

Abstract
New protein-coding genes can originate either through modification of existing genes or de novo. Recently, the importance of de novo origination has been recognized in eukaryotes, although eukaryotic genes originated de novo are relatively rare and difficult to identify. In contrast, viruses contain many de novo genes, namely those in which an existing gene has been “overprinted” by a new open reading frame, a process that generates a new protein-coding gene overlapping the ancestral gene. We analyzed the evolution of 12 experimentally validated viral genes that originated de novo and estimated their relative ages. We found that young de novo genes have a different codon usage from the rest of the genome. They evolve rapidly and are under positive or weak purifying selection. Thus, young de novo genes might have strain-specific functions, or no function, and would be difficult to detect using current genome annotation methods that rely on the sequence signature of purifying selection. In contrast to young de novo genes, older de novo genes have a codon usage that is similar to the rest of the genome. They evolve slowly and are under stronger purifying selection. Some of the oldest de novo genes evolve under stronger selection pressure than the ancestral gene they overlap, suggesting an evolutionary tug of war between the ancestral and the de novo gene.
Keywords: overlapping genes, de novo origin, new genes
Introduction
Novel protein-coding genes can have two fundamental origins (reviewed in Long et al. 2003; Babushok et al. 2007; Zhou and Wang 2008; Bornberg-Bauer et al. 2010; Kaessmann 2010; Tautz and Domazet-Loso 2011). In the first, a gene originates by modification of an existing gene, for example, through gene duplication, exon shuffling, gene fusion, horizontal gene transfer, or transposition. In the second, a gene originates de novo. This mechanism was thought to be highly improbable (Ohno 1970; Jacob 1977), but recent studies have provided experimental evidence that it may be frequent. De novo origination can take place in a previously noncoding region, such as an intergenic region (Cai et al. 2008; Toll-Riera et al. 2009b; Li, Zhang, et al. 2010), or an intron (Sorek 2007). However, a gene can also originate de novo from an open reading frame that already encodes a protein, by a mechanism called “overprinting”, in which mutations lead to the expression of a second reading frame overlapping the first one (Ohno 1984; Keese and Gibbs 1992; Rancurel et al. 2009; Li, Dong, et al. 2010). Genome-scale computational analyses or experimental analyses of RNA transcripts have proposed many candidate genes originated de novo through these mechanisms (Levine et al. 2006; Begun et al. 2007; Zhou et al. 2008; Knowles and McLysaght 2009; Chen et al. 2010; Wu et al. 2011; Yang and Huang 2011).
Most studies in this area have focused on eukaryotes, which are not necessarily the best organisms to study de novo genes. First, the incidence of de novo gene origination may be relatively low in eukaryotes, ranging from 2 to 12% of all new gene origination events according to recent estimates (Zhou et al. 2008; Toll-Riera et al. 2009a; Ekman and Elofsson 2010). Second, direct experimental evidence for the expression of the proteins encoded by candidate de novo genes is not always available in eukaryotes—some might be artifacts of genome annotation (Wang et al. 2003). Third, most eukaryotic candidate genes are structurally and functionally poorly characterized. Finally, current protocols to identify genes created de novo from noncoding sequences in eukaryotic genomes focus on genes with similarity to genes already annotated in the genome sequence, whereas some de novo genes may not be currently annotated, even as hypothetical, which would preclude their discovery (Guerzoni and McLysaght 2011).
The identification of de novo genes in viruses does not suffer from these problems or to a much lesser extent. This holds especially for genes generated by overprinting. Overlapping genes are very common in viral genomes (Belshaw et al. 2007; Chirico et al. 2010), providing an abundant source of such de novo genes. In addition, in most cases, the expression of their protein product has been proven, and their function is at least partly known (Rancurel et al. 2009). Finally, using overlapping genes allows the identification of proteins originated de novo with high reliability (see later), by avoiding the confounding factors that limit current approaches to identify proteins generated de novo from noncoding sequences (Guerzoni and McLysaght 2011). For brevity, we will refer here to de novo genes as genes that originated through overprinting.
Viral de novo genes often encode proteins that play a role in viral pathogenicity or spreading, rather than proteins central to viral replication or structure (Li and Ding 2006; Rancurel et al. 2009). The majority of these proteins are predicted to be structurally disordered, i.e., they lack a stable three-dimensional structure (Dyson and Wright 2005; Tompa 2005; Sickmeier et al. 2007), but those that are ordered have intriguing structural features (Rancurel et al. 2009). For instance, the protein p19, originated de novo in the plant virus family Tombusviridae (Rancurel et al. 2009), has a previously unknown tertiary structure and a previously unknown mode of binding to small interfering RNAs (Vargason et al. 2003). This suggests that de novo gene origination can lead to evolutionary innovations in protein structure and function (Rancurel et al. 2009; Bornberg-Bauer et al. 2010; Kaessmann 2010; Abroi and Gough 2011).
A prerequisite to identify a de novo gene is that it must have a monophyletic distribution in one clade—the “focal” clade (fig. 1a, taxa T1, T2, and T3)—while being absent from organisms outside this clade (fig. 1a, taxa T4 and T5). We note that this prerequisite is necessary but not sufficient. Genes fulfilling it may have an ancient origin, older than the focal clade, but they might have diverged beyond recognition outside this clade (Elhaik et al. 2006). Alternatively, they may have entered the focal clade through horizontal gene transfer. These confounding factors can be easily excluded for genes that arose by overprinting (fig. 1b, blue arrows) within a pre-existing “ancestral” reading frame (red arrows). Specifically, if the ancestral gene is present outside the focal clade (e.g., taxa T4 and T5 in fig. 1b), one can exclude divergence beyond recognition and horizontal gene transfer, because in either case, the ancestral gene would not be present outside the focal clade.
Fig. 1.
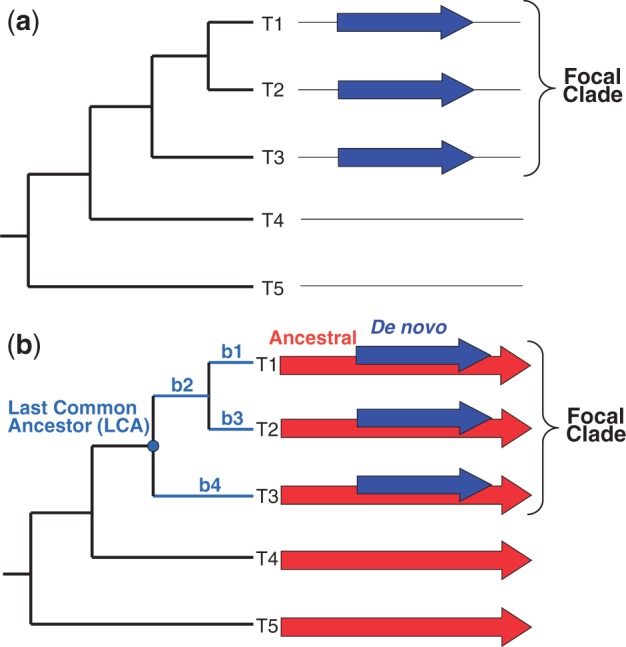
Monophyletic distribution of genes originated de novo. (a) A gene that originated de novo (blue arrows) will exhibit a monophyletic distribution among related taxa. However, this distribution could also be the result of divergence of the gene beyond recognition or of acquisition of the gene through horizontal gene transfer (HGT). (b) For a gene that originated de novo (blue arrows) by overprinting an ancestral reading frame (red arrows), these confounding factors can be excluded (see Introduction). Colors are displayed in the electronic version of the article.
Taking the above considerations into account, we ask the following questions about the evolutionary dynamics of de novo genes: Do de novo genes adapt to their genome, and if so, how rapidly? What is their rate of evolution? How do the de novo genes influence the genes that they overlap? Do de novo genes contribute to viral fitness? To answer these questions, we analyzed the evolution of 12 independent, experimentally validated de novo genes in RNA viruses. We estimated their relative age and compared their evolutionary dynamics to that of the ancestral gene from which they originated.
Results
As described in Materials and Methods, we assembled a data set of 12 experimentally validated pairs of overlapping protein-coding genes (table 1), in which the ancestral and the de novo genes could be unambiguously identified from the phylogenetic distribution of their homologs. We predicted the structural and functional organization of their protein products (fig. 2, discussed further later, see also Materials and Methods). All overlapping regions were longer than 220 nucleotides (table 1). Among the 12 de novo genes in our data set, nine genes overlap completely with their ancestral gene, whereas three overlap only partially: the machlomovirus p31 gene, the omegatetravirus p17 gene, and the ilarvirus 2 b gene (fig. 2).
Table 1.
Overlapping Genes in the Study.
| Clade | Genome Accession Number | Family | Genus | Species | Taxonomic Distribution of the Overlap | Ancestral Frame | De Novo Frame | Number of Sequences (or Sequence Pairs) in the Analyses (divergence, dN/dS, and CSI) | Length of the Overlapping Region (nt) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | NC_007358 | Orthomyxoviridae | Influenzavirus A | Influenza A virus H5N1 | Single species | PB1 | PB1-F2 | 10, 9, and 5 | 273 |
| 2 | NC_001366 | Picornaviridae | Cardiovirus | Theilovirus | 3 species in same genus | Polyprotein | Protein L* | 3, 2, and 3 | 468 |
| 3 | NC_003045 | Coronaviridae | Betacoronavirus | SARS coronavirus | 2 genotype groups | Nucleocapsid (N) | Protein I | 70, 33, and 11 | 624 |
| 4 | NC_005899 | Tetraviridae | Omegatetravirus | Dendrolimus punctatus tetravirus | Whole genus | Capsid protein | p17 | 3, 1, and 3 | 382 |
| 5 | NC_004063 | Tymoviridae | Tymovirus | Turnip yellow mosaic virus | Whole genus | Replicase | ORF69 (movement protein) | 120, 1, and 16 | 1,880 |
| 6 | NC_004366 | — | Umbravirus | Tobacco bushy top virus | Whole genus | ORF3 (movement protein) | ORF3 (long distance movement protein) | 10, 0, and 5 | 698 |
| 7 | NC_003809 | Bromoviridae | Ilarvirus | Spinach latent virus | Whole genus | Polymerase | Protein 2 b | 15, 6, and 6 | 308 |
| 8 | NC_003627 | Tombusviridae | Machlomovirus | Maize chlorotic mottle virus | Whole genus (contains a single species | Coat protein | p31 | 3, 0, and 3 | 451 |
| 9 | NC_009025 | Dicistroviridae | Aparavirus | Israel acute paralysis virus of bees | Whole genus | Structural polyprotein | Pog | 6, 3, and 4 | 312 |
| 10 | NC_001498 | Paramyxoviridae | Morbillivirus | Measles virus | 2 genera in same family | Phosphoprotein (P) | Protein C | 10, 1, and 5 | 561 |
| 11 | NC_003977 | Hepadnaviridae | Orthohepadnavirus | Hepatitis B virus | Whole family (contains 2 genera) | Polymerase (P) | Large envelope protein (L) | 28, 13, and 8 | 834 |
| 12 | NC_003448 | Nodaviridae | Betanodavirus | Striped Jack nervous necrosis virus | Whole genus | Protein A | Protein B | 10, 9, and 6 | 228 |
Fig. 2.

Structural and functional organization of the overlapping genes we studied. Proteins encoded by overlapping genes are shown to scale. For each protein pair, the ancestral protein is shown on the bottom and the de novo protein on top. B1, base domain 1; cc, coiled coil; Le, Leader region; PA2, phospholipase A2 domain; RdRP, RNA-dependent RNA polymerase domain; tm, transmembrane segment; z, zinc-binding region.
Three Quantifiers Are Useful to Describe the Evolutionary Dynamics of Overlapping Genes
We investigated three properties of the ancestral and de novo genes, and the proteins they encode (see Materials and Methods). The first property is the relative sequence divergence, a proxy for the rate at which a protein changes its sequence. Relative divergence values above 1 indicate that a coding region evolves faster than a reference sequence, in our case the full length sequence of the ancestral gene of the pair considered.
The second property is the selective constraint (dN/dS), which estimates the strength of purifying selection on a gene by its ratio of nonsynonymous to synonymous nucleotide substitutions. Values of dN/dS below 1 are evidence of purifying selection whose strength increases with decreasing dN/dS. In principle, values of dN/dS exceeding 1 might suggest that a gene evolves under positive selection (Nei and Gojobori 1986). However, the method we used to calculate dN/dS (Sabath et al. 2008) does not test statistically for positive selection, which is often limited to few sites within the gene (Nielsen and Yang 1998; Zhang et al. 2005). Therefore, values of dN/dS above 1 should be taken to indicate either neutral evolution or positive selection.
The third and final property is the Codon Similarity Index (CSI), which measures the similarity between the codon usage of a gene and that of the rest of the genome containing it. CSI is based on the same calculations as the Codon Adaptation Index (CAI), a commonly used measure of codon usage bias (Sharp and Li 1987) but uses the rest of the genome as a reference set instead of a data set of highly expressed genes (see Materials and Methods).
We first compared these three properties for all ancestral/de novo gene pairs. Table 2 summarizes the results of this comparison. Overall, de novo genes evolve significantly faster (paired two-sided Wilcoxon signed rank test, P < 7.1 × 10−34), are under stronger selective constraint (P < 2.9 × 10−4), and exhibit lower CSI values (P < 5.4 × 10−5) than ancestral genes. The magnitude of the difference between the mean CSI of ancestral genes (0.66) and that of de novo genes (0.62) is low (6%), whereas the difference between the mean relative divergence of the ancestral and de novo genes is high (43%), as is the difference between the selection intensity of ancestral and de novo genes (47%).
Table 2.
Mean Values of Three Evolutionary Properties for Ancestral and De Novo Genes.
| Ancestrala | De Novoa | P | |
|---|---|---|---|
| CSI | 0.66 (0.09) | 0.62 (0.11) | 5.4 × 10−5 |
| Relative divergence | 1.06 (0.52) | 1.85 (1.20) | 7.1 × 10−34 |
| Selection intensity | 0.40 (0.40) | 0.75 (0.55) | 2.9 × 10−4 |
aNumbers in parentheses are standard deviations.
We asked how these properties depend on the time since a de novo gene originated. To this end, we estimated this time by using the sequence divergence of the RNA-dependent RNA polymerase domain of each genome (see Materials and Methods). We then plotted the three properties against this estimated age in figures 3 and 4. The horizontal axis of figures 3 and 4 corresponds to the origination time of de novo genes (labeled by viral genus), with the most recently originated de novo genes shown to the left. The vertical axis shows the relative divergence (fig. 3a), selective constraint (fig. 3b), and CSI (fig. 4). Note that the relative divergence and the selective constraints are calculated for pairs of homologous genes, whereas the CSI is calculated for single genes (see Materials and Methods). Thus, dots in figure 3a and b correspond to values obtained for pairs, whereas dots in figure 4 correspond to values for single genes. Regression lines for ancestral genes (red) and de novo genes (blue) indicate general trends. We used analysis of covariance (ANCOVA) to test whether the slopes of the two regression lines in each panel were different.
Fig. 3.

Evolutionary dynamics of ancestral (red) and de novo genes (blue). The vertical axes show (a) relative divergence and (b) selective constraint (dN/dS) for the 12 taxa. The horizontal axis represents the evolutionary distance from the origin of each de novo gene (i.e., the estimated age of genes within the clade). Regression lines are plotted for visualization of general trends. Low dN/dS values represent strong selective constraints (see text). Note that dN/dS in (b) could only be calculated for gene pairs that have less than 50% amino acid divergence at the amino acid level (see Materials and Methods). No selective constraint data could be calculated for cases 6 and 8 (bottom panel) as the sequence pairs in these clades have all diverged beyond 50%. Where neighboring groups had similar ages, we shifted their position slightly for visual clarity (groups 5 and 6).
Fig. 4.

Codon Similarity Index (CSI) of ancestral (red) and de novo genes (blue). The horizontal axis represents the evolutionary distance from the origin of each de novo gene (as in fig. 3). Regression lines are plotted for visualization of general trends. High CSI values indicate high similarity between the codon usage of a gene and the codon usage of the rest of a genome. Colors are displayed in the electronic version of the paper.
Below, we first examine these three properties separately, before considering them together and synthesizing our observations.
Relative Divergence
Figure 3a presents for each overlap the relative sequence divergence for the pairs of ancestral proteins (red) and the pairs of de novo proteins (blue). The regression line of the ancestors (red) is nearly horizontal (although the regression coefficient is significantly different from zero, P < 0.0012) and equal to one, suggesting that on average the overlapping regions of the ancestral proteins evolve at a similar rate as the full-length ancestral proteins. In contrast, de novo proteins (blue) show a higher relative divergence than their ancestral overlapping proteins. Accordingly, the slopes of the two regression lines are significantly different (P < 3.2 × 10−25). The range of values of the relative divergences between pairs of ancestral proteins is generally narrow or moderate. For instance, the relative divergences of the de novo gene of tymovirus (taxon 11), which encodes the movement protein, range between 1.2 and 2.8 and thus vary by less than a factor three. Three notable exceptions are the youngest de novo proteins of taxa 1 and 3 (respectively, influenzavirus A PB1-F2 and betacoronavirus protein I), which exhibit a considerable range of divergences (from 0 up to 10.4).
Selective Constraint
Figure 3b presents the selective constraint (dN/dS) for the ancestral and de novo genes in each group of viruses we studied. The ancestral and de novo genes are subject to very different constraints, as attested by the significant difference (P < 4.9 × 10−13) between the slopes of their respective regression lines, and the fact that the ancestral regression line has a positive slope, whereas the de novo regression line has a negative slope. The low values of dN/dS < 1 below for most genes indicate that they are under strong selective constraints (purifying selection), i.e., mutations that change the protein sequence are likely to be selected against. In several viruses, the values of dN/dS for the ancestral genes are particularly low, suggesting an extreme functional or structural constraint. For example, the ratio dN/dS of the ancestral gene in viruses 1, 4, and 5 is below 0.05. In contrast, in some cases, either the de novo gene (the influenzavirus A PB1-F2, the betacoronavirus protein I, and the tymovirus movement protein, respectively, from groups 1, 3, and 5) or the ancestral gene (the ilavirus polymerase and the betanodavirus protein A, from groups 7 and 12, respectively) exhibits dN/dS ≥ 1, which indicates neutral evolution or positive selection. As explained at the beginning of the Results section, the test we employ cannot distinguish between them. For all groups, the trends in relative divergence (fig. 3a) and selective constraints (fig. 3b) were consistent with one another. For example, the de novo proteins of cardioviruses are more highly diverged than the ancestral proteins and they also show a lower selective constraint.
Codon Similarity Index
Figure 4 presents the CSI values of ancestral and de novo genes. The slopes of the two regression lines show a significant difference (P < 0.032). The slope of the ancestral regression line is not significantly different from zero (P > 0.96), whereas the de novo regression line has a positive slope (P < 0.003). Relative to their ancestral overlapping genes, most de novo genes show lower CSI values, although the ranges of CSI values in de novo and ancestral genes overlap markedly in most groups. An exception is omegatetraviruses (taxon 4), in which the CSI values of the de novo p17 genes are all higher than the CSI values of the ancestral capsid genes. The CSI values for homologous genes can take a wide range of values. For instance, the CSI of the betacoronavirus I gene varies from 0.27 to 0.67. Overall, the data suggest that the codon usage of de novo genes becomes slowly assimilated into that of the host genome.
De Novo Genes Have Different Properties Depending on Their Age of Origin
Overall, we found that the differences between ancestral and de novo genes appears to decrease with time. Taxa 1–5, with the shortest distance from the origin—corresponding to the youngest de novo genes—all exhibit a similar pattern (fig. 3): the de novo genes show higher relative divergence with respect to their ancestral overlapping genes and weaker selective constraint.
The pattern that contrasts most with that of taxa 1–5 occurs in taxa 7 and 12 (Ilarvirus and Betanodavirus, respectively), where the de novo genes have a more ancient origin. Here, the de novo genes evolve more slowly and exhibit stronger purifying selection relative to their ancestral overlapping genes (fig. 3). In other words, here mutations that change the sequence of the de novo proteins are more deleterious, on average, than mutations that change the sequence of the overlapping ancestral proteins.
Overall, our observations suggest that older de novo genes are more adapted to their genome and evolve under stronger purifying selection. This inference is supported by experimental data on the fitness effects of mutations in de novo genes of different ages (table 3). Mutations in the youngest de novo genes (taxa 1–3) have little to moderate effect, whereas suppression of older de novo genes (taxa 6 and 10–12) has severe effects.
Table 3.
Evidence of Expression, Function, and Fitness Effect of the De Novo Genes in the Study.
| Group | Genus | Evidence for Expression | Function(s) | Fitness Effect When the Novel Gene Is Suppressed | Description of Effect and References |
|---|---|---|---|---|---|
| 1 | Influenzavirus A | Chen et al. (2001b) | Virulence factor (Zamarin et al. 2006). Involved in regulation of polymerase activity (Mazur et al. 2008). Function seems strain-specific and host-specific and is disputed (Krumbholz et al. 2011). | Little or no effect | Suppression of PB1-F2 neither affected viral replication nor virus loads in the lungs of mice (McAuley et al. 2010). |
| 2 | Cardiovirus | van Eyll and Michiels (2002) | Involved in the establishment of permanent infections of the central nervous system (Chen et al. 1995); antiapoptotic effect in cell culture (Ghadge et al. 1998). | Moderate effect | Suppression of L* decreases the ability of Theiler’s virus to induce a chronic infection of the central nervous system (Stavrou et al. 2010). |
| 3 | Betacoronavirus | Senanayake et al. (1992) and Chen et al. (2001a) | Unknown | Little or no effect | Suppression of Protein I expression lead only to a reduced plaque size, suggesting a minor effect on fitness (Fischer et al. 1997). |
| 4 | Omegatetravirus | Hanzlik et al. (1995) | Unknown | Unknown | Unknown |
| 5 | Tymovirus | Weiland and Dreher (1989) | Viral movement through the plant (Bozarth et al. 1992). | Severe effect | A knock-out mutant of the movement protein replicates only at low levels in protoplasts (Weiland and Dreher 1989). |
| 6 | Umbravirus | Ryabov et al. (1998) | Long-distance (systemic) movement in plants (Ryabov et al. 1999); stabilizes viral genomic RNA. | Severe effect | Long-distance movement is abolished in the absence of ORF4 plants (Ryabov et al. 1999) |
| 7 | Ilarvirus | Xin et al. (1998) | Unknown | Unknown | Unknown |
| 8 | Machlomovirus | Scheets (2000) | Unknown | Unknown | Unknown |
| 9 | Aparavirus | Unknown | Unknown | Unknown | |
| 10 | Morbillivirus | Wardrop and Briedis (1991) | Virulence factor (Patterson et al. 2000). | Severe effect | Suppression of C results in much milder symptoms and lower mortality in mice (Patterson et al. 2000). |
| 11 | Orthohepadnavirus | Peterson (1981) | Viral envelope glycoprotein (Beck and Nassal 2007). | Severe effect | Deletions within the S domain of the envelope protein drastically reduce infectivity (Le Duff et al. 2009). |
| 12 | Betanodavirus | Iwamoto et al. (2005) | Blocks RNA interference (Fenner et al. 2006). | Severe effect | Suppression of B2 causes a severe impairment in the intracellular accumulation of viral RNA in cell culture (Fenner et al. 2006). |
Discussion
Several previous studies have examined the codon usage of individual or small groups of overlapping genes (McGeoch et al. 1985; Keese and Gibbs 1992; Pavesi et al. 1997; McVeigh et al. 2000; Lee et al. 2010), their rates of evolution (Mizokami et al. 1997; Sanz et al. 1999; Jordan et al. 2000; Fujii et al. 2001; Nekrutenko et al. 2005; McGirr and Buehuring 2006; Hernandez et al. 2010), and selective constraints (Fujii et al. 2001; Hughes et al. 2001; Guyader and Ducray 2002; Li et al. 2004; Hughes and Hughes 2005; Narechania et al. 2005; Campitelli et al. 2006; Holmes et al. 2006; McGirr and Buehuring 2006; Obenauer et al. 2006; Pavesi 2006; Suzuki 2006; Pavesi 2007; Zaaijer et al. 2007). Overall, our observations agree with those of previous studies—ancestral and de novo genes differ in these properties. Nevertheless, we were able to improve on these studies in several ways. First, by examining the phylogenetic distribution of overlapping gene pairs, we were able to identify reliably (see later) which of the two genes is ancestral and which one is the de novo gene. Second, our new method allowed us to estimate the relative age of origin of de novo genes and to correlate this age with several quantifiers of their evolutionary dynamics. Thus, it allowed us to analyze how the evolutionary forces affecting de novo genes change over time. Third, we used a larger data set than most studies mentioned earlier, which were carried out on individual genes. Fourth, we used a method specifically tailored to overlapping genes (Sabath et al. 2008) to study the selective constraints these genes are subjected to. Other methods can give misleading results when applied to overlapping genes (Holmes et al. 2006; Suzuki 2006; Pavesi 2007; Sabath et al. 2008).
De Novo Genes Adapt to Their Genomes
Our results suggest that de novo genes do adapt to their genome. More specifically, de novo genes evolve very rapidly shortly after their origin. As they age, they tend to experience increasingly severe selective constraints, and their codon usage tends to approach that of the ancestral gene from which they originate.
Our results are consistent with population genetics theory (Hartl and Clark 1997). Viruses have large population sizes. At such large sizes, natural selection is highly efficient, which has two consequences regarding de novo genes. First, they are likely to become fixed in a population only if they provide some selective advantage. Second, even though a de novo gene might initially only provide a very small fitness benefit, in a large population this fitness benefit can be sufficient to cause the gene’s fixation. Immediately after its origin, the sequence of a de novo gene will typically be far from optimal for the (rudimentary) function it provides, unlike a gene originated through modification of an existing gene. Consequently, one would expect a de novo gene to evolve rapidly shortly after its origin and to become better adapted as it ages, resulting in increased selective constraints and a decreased rate of sequence change.
Our results also suggest that in general, the ancestral genes are more constrained in sequence than the de novo genes which overlap them. However, this pattern can be reversed in old de novo genes. In particular, three de novo genes of our data set, which encode the Ilarvirus 2 b protein, the morbillivirus C protein, and betanodavirus B protein (taxa 7, 10 and 12), are subject to stronger selective constraints than their ancestral genes (fig. 3b). We speculate that this reversal could reflect an evolutionary “tug of war” between the two genes over the dominance of the sequence. An ultimate “victory” of a de novo gene in this tug of war would consist in the disappearance of the ancestral gene. We speculate that some viral genes that do not overlap any other gene today may have originated through overprinting but have eventually lost the overlap. A similar scenario has been proposed for an overlapping region of two protein-coding genes within the genome of archaeal Thermoplasma (Rogozin et al. 2002). These overlapping genes are nonoverlapping in other related genomes, possibly because of duplication and consequent loss (Rogozin et al. 2002).
Our Evolutionary Inferences Are Robust and Biologically Coherent
For most overlapping gene pairs in our data set, the identification of the de novo gene is highly reliable, as the ancestral gene has a much wider phylogenetic distribution than the de novo gene. For instance, the de novo movement protein of Tymoviruses (taxon 5) has homologs only in this genus, whereas its ancestor, the methyltransferase-guanylyltransferase has homologs in over a dozen families (Rozanov et al. 1992).
One important caveat of our analysis is that we are unable to estimate the absolute age of de novo genes but only their age relative to the divergence of a viral housekeeping gene that encodes the RNA-dependent RNA polymerase. Our relative age estimates of de novo genes, however, are broadly consistent with their taxonomic distribution (table 1, column 6): as expected, young de novo genes show a more restricted distribution than older de novo genes. The youngest genes (groups 1–3 in fig. 3) are found in less than one genus, with gene 1 occurring only in a single species (these observations are not due to sequencing bias, as each taxon considered contains several species or genera). Conversely, all oldest de novo genes (6–12) are found at least throughout a whole genus (in two genera for cases 10 and 11).
A case where gene age may have been overestimated is that of taxon 3 (betacoronavirus I gene). Our analysis suggested that the I and ORF9b genes have a common origin (see supplementary information, Supplementary Material online, case 3) despite their different lengths (98aa and 207aa, respectively) and lack of significant sequence similarity (not shown). This inference is based on assuming functionality for the unannotated ORFs in five closely related genomes (supplementary fig. S4, Supplementary Material online). Consequently, we calculated the age of the I gene by considering the node common to I and ORF9b (marked with blue line, supplementary fig. S5, Supplementary Material online, group 3). In the alternative scenario where these two genes have independent origins, their estimated age would be reduced. Nevertheless, the overall pattern of the results would remain unchanged (not shown). Another caveat to our study is that RNA secondary structure, and selection pressure for high protein expression level may be partly responsible for the differences we observed in codon usage (Plotkin and Kudla 2011).
Finally, the estimated rate of evolution of most ancestral and de novo proteins (fig. 3) is generally coherent with their function or effect on viral fitness (table 3). For instance, the ancestral methyltransferase-guanyltransferase of tymoviruses experiences severe constraints, as expected from an enzyme, whereas the de novo protein I of betacoronaviruses, which is dispensable for replication, experiences low or no constraints. We note two exceptions: first, the orthohepadnavirus replicase gene, encoding an essential reverse transcriptase function, is not subject to very strong selective constraint (e.g., dN/dS between 0.28 and 0.78, taxon 11 in fig. 3b). However, this discrepancy is readily explained. The overlapping region of the replicase gene in fact encodes two domains (fig. 1): a disordered, hypervariable linker and the reverse transcriptase domain. The relaxed selective constraint is the result of a high dN/dS for the linker (average of 0.94) and a very low dN/dS for the reverse transcriptase domain (average of 0.15). Second, the tymovirus movement protein gene has a dN/dS of 1, suggesting an absence of selective pressure, despite encoding an important function that allows the spread of viral RNA between cells. Again, this discrepancy can be attributed to the fact that the movement protein consists of a slowly evolving region (around aa 1–400) and a fast-evolving region (C-terminal 200aa) (results not shown).
Young De Novo Genes Might Have Strain-Specific Functions and Be Difficult to Detect by Sequence Analysis
Our results have two practical implications. First, they suggest that recently evolved genes might have strain-specific functions, or possibly no function, as suggested previously (Trifonov and Rabadan 2009) for PB1-F2 (taxon 1), the youngest de novo genes in our data set. Experimental studies should thus take this possibility into account. Our current method to estimate dN/dS does not tell us whether the elevated dN/dS values observed in some young de novo genes indicate neutral evolution or positive selection. However, it is possible that they come not only from neutral mutations but also from beneficial mutations subject to positive selection, which would reflect evolutionary adaptations.
Second, our results have implications for the identification of overlapping genes. Current bioinformatics methods to detect overlapping genes use the signature of purifying selection (Firth and Brown 2005, 2006; Sabath et al. 2009; Sabath and Graur 2010). These recently developed methods have had great success and lead to discoveries in many viral taxa (Chung et al. 2008; Firth 2008; Firth and Atkins 2008a, b; Sabath et al. 2009; Firth and Atkins 2009a, b, c; Firth and Atkins 2010, Firth et al. 2010). However, the signature of purifying selection is mostly absent in young de novo genes. Thus, the number of young de novo genes may be much larger than it appears, because these methods can simply not detect many such genes.
Conclusion and Perspectives
In closing, we point to several directions for future research. Approaches to estimate selection pressures in overlapping genes (e.g., Sabath et al. 2008) lag behind those for nonoverlapping genes (reviewed in Anisimova and Kosiol 2009), which can detect lineage-specific and site-specific selection pressures. The development of advanced methods could, for instance, reveal the role of positive selection in de novo gene origination and perhaps predict interactions between proteins encoded by overlapping genes, such as the Rz and Rz1 genes of bacteriophage lambda (Zhang and Young 1999). Finally, further research is needed to shed light on how exactly de novo overlapping genes originate and become established, i.e., the mutational events that result in their expression, their frequency, and their effects on viral fitness.
Materials and Methods
Sequence Analyses
We extracted all fully sequenced viral genomes from the NCBI viral genome database (Bao et al. 2004) in June 2011 and identified all viral proteins annotated in these genomes. All homology searches were carried out against a database of these proteins, using PSI-basic local alignment search tool (BLAST) (Altschul et al. 1997) with an E value cutoff of 10−6. We performed all multiple sequence alignments using MAFFT (Katoh et al. 2002) and constructed phylogenetic trees with the BIONG method (Gascuel 1997). We rooted these trees with the mid-point rooting method (Farris 1972). We predicted the domain organization of proteins encoded by overlapping genes using ANNIE (Ooi et al. 2009).
Collection of Viral Overlapping Genes
We identified from the literature a set of 40 overlapping gene pairs for which the expression of a protein product from two reading frames had been experimentally verified. All gene pairs in this data set come from viruses that infect eukaryotes. Among these gene pairs, we selected 29 pairs coming from viruses whose genome encodes an RNA-dependent RNA polymerase (RdRP), to facilitate comparison among clades (see later). We further narrowed the data set to overlapping gene pairs in which we could identify which gene had originated de novo (see procedure described later). In total, we obtained 12 gene pairs that correspond to 12 cases of de novo origin, stemming from 12 families of RNA viruses that met these criteria. The data set shares some genes with a previously published data set (4 cases out of 12: groups 4, 6, 8, and 11 below) (Rancurel et al. 2009). The reason why we could include only a minority of the genes published in the Rancurel data set (4 out of 17) is that we restricted ourselves to considering pairs in which both ancestral and de novo proteins had less than 50% amino acid divergence (percentage of identity). Table 1 lists, for each gene pair, the species taxonomy, the genome accession number, the names of the overlapping genes, and their lengths. In the rest of the article, we will refer to each case either by its genus or by the number of its clade, as listed in table 1. Table 3 lists bibliographical evidence about the expression, function, and fitness effect of mutations in the de novo gene.
Identifying De Novo and Ancestral Genes
To identify de novo gene candidates, we applied the criterion of monophyly stated in the introduction: one of the genes in an overlapping pair—the ancestral gene—must occur in each member of a viral clade, whereas the other gene—the de novo gene—must be restricted to a single subclade, the focal clade (fig. 1b). To find genes that meet this criterion, we first identified, for each gene pair, homologous protein products in related genomes. (We found no evidence of duplicated genes in the genomes under study, hence all our homologs are orthologs.) We aligned the homologous protein sequences of the ancestral protein (which is more phylogenetically widespread) and constructed their phylogenetic tree. By manual examination of these trees, we identified 12 cases of de novo origin (tables 1 and 3) that met the criterion of monophyly. We scanned the related genomes of the focal clade for unannotated ORFs to overcome missing genes due to fault annotation. These trees also allowed us to infer the internal node of a tree closest to the origin of the de novo gene. We call this node the last common ancestor (LCA, marked with a blue circle in the hypothetical example of fig. 1b). To ensure that the identification of the LCA is not biased by genome annotation, we manually examined the related genomes for presence of homologous unannotated ORFs. We provide detailed explanations of the challenges in de novo gene and LCA identification in the supplementary information, Supplementary Material online (supplementary figs. S1–S4, Supplementary Material online). The phylogenetic tree and the corresponding genomic maps of the 12 cases are presented in supplementary figure S5, Supplementary Material online. For all gene pairs that met the monophyly criterion, we also determined the DNA sequence alignments corresponding to the amino acid sequence alignments (of the ancestral proteins), to enable the calculations described later.
Estimation of the Relative Age of Origin of the De Novo Genes
To understand the evolutionary dynamics of de novo genes, one needs to estimate their age of origin and to compare this age among different clades. This estimation is made difficult by differences in mutation rate, population dynamics, and selection pressures among different viral genomes and genes (Duffy et al. 2008). To alleviate these difficulties, we calibrated our estimates with a reference molecule, the RdRP protein domain, which is common to all clades in the study (Bruenn 2003). The RdRP domain has a common origin and similar tertiary structure in the clades we study (Bruenn 2003), and thus we assume that as a first approximation it is subject to similar functional and structural constraints. In each genome listed in table 1, we identified the RdRP domain by using HHpred (Soding et al. 2005) against the PFAM database (Finn et al. 2008) with an E-value cutoff of 10−10. We identified the orthologous RdRP domains within the other genomes of each clade using PSI-BLAST, aligned them, and constructed their phylogenetic trees. Supplementary figure S6, Supplementary Material online, presents these “RdRP trees.”
We defined the focal clade of the RdRP tree as the smallest clade that contains all the taxa found within the focal clade of the phylogenetic tree of the overlapping genes. The LCA for the RdRP tree was defined as earlier. We compared the focal clades in the RdRP tree and the tree of overlapping genes and found that in 9 of 12 cases the focal clades were identical, whereas in three cases (2, 5, and 12, within the genera Cardiovirus, Tymovirus, and Betanodavirus, respectively), we found minor differences (supplementary fig. S6, Supplementary Material online). Overall, this comparison suggested that the RdRP genes and the overlapping gene pairs have similar evolutionary histories. On the basis of the RdRP tree, we thus estimated as a proxy for the age of a de novo gene the sequence divergence of the RdRP domain in each focal clade since the origin of the de novo gene, i.e., its accumulated genetic distance D along the tree branches since the LCA. To estimate D, we generated 100 bootstrap RdRP trees. For each tree i (1 ≤ i ≤ 100), we calculated Di, the average length of the phylogenetic tree branches between the LCA and each extant genome that contains the de novo gene. For the example in figure 1b, Di would calculate as [d(LCA, T1) + d(LCA, T2) + d(LCA, T3)]/3, where d(LCA, T1) = b1 + b2, d(LCA, T2) = b3 + b2, and d(LCA, T3) = b4, and b1, b2, b3, and b4 are the branch lengths shown in the figure. Finally, we estimate D as the average over all the bootstrap trees, 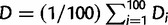 . We estimated all branch lengths by the BIONG method (Gascuel 1997). In supplementary figure S7, Supplementary Material online, we present D and the standard deviation within each group. For convenience, we ordered the clades in table 1 according to increasing D.
. We estimated all branch lengths by the BIONG method (Gascuel 1997). In supplementary figure S7, Supplementary Material online, we present D and the standard deviation within each group. For convenience, we ordered the clades in table 1 according to increasing D.
Analysis of the Evolutionary Properties of Overlapping Genes
For each of our 12 overlapping gene pairs (table 1), we collected the full sequences of the ancestral and the de novo genes, the sequence of the region of the genome where they overlapped, and the sequences of other genes annotated in the genome in which they occur. We also collected this information for homologous overlapping genes in related genomes within the focal clade (see earlier). As all subsequent analyses are carried on sequences within the focal clade, which is defined by the distribution of the de novo protein rather than the ancestral protein, it is independent of the BLAST cutoff. We used these data to study the following three properties of ancestral and de novo genes:
The “relative sequence divergence” between pairs of homologous proteins that the genes encode. We define the sequence divergence between two proteins as the proportion of amino acids in which they differ and the “relative” sequence divergence between two protein regions as their sequence divergence normalized (divided) by the sequence divergence of the corresponding ancestral proteins over their entire length. Consider, as a hypothetical example, the protein products of the overlapping genes in taxa T1 and T2 in figure 1b. The relative divergence between the ancestral proteins (red) of taxa T1 and T2 is the divergence between the protein region encoded by the part of the red gene that overlaps with the blue gene in taxa T1 and T2, divided by the divergence between the full-length red proteins of taxa T1 and T2. Analogously, the relative divergence between the de novo proteins of taxa T1–T2 is the divergence between the protein region of the blue gene that overlaps with the red gene in T1 and T2 (in this case the whole blue protein), again divided by the divergence between the full-length red proteins of taxa T1 and T2. The reason why we chose to normalize divergence in this way is to allow comparison between pairs of species that have diverged at different times (e.g., species T1–T2 diverged more recently than T1–T3 in fig. 1b). We calculated the relative sequence divergence in this way for all homologous pairs of ancestral proteins and for all homologous pairs of de novo proteins in each of our 12 clades containing de novo gene pairs. Note that if the ancestral gene overlaps the de novo gene over its entire length, the relative divergence of the ancestral protein will be 1. This happens in case 6 (umbravirus).
The “selective constraint” in the overlapping regions, estimated by the method of Sabath et al. (2008). This method, developed specifically for overlapping genes, accounts for independent selection pressures acting simultaneously on two overlapping genes by extending the single-gene model of codon evolution (Goldman and Yang 1994) to estimate the nonsynonymous/synonymous rate ratio (dN/dS) for each gene (reading frame) separately. Like the relative divergence, we calculated the selective constraint for pairs of homologous sequences. Specifically, we calculated selective constraint for all ancestral genes and de novo genes in each of our 12 cases. To prevent artifacts from saturation of synonymous substitutions, we restricted the estimation of the selective constraint to pairs in which both ancestral and de novo proteins had less than 50% amino acid divergence (percentage of identity). Because the method is inaccurate at low divergences (Sabath et al. 2008), we excluded sequence pairs with less than 1% amino acid divergence.
The CSI of the overlapping regions relative to protein coding genes in the rest of the genome. CSI is based on the algorithm used to calculate the CAI (Sharp and Li 1987), which is the most commonly used measure of codon usage bias. The CAI compares the codon usage of a gene with that of a reference set of highly expressed genes in a given genome to examine whether a protein-coding gene is subject to selection for high translation rate (and thus presumably highly expressed). Instead, in this study, we apply the CSI as a measure of the similarity between the codon usage of a gene and that of all other genes in the same genome. Therefore, we calculated the CSI of a given gene as described in Sharp and Li (1987) with the difference that the reference used was the codon usage of all other protein-coding genes in the same genome instead of the codon usage only of highly expressed genes. We performed this calculation for the overlapping regions of all 12 ancestral/de novo gene pairs. Note that unlike relative divergence and selective constraint, the CSI is calculated for single genes and not for pairs of homologous genes.
Supplementary Material
Supplementary information and figures S1–S7 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Robert Belshaw, Gkikas Magiorkinis, and Angelo Pavesi for comments on the manuscript and Andrew Firth for bibliographical references. D.K. acknowledges support by the Wellcome Trust grant number 090005. A.W. acknowledges support through Swiss National Science Foundation grant 315230-129708, as well as through the YeastX project of SystemsX.ch, and the University Priority Research Program in Systems Biology at the University of Zurich.
References
- Abroi A, Gough J. Are viruses a source of new protein folds for organisms?—virosphere structure space and evolution. Bioessays. 2011;33:626–635. doi: 10.1002/bies.201000126. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Kosiol C. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol Biol Evol. 2009;26:255–271. doi: 10.1093/molbev/msn232. [DOI] [PubMed] [Google Scholar]
- Babushok DV, Ostertag EM, Kazazian HH. Current topics in genome evolution: molecular mechanisms of new gene formation. Cellular Mol Life Sci. 2007;64:542–554. doi: 10.1007/s00018-006-6453-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y, Federhen S, Leipe D, Pham V, Resenchuk S, Rozanov M, Tatusov R, Tatusova T. National center for biotechnology information viral genomes project. J Virol. 2004;78:7291–7298. doi: 10.1128/JVI.78.14.7291-7298.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck J, Nassal M. Hepatitis B virus replication. World J Gastroenterol. 2007;13:48–64. doi: 10.3748/wjg.v13.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Lindfors HA, Kern AD, Jones CD. Evidence for de novo evolution of testis-expressed genes in the Drosophila yakuba/Drosophila erecta clade. Genetics. 2007;176:1131–1137. doi: 10.1534/genetics.106.069245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw R, Pybus OG, Rambaut A. The evolution of genome compression and genomic novelty in RNA viruses. Genome Res. 2007;17:1496–1504. doi: 10.1101/gr.6305707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornberg-Bauer E, Huylmans AK, Sikosek T. How do new proteins arise? Curr Opin Struct Biol. 2010;20:390–396. doi: 10.1016/j.sbi.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Bozarth CS, Weiland JJ, Dreher TW. Expression of Orf-69 of turnip yellow mosaic-virus is necessary for viral spread in plants. Virology. 1992;187:124–130. doi: 10.1016/0042-6822(92)90301-5. [DOI] [PubMed] [Google Scholar]
- Bruenn JA. A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucleic Acids Res. 2003;31:1821–1829. doi: 10.1093/nar/gkg277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Zhao RP, Jiang HF, Wang W. De novo origination of a new protein-coding gene in Saccharomyces cerevisiae. Genetics. 2008;179:487–496. doi: 10.1534/genetics.107.084491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campitelli L, Ciccozzi M, Salemi M, Taglia F, Boros S, Donatelli I, Rezza G. H5N1 influenza virus evolution: a comparison of different epidemics in birds and humans (1997–2004) J Gen Virol. 2006;87:955–960. doi: 10.1099/vir.0.81397-0. [DOI] [PubMed] [Google Scholar]
- Chen W, Calvo PA, Malide D, et al. (13 co-authors) A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001a;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- Chen WS, Calvo PA, Malide D, et al. (13 co-authors) A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001b;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- Chen HH, Kong WP, Zhang L, Ward PL, Roos RP. A picornaviral protein synthesized out of frame with the polyprotein plays a key role in a virus-induced immune-mediated demyelinating disease. Nat Med. 1995;1:927–931. doi: 10.1038/nm0995-927. [DOI] [PubMed] [Google Scholar]
- Chen S, Zhang YE, Long M. New genes in Drosophila quickly become essential. Science. 2010;330:1682–1685. doi: 10.1126/science.1196380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirico N, Vianelli A, Belshaw R. Why genes overlap in viruses. Proc R Soc B Biol Sci. 2010;277:3809–3817. doi: 10.1098/rspb.2010.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung BY, Miller WA, Atkins JF, Firth AE. An overlapping essential gene in the Potyviridae. Proc Natl Acad Sci U S A. 2008;105:5897–5902. doi: 10.1073/pnas.0800468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S, Shackelton LA, Holmes EC. Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet. 2008;9:267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Ekman D, Elofsson A. Identifying and quantifying orphan protein sequences in fungi. J Mol Biol. 2010;396:396–405. doi: 10.1016/j.jmb.2009.11.053. [DOI] [PubMed] [Google Scholar]
- Elhaik E, Sabath N, Graur D. The “inverse relationship between evolutionary rate and age of mammalian genes” is an artifact of increased genetic distance with rate of evolution and time of divergence. Mol Biol Evol. 2006;23:1–3. doi: 10.1093/molbev/msj006. [DOI] [PubMed] [Google Scholar]
- Farris JS. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972;106:645–668. [Google Scholar]
- Fenner BJ, Thiagarajan R, Chua HK, Kwang J. Betanodavirus B2 is an RNA interference antagonist that facilitates intracellular viral RNA accumulation. J Virol. 2006;80:85–94. doi: 10.1128/JVI.80.1.85-94.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth AE. Bioinformatic analysis suggests that the Orbivirus VP6 cistron encodes an overlapping gene. Virol J. 2008;5:48. doi: 10.1186/1743-422X-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth AE, Atkins JF. Bioinformatic analysis suggests that a conserved ORF in the waikaviruses encodes an overlapping gene. Arch Virol. 2008a;153:1379–1383. doi: 10.1007/s00705-008-0119-5. [DOI] [PubMed] [Google Scholar]
- Firth AE, Atkins JF. Bioinformatic analysis suggests that the Cypovirus 1 major core protein cistron harbours an overlapping gene. Virol J. 2008b;5:62. doi: 10.1186/1743-422X-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth AE, Atkins JF. Analysis of the coding potential of the partially overlapping 3' ORF in segment 5 of the plant fijiviruses. Virol J. 2009a;6:32. doi: 10.1186/1743-422X-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth AE, Atkins JF. A case for a CUG-initiated coding sequence overlapping torovirus ORF1a and encoding a novel 30 kDa product. Virol J. 2009b;6:136. doi: 10.1186/1743-422X-6-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth AE, Atkins JF. Evidence for a novel coding sequence overlapping the 5'-terminal approximately 90 codons of the gill-associated and yellow head okavirus envelope glycoprotein gene. Virol J. 2009c;6:222. doi: 10.1186/1743-422X-6-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth AE, Atkins JF. Candidates in Astroviruses, Seadornaviruses, Cytorhabdoviruses and Coronaviruses for +1 frame overlapping genes accessed by leaky scanning. Virol J. 2010;7:17. doi: 10.1186/1743-422X-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth AE, Blitvich BJ, Wills NM, Miller CL, Atkins JF. Evidence for ribosomal frameshifting and a novel overlapping gene in the genomes of insect-specific flaviviruses. Virology. 2010;399:153–166. doi: 10.1016/j.virol.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth AE, Brown CM. Detecting overlapping coding sequences with pairwise alignments. Bioinformatics. 2005;21:282–292. doi: 10.1093/bioinformatics/bti007. [DOI] [PubMed] [Google Scholar]
- Firth AE, Brown CM. Detecting overlapping coding sequences in virus genomes. BMC Bioinformatics. 2006;7:75. doi: 10.1186/1471-2105-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F, Peng D, Hingley ST, Weiss SR, Masters PS. The internal open reading frame within the nucleocapsid gene of mouse hepatitis virus encodes a structural protein that is not essential for viral replication. J Virol. 1997;71:996–1003. doi: 10.1128/jvi.71.2.996-1003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii Y, Kiyotani K, Yoshida T, Sakaguchi T. Conserved and non-conserved regions in the Sendai virus genome: evolution of a gene possessing overlapping reading frames. Virus Genes. 2001;22:47–52. doi: 10.1023/a:1008130318633. [DOI] [PubMed] [Google Scholar]
- Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14:685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- Ghadge GD, Ma L, Sato S, Kim J, Roos RP. A protein critical for a Theiler's virus-induced immune system-mediated demyelinating disease has a cell type-specific antiapoptotic effect and a key role in virus persistence. J Virol. 1998;72:8605–8612. doi: 10.1128/jvi.72.11.8605-8612.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Guerzoni D, McLysaght A. De novo origins of human genes. Plos Genet. 2011;7:e1002381. doi: 10.1371/journal.pgen.1002381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyader S, Ducray DG. Sequence analysis of Potato leafroll virus isolates reveals genetic stability, major evolutionary events and differential selection pressure between overlapping reading frame products. J Gen Virol. 2002;83:1799–1807. doi: 10.1099/0022-1317-83-7-1799. [DOI] [PubMed] [Google Scholar]
- Hanzlik TN, Dorrian SJ, Johnson KN, Brooks EM, Gordon KH. Sequence of RNA2 of the Helicoverpa armigera stunt virus (Tetraviridae) and bacterial expression of its genes. J Gen Virol. 1995;76(Pt 4):799–811. doi: 10.1099/0022-1317-76-4-799. [DOI] [PubMed] [Google Scholar]
- Hartl DL, Clark AG. Principles of population genetics. Sunderland (MA): Sinauer Associates; 1997. [Google Scholar]
- Hernandez M, Villegas P, Hernandez D, Banda A, Maya L, Romero V, Tomas G, Perez R. Sequence variability and evolution of the terminal overlapping VP5 gene of the infectious bursal disease virus. Virus Genes. 2010;41:59–66. doi: 10.1007/s11262-010-0485-4. [DOI] [PubMed] [Google Scholar]
- Holmes EC, Lipman DJ, Zamarin D, Yewdell JW. Comment on “Large-scale sequence analysis of avian influenza isolates.”. Science. 2006;313:1573. doi: 10.1126/science.1131729. author reply 1573. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Hughes MA. Patterns of nucleotide difference in overlapping and non-overlapping reading frames of papillomavirus genomes. Virus Res. 2005;113:81–88. doi: 10.1016/j.virusres.2005.03.030. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Westover K, da Silva J, O'Connor DH, Watkins DI. Simultaneous positive and purifying selection on overlapping reading frames of the tat and vpr genes of simian immunodeficiency virus. J Virol. 2001;75:7966–7972. doi: 10.1128/JVI.75.17.7966-7972.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto T, Mise K, Takeda A, Okinaka Y, Mori K, Arimoto M, Okuno T, Nakai T. Characterization of striped jack nervous necrosis virus subgenomic RNA3 and biological activities of its encoded protein B2. J Gen Virol. 2005;86:2807–2816. doi: 10.1099/vir.0.80902-0. [DOI] [PubMed] [Google Scholar]
- Jacob F. Evolution and tinkering. Science. 1977;196:1161–1166. doi: 10.1126/science.860134. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Sutter BA, McClure MA. Molecular evolution of the Paramyxoviridae and Rhabdoviridae multiple-protein-encoding P gene. Mol Biol Evol. 2000;17:75–86. doi: 10.1093/oxfordjournals.molbev.a026240. [DOI] [PubMed] [Google Scholar]
- Kaessmann H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010;20:1313–1326. doi: 10.1101/gr.101386.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keese PK, Gibbs A. Origins of genes: “big bang” or continuous creation? Proc Natl Acad Sci U S A. 1992;89:9489–9493. doi: 10.1073/pnas.89.20.9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles DG, McLysaght A. Recent de novo origin of human protein-coding genes. Genome Res. 2009;19:1752–1759. doi: 10.1101/gr.095026.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumbholz A, Philipps A, Oehring H, Schwarzer K, Eitner A, Wutzler P, Zell R. Current knowledge on PB1-F2 of influenza A viruses. Med Microbiol Immunol. 2011;200:69–75. doi: 10.1007/s00430-010-0176-8. [DOI] [PubMed] [Google Scholar]
- Le Duff Y, Blanchet M, Sureau C. The Pre-S1 and antigenic loop infectivity determinants of the hepatitis B virus envelope proteins are functionally independent. J Virol. 2009;83:12443–12451. doi: 10.1128/JVI.01594-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Weon S, Lee S, Kang C. Relative codon adaptation index, a sensitive measure of codon usage bias. Evol Bioinform Online. 2010;6:47–55. doi: 10.4137/ebo.s4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MT, Jones CD, Kern AD, Lindfors HA, Begun DJ. Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. Proc Natl Acad Sci U S A. 2006;103:9935–9939. doi: 10.1073/pnas.0509809103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Ding SW. Virus counterdefense: diverse strategies for evading the RNA-silencing immunity. Annu Rev Microbiol. 2006;60:503–531. doi: 10.1146/annurev.micro.60.080805.142205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Dong Y, Jiang Y, Jiang HF, Cai J, Wang W. A de novo originated gene depresses budding yeast mating pathway and is repressed by the protein encoded by its antisense strand. Cell Res. 2010;20:408–420. doi: 10.1038/cr.2010.31. [DOI] [PubMed] [Google Scholar]
- Li KS, Guan Y, Wang J, et al. (22 co-authors) Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature. 2004;430:209–213. doi: 10.1038/nature02746. [DOI] [PubMed] [Google Scholar]
- Li C-Y, Zhang Y, Wang Z, Zhang Y, Cao C, et al. (14 co-authors) A human-specific de novo protein-coding gene associated with human brain functions. Plos Comput Biol. 2010 doi: 10.1371/journal.pcbi.1000734. (3):e1000734. doi:10.1371/journal.pcbi.1000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M, Betran E, Thornton K, Wang W. The origin of new genes: glimpses from the young and old. Nat Rev Genet. 2003;4:865–875. doi: 10.1038/nrg1204. [DOI] [PubMed] [Google Scholar]
- Mazur I, Anhlan D, Mitzner D, Wixler L, Schubert U, Ludwig S. The proapoptotic influenza A virus protein PB1-F2 regulates viral polymerase activity by interaction with the PB1 protein. Cell Microbiol. 2008;10:1140–1152. doi: 10.1111/j.1462-5822.2008.01116.x. [DOI] [PubMed] [Google Scholar]
- McAuley JL, Zhang K, McCullers JA. The effects of influenza A virus PB1-F2 protein on polymerase activity are strain specific and do not impact pathogenesis. J Virol. 2010;84:558–564. doi: 10.1128/JVI.01785-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch DJ, Dolan A, Donald S, Rixon FJ. Sequence determination and genetic content of the short unique region in the genome of herpes simplex virus type 1. J Mol Biol. 1985;181:1–13. doi: 10.1016/0022-2836(85)90320-1. [DOI] [PubMed] [Google Scholar]
- McGirr KM, Buehuring GC. Tax & rex: overlapping genes of the Deltaretrovirus group. Virus Genes. 2006;32:229–239. doi: 10.1007/s11262-005-6907-z. [DOI] [PubMed] [Google Scholar]
- McVeigh A, Fasano A, Scott DA, Jelacic S, Moseley SL, Robertson DC, Savarino SJ. IS1414, an Escherichia coli insertion sequence with a heat-stable enterotoxin gene embedded in a transposase-like gene. Infect Immun. 2000;68:5710–5715. doi: 10.1128/iai.68.10.5710-5715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizokami M, Orito E, Ohba K, Ikeo K, Lau JY, Gojobori T. Constrained evolution with respect to gene overlap of hepatitis B virus. J Mol Evol. 1997;44(Suppl 1):S83–S90. doi: 10.1007/pl00000061. [DOI] [PubMed] [Google Scholar]
- Narechania A, Terai M, Burk RD. Overlapping reading frames in closely related human papillomaviruses result in modular rates of selection within E2. J Gen Virol. 2005;86:1307–1313. doi: 10.1099/vir.0.80747-0. [DOI] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nekrutenko A, Wadhawan S, Goetting-Minesky P, Makova KD. Oscillating evolution of a mammalian locus with overlapping reading frames: an XLalphas/ALEX relay. PLoS Genet. 2005;1:e18. doi: 10.1371/journal.pgen.0010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer JC, Denson J, Mehta PK, et al. (17 co-authors) Large-scale sequence analysis of avian influenza isolates. Science. 2006;311:1576–1580. doi: 10.1126/science.1121586. [DOI] [PubMed] [Google Scholar]
- Ohno S. Birth of a unique enzyme from an alternative reading frame of the preexisted, internally repetitious coding sequence. Proc Natl Acad Sci U S A. 1984;81:2421–2425. doi: 10.1073/pnas.81.8.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Evolution by gene duplication. New York: Springer; 1970. [Google Scholar]
- Ooi HS, Kwo CY, Wildpaner M, Sirota FL, Eisenhaber B, Maurer-Stroh S, Wong WC, Schleiffer A, Eisenhaber F, Schneider G. ANNIE: integrated de novo protein sequence annotation. Nucleic Acids Res. 2009;37:W435–W440. doi: 10.1093/nar/gkp254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson JB, Thomas D, Lewicki H, Billeter MA, Oldstone MBA. V and C proteins of measles virus function as virulence factors in vivo. Virology. 2000;267:80–89. doi: 10.1006/viro.1999.0118. [DOI] [PubMed] [Google Scholar]
- Pavesi A. Origin and evolution of overlapping genes in the family Microviridae. J Gen Virol. 2006;87:1013–1017. doi: 10.1099/vir.0.81375-0. [DOI] [PubMed] [Google Scholar]
- Pavesi A. Pattern of nucleotide substitution in the overlapping nonstructural genes of influenza A virus and implication for the genetic diversity of the H5N1 subtype. Gene. 2007;402:28–34. doi: 10.1016/j.gene.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Pavesi A, De Iaco B, Granero MI, Porati A. On the informational content of overlapping genes in prokaryotic and eukaryotic viruses. J Mol Evol. 1997;44:625–631. doi: 10.1007/PL00006185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DL. Isolation and characterization of the major protein and glycoprotein of hepatitis B surface antigen. J Biol Chem. 1981;256:6975–6983. [PubMed] [Google Scholar]
- Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011;12:32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancurel C, Khosravi M, Dunker AK, Romero PR, Karlin D. Overlapping genes produce proteins with unusual sequence properties and offer insight into de novo protein creation. J Virol. 2009;83:10719–10736. doi: 10.1128/JVI.00595-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozin IB, Spiridonov AN, Sorokin AV, Wolf YI, Jordan IK, Tatusov RL, Koonin EV. Purifying and directional selection in overlapping prokaryotic genes. Trends Genet. 2002;18:228–232. doi: 10.1016/s0168-9525(02)02649-5. [DOI] [PubMed] [Google Scholar]
- Rozanov MN, Koonin EV, Gorbalenya AE. Conservation of the putative methyltransferase domain: a hallmark of the “Sindbis-like” supergroup of positive-strand RNA viruses. J Gen Virol. 1992;Pt 8(73):2129–2134. doi: 10.1099/0022-1317-73-8-2129. [DOI] [PubMed] [Google Scholar]
- Ryabov EV, Oparka KJ, Santa Cruz S, Robinson DJ, Taliansky ME. Intracellular location of two groundnut rosette umbravirus proteins delivered by PVX and TMV vectors. Virology. 1998;242:303–313. doi: 10.1006/viro.1997.9025. [DOI] [PubMed] [Google Scholar]
- Ryabov EV, Robinson DJ, Taliansky ME. A plant virus-encoded protein facilitates long-distance movement of heterologous viral RNA. Proc Natl Acad Sci U S A. 1999;96:1212–1217. doi: 10.1073/pnas.96.4.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabath N, Graur D. Detection of functional overlapping genes: simulation and case studies. J Mol Evol. 2010;71:308–316. doi: 10.1007/s00239-010-9386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabath N, Landan G, Graur D. A method for the simultaneous estimation of selection intensities in overlapping genes. PLoS One. 2008;3:e3996. doi: 10.1371/journal.pone.0003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabath N, Price N, Graur D. A potentially novel overlapping gene in the genomes of Israeli acute paralysis virus and its relatives. Virol J. 2009;6:144. doi: 10.1186/1743-422X-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz AI, Fraile A, Gallego JM, Malpica JM, Garcia-Arenal F. Genetic variability of natural populations of cotton leaf curl geminivirus, a single-stranded DNA virus. J Mol Evol. 1999;49:672–681. doi: 10.1007/pl00006588. [DOI] [PubMed] [Google Scholar]
- Scheets K. Maize chlorotic mottle machlomovirus expresses its coat protein from a 1.47-kb subgenomic RNA and makes a 0.34-kb subgenomic RNA. Virology. 2000;267:90–101. doi: 10.1006/viro.1999.0107. [DOI] [PubMed] [Google Scholar]
- Senanayake SD, Hofmann MA, Maki JL, Brian DA. The nucleocapsid protein gene of bovine coronavirus is bicistronic. J Virol. 1992;66:5277–5283. doi: 10.1128/jvi.66.9.5277-5283.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH. The Codon Adaptation Index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmeier M, Hamilton JA, LeGall T, et al. (12 co-authors) DisProt: the database of disordered proteins. Nucleic Acids Res. 2007;35:D786–D793. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R. The birth of new exons: mechanisms and evolutionary consequences. RNA. 2007;13:1603–1608. doi: 10.1261/rna.682507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrou S, Baida G, Viktorova E, Ghadge G, Agol VI, Roos RP. Theiler's murine encephalomyelitis virus L* amino acid position 93 is important for virus persistence and virus-induced demyelination. J Virol. 2010;84:1348–1354. doi: 10.1128/JVI.01585-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y. Natural selection on the influenza virus genome. Mol Biol Evol. 2006;23:1902–1911. doi: 10.1093/molbev/msl050. [DOI] [PubMed] [Google Scholar]
- Tautz D, Domazet-Loso T. The evolutionary origin of orphan genes. Nat Rev Genet. 2011;12:692–702. doi: 10.1038/nrg3053. [DOI] [PubMed] [Google Scholar]
- Toll-Riera M, Bosch N, Bellora N, Castelo R, Armengol L, Estivill X, Alba MM. Origin of primate orphan genes: a comparative genomics approach. Mol Biol Evol. 2009a;26:603–612. doi: 10.1093/molbev/msn281. [DOI] [PubMed] [Google Scholar]
- Toll-Riera M, Bosch N, Bellora N, Castelo R, Armengol L, Estivill X, Alba MM. Origin of primate orphan genes: a comparative genomics approach. Mol Biol Evol. 2009b;26:603–612. doi: 10.1093/molbev/msn281. [DOI] [PubMed] [Google Scholar]
- Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- Trifonov V, Rabadan R. The Contribution of the PB1-F2 protein to the fitness of Influenza A viruses and its recent evolution in the 2009 Influenza A (H1N1) pandemic virus. PLoS Curr. 2009;1:RRN1006. doi: 10.1371/currents.RRN1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eyll O, Michiels T. Non-AUG-initiated internal translation of the L* protein of Theiler's virus and importance of this protein for viral persistence. J Virol. 2002;76:10665–10673. doi: 10.1128/JVI.76.21.10665-10673.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargason JM, Szittya G, Burgyan J, Hall TM. Size selective recognition of siRNA by an RNA silencing suppressor. Cell. 2003;115:799–811. doi: 10.1016/s0092-8674(03)00984-x. [DOI] [PubMed] [Google Scholar]
- Wang J, Li S, Zhang Y, Zheng H, Xu Z, Ye J, Yu J, Wong GK. Vertebrate gene predictions and the problem of large genes. Nat Rev Genet. 2003;4:741–749. doi: 10.1038/nrg1160. [DOI] [PubMed] [Google Scholar]
- Wardrop EA, Briedis DJ. Characterization of V protein in measles virus-infected cells. J Virol. 1991;65:3421–3428. doi: 10.1128/jvi.65.7.3421-3428.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland JJ, Dreher TW. Infectious TYMV RNA from cloned cDNA: effects in vitro and in vivo of point substitutions in the initiation codons of two extensively overlapping ORFs. Nucleic Acids Res. 1989;17:4675–4687. doi: 10.1093/nar/17.12.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DD, Irwin DM, Zhang YP. De novo origin of human protein-coding genes. PLoS Genet. 2011;7:e1002379. doi: 10.1371/journal.pgen.1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin HW, Ji LH, Scott SW, Symons RH, Ding SW. Ilarviruses encode a cucumovirus-like 2 b gene that is absent in other genera within the Bromoviridae. J Virol. 1998;72:6956–6959. doi: 10.1128/jvi.72.8.6956-6959.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Huang J. De novo origin of new genes with introns in Plasmodium vivax. FEBS Lett. 2011;585:641–644. doi: 10.1016/j.febslet.2011.01.017. [DOI] [PubMed] [Google Scholar]
- Zaaijer HL, van Hemert FJ, Koppelman MH, Lukashov VV. Independent evolution of overlapping polymerase and surface protein genes of hepatitis B virus. J Gen Virol. 2007;88:2137–2143. doi: 10.1099/vir.0.82906-0. [DOI] [PubMed] [Google Scholar]
- Zamarin D, Ortigoza MB, Palese P. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J Virol. 2006;80:7976–7983. doi: 10.1128/JVI.00415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- Zhang N, Young R. Complementation and characterization of the nested Rz and Rz1 reading frames in the genome of bacteriophage lambda. Mol Gen Genet. 1999;262:659–667. doi: 10.1007/s004380051128. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Wang W. On the origin and evolution of new genes—a genomic and experimental perspective. J Genet Genomics. 2008;35:639–648. doi: 10.1016/S1673-8527(08)60085-5. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Zhang G, Zhang Y, Xu S, Zhao R, Zhan Z, Li X, Ding Y, Yang S, Wang W. On the origin of new genes in Drosophila. Genome Res. 2008;18:1446–1455. doi: 10.1101/gr.076588.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.