reimer R., Hohenberg H., Ittrich H., Peldschus K., Kaul M. G., Tromsdorf U. I., Weller H., Waurisch C., et al. 2011. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 17: 200–205 [DOI] [PubMed] [Google Scholar]
- 28.Zeng Y., Tao N., Chung K. N., Heuser J. E., Lublin D. M. 2003. Endocytosis of oxidized low density lipoprotein through scavenger receptor CD36 utilizes a lipid raft pathway that does not require caveolin-1. J. Biol. Chem. 278: 45931–45936 [DOI] [PubMed] [Google Scholar]
- 29.Peterson J., Bihain B. E., Bengtsson-Olivecrona G., Deckelbaum R. J., Carpentier Y. A., Olivecrona T. 1990. Fatty acid control of lipoprotein lipase: a link between energy metabolism and lipid transport. Proc. Natl. Acad. Sci. USA. 87: 909–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olafsen T., Young S. G., Davies B. S., Beigneux A. P., Kenanova V. E., Voss C., Young G., Wong K. P., Barnes R. H., 2nd, Tu Y., et al. 2010. Unexpected expression pattern for glycosylphosphatidylinositol-anchored HDL-binding protein 1 (GPIHBP1) in mouse tissues revealed by positron emission tomography scanning. J. Biol. Chem. 285: 39239–39248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cazes A., Galaup A., Chomel C., Bignon M., Brechot N., Le Jan S., Weber H., Corvol P., Muller L., Germain S., et al. 2006. Extracellular matrix-bound angiopoietin-like 4 inhibits endothelial cell adhesion, migration, and sprouting and alters actin cytoskeleton. Circ. Res. 99: 1207–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon J. C., Chickering T. W., Rosen E. D., Dussault B., Qin Y., Soukas A., Friedman J. M., Holmes W. E., Spiegelman B. M. 2000. Peroxisome proliferator-activated receptor gamma target gene encoding a novel angiopoietin-related protein associated with adipose differentiation. Mol. Cell. Biol. 20: 5343–5349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida K., Shimizugawa T., Ono M., Furukawa H. 2002. Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J. Lipid Res. 43: 1770–1772 [DOI] [PubMed] [Google Scholar]
- 34.Nilsson S. K., Anderson F., Ericsson M., Larsson M., Makoveichuk E., Lookene A., Heeren J., Olivecrona G. 2012. Triacylglycerol-rich lipoproteins protect lipoprotein lipase from inactivation by ANGPTL3 and ANGPTL4. Biochim. Biophys. Acta. 1821: 1370–1378 [DOI] [PubMed] [Google Scholar]
- 35.Robciuc M. R., Tahvanainen E., Jauhiainen M., Ehnholm C. 2010. Quantitation of serum angiopoietin-like proteins 3 and 4 in a Finnish population sample. J. Lipid Res. 51: 824–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smart-Halajko M. C., Robciuc M. R., Cooper J. A., Jauhiainen M., Kumari M., Kivimaki M., Khaw K. T., Boekholdt S. M., Wareham N. J., Gaunt T. R., et al. 2010. The relationship between plasma angiopoietin-like protein 4 levels, angiopoietin-like protein 4 genotype, and coronary heart disease risk. Arterioscler. Thromb. Vasc. Biol. 30: 2277–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gin P., Beigneux A. P., Voss C., Davies B. S., Beckstead J. A., Ryan R. O., Bensadoun A., Fong L. G., Young S. G. 2011. Binding preferences for GPIHBP1, a glycosylphosphatidylinositol-anchored protein of capillary endothelial cells. Arterioscler. Thromb. Vasc. Biol. 31: 176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Howes M. T., Mayor S., Parton R. G. 2010. Molecules, mechanisms, and cellular roles of clathrin-independent endocytosis. Curr. Opin. Cell Biol. 22: 519–527 [DOI] [PubMed] [Google Scholar]
- 39.Lamaze C., Dujeancourt A., Baba T., Lo C. G., Benmerah A., Dautry-Varsat A. 2001. Interleukin 2 receptors and detergent-resistant membrane domains define a clathrin-independent endocytic pathway. Mol. Cell. 7: 661–671 [DOI] [PubMed] [Google Scholar]
- 40.Davies B. S., Beigneux A. P., Fong L. G., Young S. G. 2012. New wrinkles in lipoprotein lipase biology. Curr. Opin. Lipidol. 23: 35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.