

Abstract
Gene expression levels correlate with multiple aspects of gene sequence and gene structure in phylogenetically diverse taxa, suggesting an important role of gene expression levels in the evolution of protein-coding genes. Here we present results of a genome-wide study of the influence of gene expression on synonymous codon usage, amino acid composition, and gene structure in the red flour beetle, Tribolium castaneum. Consistent with the action of translational selection, we find that synonymous codon usage bias increases with gene expression. However, the correspondence between tRNA gene copy number and optimal codons is weak. At the amino acid level, translational selection is suggested by the positive correlation between tRNA gene numbers and amino acid usage, which is stronger for highly expressed genes. In addition, there is a clear trend for increased use of metabolically cheaper, less complex amino acids as gene expression increases. tRNA gene numbers also correlate negatively with amino acid size/complexity (S/C) score indicating the coupling between translational selection and selection to minimize the use of large/complex amino acids. Interestingly, the analysis of 10 additional genomes suggests that the correlation between tRNA gene numbers and amino acid S/C score is widespread and might be explained by selection against negative consequences of protein misfolding. At the level of gene structure, three major trends are detected: 1) complete coding region length increases across low and intermediate expression levels but decreases in highly expressed genes; 2) the average intron size shows the opposite trend, first decreasing with expression, followed by a slight increase in highly expressed genes; and 3) intron density remains nearly constant across all expression levels. These changes in gene architecture are only in partial agreement with selection favoring reduced cost of biosynthesis.
Keywords: Tribolium castaneum, expression, translational selection, size/complexity score, tRNA abundance, gene structure
Introduction
Levels of mRNA and protein expression between different genes vary across three and five orders of magnitude, respectively (Ghaemmaghami et al. 2003; Schwanhausser et al. 2011). This variation highlights the differences in energy and time that cells must allocate to the expression of genes whose products are required in different amounts. Such variation in gene expression levels provides an opportunity for selection to act on gene features that are not directly tied to gene function. For instance, changes in gene sequence and gene structure that increase the efficiency of steps involved in transcription, translation, transcript processing, and protein folding, or changes that reduce negative fitness consequences associated with errors that occur during these steps are expected to be especially favored in highly expressed genes. Over the last three decades, multiple studies have demonstrated that several gene features including synonymous codon usage, amino acid composition, rates of protein evolution, coding sequence (CDS) length, intron size, and intron density correlate with expression levels in prokaryotes, as well as in unicellular and multicellular eukaryotes. The phylogenetically widespread occurrence of these associations exposes the influential role of gene expression in the evolution of protein-coding genes.
The increase in synonymous codon usage bias with gene expression levels has been observed in organisms from all domains of life (Ikemura 1985; Duret and Mouchiroud 1999; Coghlan and Wolfe 2000; Ghaemmaghami et al. 2003; Urrutia and Hurst 2003; Comeron 2004; Cutter et al. 2006; Drummond and Wilke 2008; Ingvarsson 2008; Qiu et al. 2011). This pattern is attributed to the action of weak selection on synonymous sites that generate translationally optimal codons (Bulmer 1991; Kliman and Hey 1993; Akashi 1995; Comeron 2006). Such selection for increased efficiency of translation is invoked because, in a number of species, the set of synonymous codons that is preferentially used in highly expressed genes corresponds to most abundant tRNAs (Ikemura 1985; Moriyama and Powell 1997; Duret 2000; Kanaya et al. 2001; Comeron 2004; Ingvarsson 2007). Correspondence between synonymous codons and their decoding tRNAs increases the speed and accuracy of translation and may also reduce the cost associated with proofreading during protein synthesis (reviewed in Andersson and Kurland 1990; Powell and Moriyama 1997; Akashi 2001; Duret 2002; Hershberg and Petrov 2008; but see Shah and Gilchrist 2010). However, correspondence between tRNA abundance and codon usage is not always strong, and selection pressures other than, or in addition to, translational efficiency are likely to contribute to codon usage patterns (reviewed in Duret 2002; Chamary et al. 2006; Plotkin and Kudla 2011).
The amino acid composition of proteins also varies with increasing levels of gene expression (Duret 2000; Jansen and Gerstein 2000; Akashi and Gojobori 2002; Akashi 2003; Seligmann 2003; Cutter et al. 2006; Heizer et al. 2006; Raiford et al. 2008). Amino acids that are used more frequently in highly expressed genes tend to correspond to most abundant tRNAs, suggesting the action of translational selection at the amino acid level (Lobry and Gautier 1994; Percudani et al. 1997; Duret 2000; Akashi 2003). Several studies also demonstrate that amino acid composition is influenced by selection to reduce the metabolic cost of protein production (Akashi and Gojobori 2002; Heizer et al. 2006; Swire 2007; Raiford et al. 2008). Using species-specific estimates of the chemical energy cost required to produce each amino acid, these studies show an increase in the frequency of biosynthetically cheaper amino acids in highly expressed proteins (see also Swire 2007 for expression-independent ways to detect selection for metabolic efficiency). Similarly, using the molecular weight (or a combination of weight and complexity) as a proxy for cost, other studies demonstrate that proteins tend to minimize the use of large/complex amino acids (Dufton 1997; Seligmann 2003; Urrutia and Hurst 2003; Kahali et al. 2007) resulting in a negative correlation between gene expression levels and the average protein cost (Seligmann 2003; Urrutia and Hurst 2003). It is notable that cost measures based on the size and complexity of amino acids reflect not only chemical energy investment but also costs associated with the stability of a protein’s final conformation (Dufton 1997). Thus, the observation that highly expressed proteins use fewer large/complex amino acids suggests that selection may act to maximize metabolic efficiency, as well as stability of protein folding (Dufton 1997).
Aspects of gene structure such as protein size, intron size, and intron density also show associations with gene expression levels. Protein size and expression levels are negatively correlated in yeast (Coghlan and Wolfe 2000; Jansen and Gerstein 2000; Akashi 2003), Caenorhabditis elegans (Castillo-Davis et al. 2002; Fahey and Higgins 2007), Drosophila melanogaster (Lemos et al. 2005; Fahey and Higgins 2007), and vertebrates (Urrutia and Hurst 2003; Comeron 2004; Subramanian and Kumar 2004), although Duret and Mouchiroud (1999) report the opposite trends for C. elegans and D. melanogaster. Intron sizes also decline with increasing expression levels (Castillo-Davis et al. 2002; Urrutia and Hurst 2003; Comeron 2004). Both trends are consistent with selection acting to minimize the cost of transcription and translation in highly expressed genes. However, intron density (number of introns per kb of CDS) increases with gene expression level in D. melanogaster, Arabidopsis thaliana and humans (Comeron 2004; Carmel and Koonin 2009). This finding indicates that forces other than selection for reduced cost of biosynthesis must be acting to maintain intron presence in highly expressed genes.
In this article, we examine evidence for expression-mediated selection in the red flour beetle, Tribolium castaneum, the first fully sequenced representative of the most species-rich metazoan order, Coleoptera. We use genome sequence and genome-wide expression data to investigate the relationship between gene expression and synonymous codon usage, amino acid composition, and intron–exon gene structure. With regard to gene sequence, we show that 1) the bias in synonymous codon usage increases with expression levels but selection for increased efficiency of translation does not appear to be responsible for this trend; and 2) highly expressed proteins tend to contain higher proportions of small, structurally simple amino acids that also correspond to more abundant tRNAs. With regard to gene structure, we observe that 1) protein size increases across low and intermediate expression levels but decreases in highly expressed genes; 2) the average intron size shows the opposite trend, first decreasing with expression, followed by a slight increase in highly expressed genes; and 3) intron density remains nearly constant across all expression levels.
Materials and Methods
Sequence Data
Genome sequence information for T. castaneum (T.cas 3.0 assembly) was obtained from BeetleBase (http://beetlebase.org/). Analyzed sequences include CDSs and introns of the genes included in the official gene set (OGS, Kim et al. 2010) specified in the Official_Gene_GFF3 folder (ftp://bioinformatics.ksu.edu/pub/BeetleBase/3.0/GFF3).
Sequences with problematic open reading frames (CDSs that lack start or stop codons, have internal stop codons, and those with sequence lengths that are not a multiple of three) and/or suspicious exon–intron structure (exon or intron length <20 bp) were removed from the dataset. We excluded all sequences with matches to transposable elements based on BLAST searches (tblastx, E = 1e-5) against the database of TEs available at RepBase (http://www.girinst.org/repbase/). Genes with multiple splice form annotations (only two genes have annotated splice forms in the official gene set) and introns with embedded exons of other genes were also removed. Our final data sets contained 13,630 CDSs (∼18 Mb) and 44,844 introns (∼32 Mb).
We approximated the GC content of putatively neutral sequences (GCi_neut) using the GC content of introns between 150 and 2,000 bp with 50 bp from each end removed. We limited intron sequences to this range in an effort to exclude sites that potentially evolve under selection (e.g., splice sites and regulatory regions in long introns). The final set of “neutral” sequences contains 9,309 introns (∼6 Mb). This set comprises only ∼20% of all introns because most introns in T. castaneum are very short (average size 812 bp and median 53 bp). Intron GC content for each gene was calculated using the concatenated sequences of individual introns.
For the analysis of intron–exon gene structure, we limited our dataset to genes with introns. We further excluded genes containing introns longer than 10,000 bp to avoid incorporating genes with potentially missannotated intron–exon structure that can have a large outlier effect on the analysis of intron sizes. The final data set for the analysis of gene structure included 8,689 genes (∼64% of all genes).
Expression Data
Gene expression data for whole-body beetles and reproductive tracts (RT) of both sexes was obtained using custom-designed microarrays as described previously (Prince et al. 2010). The array contains probes for ∼98% of genes (16,130/16,434) based on the initial annotation (genome release T.cas_1.0). We retained probes for genes included in the T. castaneum Official Gene Set that was generated using the latest genome assembly (genome release T.cas_3.0) and updated annotation (Kim et al. 2010). In our final data set, expression data were available for 12,946 genes. For whole body expression data, we used the average of male and female expression. All three expression data sets (whole-body, male RT, female RT) were used for the analysis of codon bias. Because little difference was observed between the results using different expression data sets, we report the rest of the analyses using whole-body expression.
For the analysis of the correlations between gene structure and expression levels, we examined trends within and between three expression classes. We defined low (25% of genes), intermediate (50% of genes), and high (25% of genes) expression categories based on the complete set of genes in the expression data set (N = 12,946). However, because genes lacking introns (which are excluded from gene structure analyses) are not uniformly distributed across these classes, the set of analyzed genes contained 1,758 genes in the low expression class, 4,405 genes in intermediate expression class, and 2,526 genes in high expression class.
Identification of Optimal Codons
We used the program CodonW (Peden 1999, http://codonw.sourceforge.net/) to obtain the raw usage of synonymous codons for each amino acid in each gene. Optimal codons, that is codons that increase in frequency with gene expression levels (Lloyd and Sharp 1991; Peden 1999) were identified by a significant positive correlation between codon frequency and expression levels obtained from whole body and reproductive tracts of males and females, as described above. For our final set of optimal codons, we retained codons that increase in frequency with gene expression levels obtained from both male and female reproductive tracts since gene expression data from fewer tissues is more likely to reflect “magnitude” rather than “breadth” of expression. The set of optimal codons was then used to calculate the “frequency of optimal codons” (FOP, Ikemura 1981), a measure of codon bias for each gene, using the program CodonW (Peden 1999, http://codonw.sourceforge.net/).
Analysis of tRNA Genes
The number of genes encoding tRNAs was used as a proxy for tRNA abundance (Percudani et al. 1997; Kanaya et al. 1999; Duret 2000; Akashi 2003). We scanned the genome of T. castaneum for tRNA genes with tRNAscan-SE software (Lowe and Eddy 1997). Using default settings, we obtained 436 tRNA genes, 200 of which were classified as pseudogenes by tRNAscan-SE. Out of 236 remaining tRNA genes, 11 encode selenocysteine tRNAs and 255 specify tRNAs for 20 standard amino acids. All 225 tRNA genes encoding tRNAs for standard amino acids had a Cove score above 58 bits (far above default 20) and were included in the final set of functional tRNA genes.
Measures of Protein Cost
We used two measures reflecting the cost of amino acid synthesis. The first measure is the number of high-energy phosphate bonds (∼PO4) required to synthesize each amino acid in yeast under aerobic conditions (Wagner 2005). We abbreviate this measure as HEB (high energy bond). The second measure is the size/complexity (S/C) score assigned to each amino acid on the bases of its molecular weight and overall shape (Dufton 1997). Each of these proxies for amino acid cost was used to calculate the average (per amino acid) cost of protein synthesis.
Statistical Analyses
Statistical analyses were carried out using Jump 9.0.2 software. Whenever the trends are illustrated by binning the data points, all Spearman’s correlation coefficients were calculated using all data points independently, not the bin averages. For multiple regression analyses, expression levels, CDS length and intron sizes were log10 transformed. Where necessary, correction for multiple tests was carried out using the sequential Bonferroni procedure (Holm 1979; Rice 1989).
Results
Gene Expression and Synonymous Codon Usage
Two lines of evidence suggest that selection influences synonymous nucleotide composition in T. castaneum. First, the GC content of synonymous sites is substantially higher than that of introns: The GC contents of the third codon positions (GC3) and of 4-fold degenerate third codon positions (GC4) are 51.9% and 55.3%, respectively, whereas the GC content of introns (GCi) is 32.4%, close to the 33% genome average (Richards et al. 2008). Second, although there is a positive correlation between GC4 and GC content of the same-gene introns (rS = 0.263, P < 0.0001), regression analysis shows that variation in GCi accounts only for ∼8% of variation in GC4. Furthermore, when we limit the analysis to putatively neutral intron sequences (see Materials and Methods), the correlation between GC4 and GC content of the same-gene introns is much weaker (rS = 0.046, P = 0.0012). Together, these results provide the first indication that neutral mechanisms such as regional variation in mutation bias or GC-biased gene conversion (Marais 2003) cannot fully account for the observed base composition at synonymous sites. The alternative explanation involves selection on synonymous sites that may operate at different levels including translational efficiency, mRNA stability, splicing efficiency, and transcriptional efficiency (Duret 2002; Chamary et al. 2006; Plotkin and Kudla 2011; Trotta 2011). As selection for translational efficiency has been invoked most frequently, we investigated if this selective force is also relevant in T. castaneum genome.
Typically, selection for increased translational efficiency is inferred when 1) there is a positive correlation between gene expression levels and codon usage bias, and 2) codons that increase in frequency with expression (optimal codons) correspond to most abundant tRNAs (reviewed in Plotkin and Kudla 2011, but see Shah and Gilchrist 2010). We find that in T. castaneum, codon bias increases with expression, but correspondence between tRNA abundance (as measured by tRNA gene copy number) and the optimal codons is weak. We identified 25 optimal codons on the basis of the positive correlation between codon usage and gene expression levels (table 1). As expected, the per gene summary of codon bias measured by the frequency of optimal codons (FOP) is also positively correlated with gene expression levels from all three expression datasets (rS = 0.256, 0.269, 0.255 for male RT, female RT, and whole body, respectively, all P < 0.0001; fig. 1). As 24 of 25 optimal codons in T. castaneum are G/C-ending and FOP increases with expression, there is also a positive correlation between GC3 and expression levels (rS = 0.195, 0.214, 0.202 for male RT, female RT, and whole body, respectively, all P < 0.0001; N = 12,946). As transcription itself can be mutagenic (Beletskii and Bhagwat 1996; Kim et al. 2007), positive association between GC3 and expression may result from transcription-associated mutation bias. In this case, we would also expect to see a positive correlation between gene expression and the GC content of neutral introns (GCi_neut). However, we observe a negative correlation between GCi_neut and gene expression (rS = −0.224, −0.175, and −0.168 for male RT, female RT, and whole body, respectively, all P < 0.0001; N = 4,932), whereas the correlation between GC3 and expression remains positive in this set of genes. Thus, the increase in GC3 with expression is observed despite the transcription-mediated mutation pressure to reduce GC content in highly expressed genes. These results suggest that expression-mediated selection rather than mutational bias is responsible for the positive correlation between synonymous codon usage bias (FOP) and gene expression.
Table 1.
Optimal Codons and tRNA Genes in T. castaneum.
| AA | Optimal | Anticodon | tRNAb | tRNAc | Male | Female | Whole |
|---|---|---|---|---|---|---|---|
| Codona | 5′-3′ | number | decoding OC | RTd | RTe | bodyf | |
| Ala | GCU | AGC | 14 | −0.014g | −0.028 | 0.005g | |
| GCC | GGC | 0 | AGC | 0.16 | 0.176 | 0.184 | |
| GCA | UGC | 2 | −0.15 | −0.17 | −0.155 | ||
| GCG | CGC | 3 | CGC/UGC | 0.061 | 0.078 | 0.017g | |
| Pro | CCU | AGG | 7 | 0.004g | −0.001g | 0.011g | |
| CCC | GGG | 0 | AGG | 0.185 | 0.186 | 0.17 | |
| CCA | UGG | 13 | −0.075 | −0.078 | −0.059 | ||
| CCG | CGG | 1 | 0.001g | 0.015g | −0.004g | ||
| Val | GUU | AAC | 7 | −0.031 | −0.049 | −0.038 | |
| GUC | GAC | 0 | AAC | 0.077 | 0.09 | 0.106 | |
| GUA | UAC | 5 | −0.069 | −0.057 | −0.058 | ||
| GUG | CAC | 3 | CAC /UAC | 0.112 | 0.104 | 0.081 | |
| Thr | ACU | AGU | 5 | 0.031 | 0.004g | 0.025 | |
| ACC | GGU | 0 | AGU | 0.097 | 0.111 | 0.142 | |
| ACA | UGU | 3 | −0.119 | −0.12 | −0.123 | ||
| ACG | CGU | 2 | CGU/UGU | 0.079 | 0.088 | 0.039 | |
| Gly | GGU | ACC | 0 | 0.022g | 0.008g | 0.03 | |
| GGC | GCC | 8 | GCC | 0.069 | 0.095 | 0.067 | |
| GGA | UCC | 15 | −0.203 | −0.196 | −0.145 | ||
| GGG | CCC | 1 | CCC/UCC | 0.205 | 0.196 | 0.157 | |
| Ile | AUU | AAU | 7 | −0.059 | −0.089 | −0.087 | |
| AUC | GAU | 0 | AAU | 0.137 | 0.148 | 0.171 | |
| AUA | UAU | 2 | −0.065 | −0.043 | −0.078 | ||
| Arg | CGU | ACG | 5 | 0.008g | 0.016g | 0.0002g | |
| CGC | GCG | 0 | ACG | 0.075 | 0.068 | 0.052 | |
| CGA | UCG | 4 | −0.154 | −0.15 | −0.13 | ||
| CGG | CCG | 0 | −0.029 | −0.012g | −0.038 | ||
| AGA | UCU | 3 | 0.012g | −0.006g | 0.031 | ||
| AGG | CCU | 3 | CCU/UCU | 0.225 | 0.23 | 0.209 | |
| Leu | CUU | AAG | 5 | −0.111 | −0.123 | −0.081 | |
| CUC | GAG | 0 | AAG | 0.05 | 0.076 | 0.084 | |
| CUA | UAG | 2 | −0.019g | −0.013g | 0.0003g | ||
| CUG | CAG | 2 | UAG/CAG | 0.055 | 0.059 | 0.06 | |
| UUA | UAA | 2 | −0.096 | −0.07 | −0.121 | ||
| UUG | CAA | 4 | CAA/UAA | 0.237 | 0.182 | 0.188 | |
| Ser | AGU | ACU | 0 | GCU | 0.108 | 0.071 | 0.075 |
| AGC | GCU | 3 | GCU | 0.045 | 0.056 | 0.06 | |
| UCU | AGA | 4 | −0.071 | −0.06 | −0.053 | ||
| UCC | GGA | 0 | −0.001g | 0.015g | 0.037 | ||
| UCA | UGA | 2 | −0.039 | −0.041 | −0.048 | ||
| UCG | CGA | 2 | CGA/UGA | 0.09 | 0.103 | 0.073 | |
| Asp | GAU | AUC | 0 | −0.023g | −0.028 | −0.043 | |
| GAC | GUC | 10 | |||||
| Cys | UGU | ACA | 0 | 0.009g | −0.014g | −0.011g | |
| UGC | GCA | 3 | |||||
| Asn | AAU | AUU | 0 | −0.1 | −0.112 | −0.135 | |
| AAC | GUU | 5 | GUU | ||||
| His | CAU | AUG | 0 | −0.054 | −0.043 | −0.052 | |
| CAC | GUG | 7 | GUG | ||||
| Tyr | UAU | AUA | 0 | −0.107 | −0.094 | −0.124 | |
| UAC | GUA | 13 | GUA | ||||
| Phe | UUU | AAA | 1 | −0.122 | −0.143 | −0.17 | |
| UUC | GAA | 5 | GAA/AAA | ||||
| Gln | CAA | UUG | 5 | −0.075 | −0.101 | −0.064 | |
| CAG | CUG | 3 | CUG/UUG | ||||
| Glu | GAA | UUC | 8 | −0.137 | −0.156 | −0.108 | |
| GAG | CUC | 5 | CUC/UUC | ||||
| Lys | AAA | UUU | 6 | −0.161 | −0.194 | −0.158 | |
| AAG | CUU | 5 | CUU/UUU |
aOptimal codons (underlined) identified by positive correlation between codon frequency and gene expression levels in tissues from male and female reproductive tracts.
btRNA gene copy number.
cPredicted decoding tRNA for optimal codons assigned according to classical wobble rules (first anticodon–third codon positions): G-C/U; C-G; I-U/C>A; U-A/G. Spearman’s correlation between codon frequency and expression levels in dmale reproductive tract, efemale reproductive tract, and fwhole body. gNonsignificant after Bonferroni sequential correction.
Fig. 1.

Relationship between the frequency of optimal codons (FOP) and gene expression levels. Genes are grouped into 10 bins of equal sizes (∼1,295 genes each) according to increasing levels of gene expression. Spearman’s correlation coefficient was calculated using all genes independently. Similar results are obtained using gene expression levels from reproductive tissues (see Discussion, Gene Expression and Synonymous Codon Usage).
If expression-mediated selection acts to enhance the efficiency of translation, codons that increase in frequency with expression should also be the codons that correspond to most abundant decoding tRNAs. To investigate whether such correspondence exists in T. castaneum, we used tRNA gene copy number as a proxy for tRNA abundance and assigned decoding isoaccepting tRNAs to each optimal codon according to classical wobble rules (Crick 1966; Ikemura 1985) (table 1). Overall, support for the coadaptation between tRNA abundance and synonymous codon usage is weak for two reasons. First, for non-2-fold degenerate amino acids, in cases where a single I-starting tRNA decodes two codons, it is difficult to justify systematic preference of C-ending over U-ending codons because both I-U and I-C paring involves formation of two hydrogen bonds (Percudani and Ottonello 1999). Take Ala as an example. To decode the optimal codon GCC, tRNAAGC (where A is assumed to be modified to I) must be used. As this tRNA is most abundant, the correspondence between the optimal codon and tRNA abundance seems to support translational selection. However, since tRNAAGC can decode C- and U-ending codons, translational selection should favor both GCC and GCU codons as observed in C. elegans (Duret 2000; Percudani 2001). In our data set, for each optimal C-ending codon decoded by tRNAANN, the frequency of the U-ending codon decoded by the same tRNA tends to decrease with expression level. It is difficult to account for this pattern by invoking translational selection as the choice of C-ending codon over U-ending codon would require further explanation.
Second, complementary anticodon–codon interactions between the most abundant (major) tRNA and the optimal codon are rarely observed. Among seven 2-fold degenerate amino acids with optimal codons, only four are decoded by the complementary major tRNA (Asn, His, Tyr, and Phe). For amino acids Gln, Glu, and Lys and other amino acids with G-ending optimal codons, the major decoding tRNA frequently does not have a complementary anticodon. For example, the tRNA with complementary pairing to the Glu optimal codon GAG is less abundant than the tRNA with complementary pairing to the Glu nonoptimal codon, GAA (table 1). However, as classical rules of anticodon–codon pairing permit U-G but not C-A wobble pairing, the GAG optimal codon can be favored by translational selection as it can be decoded by both tRNAUUC and tRNACUC. The same reasoning can be applied to the rest of the amino acids where two tRNAs exist that can decode the same codon (tRNAUNN/CNN decoding NNG codons). However, Percudani (2001) argued against U-G wobble pairing as such pairing generates functional redundancy. In T. castaneum, as in C. elegans, for every tRNAUNN, there is a tRNACNN and allowing tRNAUNN to read G-ending codons would make tRNACNN functionally redundant. If tRNAUNN and tRNACNN only recognize complementary codons, translational selection is expected to favor the use of codons that are complementary to the most abundant tRNA of the two, which is generally not seen in our data set. If we strictly follow the complementary anticodon–codon pairing for A/G-ending codons (following Percudani 2001), only 2 of 11 A/G-ending optimal codons correspond to most abundant tRNAs. In contrast, in C. elegans, all five A/G-ending optimal codons can be predicted from tRNA gene numbers based on complementary anticodon–codon pairing rule (Duret 2000). Together, these results provide only limited support for coadaptation between tRNA abundance and expression-linked codon usage.
Gene Expression and Amino Acid Composition
Analysis of amino acid composition in T. castaneum shows that the use of 16 amino acids changes significantly with increasing levels of gene expression (table 2). There is an overall positive correlation between amino acid usage and tRNA gene copy number (rS = 0.584, P = 0.007; fig. 2). This association is stronger for highly expressed genes (top 10%, rS = 0.652, P = 0.002) than for genes expressed at low levels (bottom 10%, rS = 0.487, P = 0.029; fig. 2). These results are consistent with translational selection at the amino acid level (Lobry and Gautier 1994; Akashi 2003).
Table 2.
Associations between Amino Acid Usage and Expression.
| AA | AA usagea | rS (Whole Body)b | HEBc | S/Cd | tRNA |
|---|---|---|---|---|---|
| Trp | 0.011 | 0.016e | 75.50 | 73.00 | 4 |
| Cys | 0.020 | −0.152 | 26.50 | 57.16 | 3 |
| Met | 0.022 | −0.005e | 36.50 | 64.68 | 6 |
| His | 0.024 | −0.055 | 29.00 | 58.70 | 7 |
| Tyr | 0.033 | 0.033 | 59.00 | 57.00 | 13 |
| Gln | 0.041 | 0.115 | 10.50 | 37.48 | 8 |
| Phe | 0.043 | −0.145 | 61.00 | 44.00 | 6 |
| Pro | 0.050 | 0.084 | 14.50 | 31.80 | 21 |
| Asn | 0.051 | 0.016e | 18.50 | 33.72 | 5 |
| Arg | 0.052 | −0.227 | 20.50 | 56.34 | 15 |
| Gly | 0.053 | 0.103 | 14.50 | 1.00 | 24 |
| Asp | 0.054 | 0.200 | 15.50 | 32.72 | 10 |
| Ile | 0.058 | −0.055 | 38.00 | 16.04 | 9 |
| Thr | 0.058 | −0.029 | 21.50 | 21.62 | 10 |
| Ala | 0.059 | 0.039 | 14.50 | 4.76 | 19 |
| Val | 0.064 | 0.074 | 29.00 | 12.28 | 15 |
| Glu | 0.067 | 0.203 | 9.50 | 36.48 | 13 |
| Lys | 0.070 | 0.129 | 36.00 | 30.14 | 11 |
| Ser | 0.075 | −0.072 | 14.50 | 17.86 | 11 |
| Leu | 0.094 | −0.019e | 37.00 | 16.04 | 15 |
aOverall amino acid usage in T. castaneum genome.
bSpearman’s correlation coefficient between amino acid usage and gene expression.
cCost of amino acid measured by the number of high-energy phosphate bonds (Wagner 2005).
dCost of amino acid measured by S/C score (Dufton 1997).
eNonsignificant after sequential Bonferroni correction.
Fig. 2.
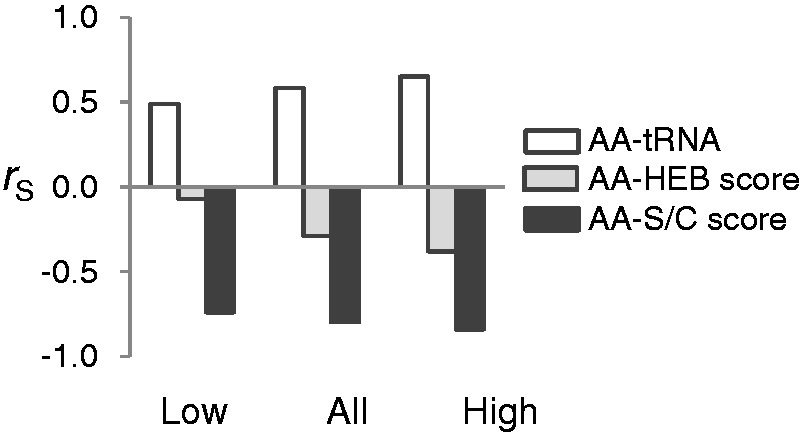
Spearman’s correlation between amino acid composition and tRNA gene copy number, HEB score, and S/C score for genes expressed at low levels (bottom 10% of genes; rS = 0.487, P = 0.029; rS = −0.073, P = 0.761 and rS = −0.740, P < 0.001, respectively), all genes (rS = 0.584, P = 0.007; rS = −0.289, P = 0.216 and rS = −0.797, P < 0.0001, respectively), and for genes expressed at high levels (top 10% of genes; rS = 0.652, P = 0.002; rS = −0.382, P = 0.097 and rS = −0.842, P < 0.0001, respectively).
To assess whether cost selection could also underlie expression-associated differences in amino acid composition in T. castaneum, we examined if changes in the amino acid usage reflect differences in amino acid cost. We employed two measures of the amino acid cost. The first measure is the number of high-energy phosphate bonds required for the synthesis of each amino acid, HEB score (Wagner 2005; Raiford et al. 2008). Note that these estimates of the amino acid cost are based on the amino acid biosynthetic pathways that are specific to yeast under aerobic conditions (Wagner 2005) and therefore, might only approximate the cost of amino acid synthesis in T. castaneum (table 2). The second measure of amino acid cost is the S/C score developed by Dufton (1997), which is independent of the biosynthetic pathways and is based on the combination of molecular weight and the complexity of each amino acid (table 2). The two cost measures are not significantly correlated with each other (rS = 0.341, P = 0.141), likely because S/C score reflects additional components of the amino acid cost not accounted for by the measure based on the investment of the chemical energy alone (Dufton 1997; Seligmann 2003).
Irrespective of which amino acid cost measure is used, average (per amino acid) protein cost is negatively correlated with gene expression levels (HEB score: rS = −0.099, P < 0.0001 and S/C score: rS = −0.165, P < 0.0001; N = 12,946). The observed decrease in the average protein cost is gradual and is present across all expression levels indicating that selection for reduced cost/complexity is not limited to highly expressed genes (fig. 3). A closer look at the contribution of each amino acid to this pattern reveals that 5 out of 7 amino acids that decrease significantly in frequency with increasing expression levels (His, Cys, Arg, Phe, Ile) have high S/C score and/or high HEB score, and 8 out of 9 amino acids that increase in frequency with expression levels (Gln, Glu, Asp, Pro, Lys, Val, Ala, Gly) have low S/C score and/or low HEB score (table 2 and supplementary fig. S1, Supplementary Material online). Three amino acids (Thr, Ser, Tyr) do not follow these trends, with Thr and Ser decreasing with expression despite low S/C and HEB scores and Tyr slightly increasing with expression despite it having high S/C and HEB scores (table 2, supplementary fig. S1, Supplementary Material online). Nevertheless, we observe a stronger negative correlation between amino acid usage and both HEB score and S/C score for highly expressed genes (top 10%) than for genes expressed at low levels (bottom 10%) although the correlations are only significant for S/C score versus amino acid usage (fig. 2). Note also that two largest/most complex amino acids (highest S/C score) that do not change in frequency with expression (Trp and Met) are present at some of the lowest frequencies in the genome (table 2 and supplementary fig. S1, Supplementary Material online). These results support the cost selection hypotheses wherein that overall amino acid usage is optimized to minimize the use of large/complex amino acids (i.e., high S/C score) and increasing levels of gene expression favor the use of less complex amino acids, some of which are also energetically inexpensive (low HEB score).
Fig. 3.

Decrease in average protein cost with increasing levels of gene expression using S/C score (light bars, rS = −0.165, P < 0.0001; N = 12,946) or HEB (dark bars, rS = −0.099, P < 0.0001; N = 12,946) measures of amino acid cost.
We also find that S/C score (but not HEB score) correlates negatively with tRNA gene copy number (rS = −0.670, P = 0.001). This association arises because S/C score and tRNA gene copy number both correlate with amino acid composition: rS = −0.797, P < 0.0001 and rS = 0.584, P = 0.007, respectively. The interesting result is the lack of correlation between tRNA gene copy number and amino acid composition when we control for variation in S/C score (B = 0.041, P = 0.893), whereas the correlation between amino acid composition and S/C score remains negative after controlling for tRNA gene copy number (B = −0.614, P = 0.001). This result indicates that amino acid composition is primarily controlled by S/C score. Selection for increased efficiency of translation regulates tRNA pools such that most abundant tRNAs correspond to most frequently used amino acids that have low S/C scores. As levels of gene expression increase, the use of amino acids with lower S/C scores is favored, thus resulting in a stronger correlation between tRNA gene number and amino acid usage (i.e., amino acids that are more frequent are also smaller and correspond to more abundant tRNAs). That is, the increase in correlation between tRNA pools and amino acid composition across expression levels is mainly the consequence of underlying selection on the amino acid composition that minimizes the use of complex amino acids.
Gene Expression and Gene Architecture
We examined the relationship between gene expression levels and various gene features (CDS length, total intron size, average intron size, and intron density) in a set of 8,689 genes with introns. Contrary to expectations, CDS length increases with expression (rS = 0.317, P < 0.0001; table 4). As longer CDSs also have more introns (rS = 0.681, P < 0.0001), total intron size also increases with expression (rS = 0.084, P < 0.0001; table 4). Since CDS length and total intron size are correlated (rS = 0.356, P < 0.0001), the positive relationship between total intron size and expression may be a byproduct of the correlation between CDS length and expression. When we account for CDS length, the correlation between total intron size and expression is negative (B = −0.05, P < 0.0001). Similarly, average intron size decreases with expression after correction for CDS length (table 4). Clearly, these overall correlations do not allow the detection of opposing trends that might be present within different expression categories. For example, Carmel and Koonin (2009) showed that CDS length and total intron size do not decrease gradually with expression, but follow a nonmonotonic trend with the increase in CDS length and total intron size at intermediate levels of expression.
Table 4.
The Average Values for Each Gene Feature in Each Expression Category and the Results of Mann–Whitney U test for Differences between Categories.
| CDS Lengtha | Total Intron Sizea | Average Intron Sizea | Intron Number | Intron Densityb | |
|---|---|---|---|---|---|
| Low (1,758) | 855.35 | 2,008.25 | 689.09 | 2.35 | 3.19 |
| Medium (4,405) | 1,495.63 | 1,847.27 | 363.25 | 4.03 | 3.07 |
| High (2,526) | 1,662.69 | 2,255.17 | 388.75 | 4.70 | 3.20 |
| Mann–Whitney U test, P value | |||||
| Low–Medium | <0.0001 | 0.132 | <0.0001 | <0.0001 | 0.014c |
| Medium–High | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.001c |
| Low–High | <0.0001 | <0.0001 | 0.055 | <0.0001 | 0.702 |
aLength is measured in number of base pairs.
bIntron density is measured as number of introns per 1 kb of CDS.
cNonsignificant after sequential Bonferroni correction.
To assess how various gene features change with expression levels in finer detail, we analyzed trends within and among low, medium, and high expression classes defined on the bases of expression levels of the complete data set (see Materials and Methods). Three important findings emerge from this analysis. First, CDS length increases with gene expression for genes in low and medium expression classes, but decreases with expression in highly expressed genes (rS = −0.097, P < 0.0001; fig. 4A and table 3). Second, total intron size and the average intron size are lowest for genes with medium expression (table 4 and fig. 4A) and the distribution of average intron sizes does not differ between low and high expression classes (Mann–Whitney U test, P = 0.055). There is also a slight positive correlation between intron size and expression within the high expression class (average intron size: B = 0.056, P = 0.004 and total intron size: B = 0.046, P = 0.009, table 3). Clearly, the negative correlations between intron sizes and expression after correcting for CDS length are largely driven by genes with intermediate expression (table 3 and fig. 4A). Finally, intron density (number of introns per kb of CDS) remains nearly constant within and among expression classes (tables 3 and 4; fig. 4B).
Fig. 4.

Relationship between gene expression levels and CDS length, total and average intron size (A), intron number, and intron density (B). For illustrative purposes, genes are grouped into 10 bins of equal sizes. For statistical analyses, the comparisons were made within and between low, medium, and high expression categories (see Results, Gene Expression and Gene Architecture). Similar trends are observed when median values are used.
Table 3.
Correlations between Expression Levels and Various Gene Features.
| Gene feature | Spearman’s Correlation with Expression and Associated P Values | Regression Coefficient (B) when Controlling for Variation in CDS Length | ||||||
|---|---|---|---|---|---|---|---|---|
| All | Low | Medium | High | All | Low | Medium | High | |
| CDS length | 0.317 | 0.175 | 0.170 | −0.097 | — | — | — | — |
| <0.0001 | <0.0001 | <0.0001 | <0.0001 | — | — | — | — | |
| Total intron size | 0.084 | 0.043 | 0.040 | 0.009 | −0.05 | 0.041 | −0.042 | 0.046 |
| <0.0001 | 0.072 | 0.009a | 0.645 | <0.0001 | 0.09 | 0.002 | 0.009 | |
| Average intron size | 0.005 | 0.000 | −0.006 | 0.034 | −0.092 | 0.026 | −0.075 | 0.056 |
| 0.665 | 0.999 | 0.678 | 0.085 | <0.0001 | 0.267 | <0.0001 | 0.004 | |
| Intron density | 0.17 | −0.013 | 0.017 | 0.040 | — | — | — | — |
| 0.124 | 0.576 | 0.257 | 0.042a | — | — | — | — | |
aNonsignificant after sequential Bonferroni correction. Bonferroni correction was applied to correlation and regression analyses separately.
Discussion
It has been emphasized for some time now that the function of the final gene product is not the only determining factor in the evolution of gene sequences (Richmond 1970). The influence of gene expression on the evolution of gene sequence and structure has been given considerable attention owing to the fact that genes vary widely in the levels of mRNA and protein expression (Ghaemmaghami et al. 2003; Schwanhausser et al. 2011). As the complex processes of transcription and translation require the commitment of time and energy, changes in gene sequence and structure that offset these costs should be particularly favored in highly expressed genes. Expression-mediated selection may act to increase the efficiency and accuracy of various steps of transcript and protein synthesis and processing, as well as to minimize deleterious consequences of the inevitable mistakes that occur during these steps. In this article, we examined evidence for expression-mediated selection in T. castaneum by examining associations between levels of gene expression and synonymous codon usage, amino acid composition, and gene architecture.
Gene Expression and Synonymous Codon Usage
In various species, bias in synonymous codon usage increases with expression levels (see Introduction). Such a pattern has been attributed to weak selection favoring changes in synonymous sites that generate translationally optimal codons. Although we find that in T. castaneum synonymous sites are under selection and codon bias is stronger in highly expressed genes, the evidence for selection at the level of translational efficiency is weak.
Our analysis of GC content of protein coding genes indicates that synonymous base composition is under selection as neither variation in local base composition nor transcription-associated mutation bias can fully account for the overall high GC content of synonymous sites and positive correlation between GC3 and expression levels. Furthermore, codon usage bias increases with expression, suggesting the action of translational selection. However, we find only weak support for the expected correspondence between optimal codons and tRNA abundance. There are several explanations that may account for this result in a way that remains consistent with the action of translational selection. First, codon–anticodon recognition depends heavily on post-transcriptional modifications of tRNAs. Specifically, in addition to almost ubiquitous modification of A to I, U at the first anticodon position also undergoes extensive modifications that can expand or restrict the number of recognized codons (Agris et al. 2007). In the absence of the experimental data on post-transcriptional modifications of tRNAs in T. castaneum, we cannot be certain about the identities of tRNAs that decode optimal codons. Second, codons that correspond to the most abundant tRNAs may not be translated most accurately (Shah and Gilchrist 2010). Consequently, if codon bias is primarily driven by selection for translational accuracy, the strong correspondence between tRNA abundance and optimal codons is no longer expected. Finally, tRNA gene copy numbers might not accurately reflect tissue-specific tRNA abundance. Although these caveats make it impossible for us to rule out translational selection, differences in correspondence patterns between T. castaneum and C. elegans (where correspondence between major tRNAs and optimal codons clearly supports translational selection (Duret 2000; Percudani 2001) are large, and unless codon–anticodon pairing or the nature of translational selection differs between the two species, our data suggest that the increase in codon usage bias with expression in T. castaneum may be driven by forces other than translational selection. For example, recent work in yeast suggests that codon bias is the result of selection favoring increased efficiency of transcription rather than translation (Trotta 2011).
Gene Expression and Amino Acid Composition
Changes in amino acid composition with expression levels have been documented in a number of prokaryotes and eukaryotes (Akashi and Gojobori 2002; Urrutia and Hurst 2003; Cutter et al. 2006; Heizer et al. 2006; Raiford et al. 2008). Selection favoring efficient protein synthesis in highly expressed genes either by increasing the correspondence between amino acid usage and tRNA abundance (translational selection), or by minimizing the cost of protein synthesis (cost selection) have both been invoked to explain this pattern.
In T. castaneum, 16 amino acids vary as a function of expression levels. We find that as levels of gene expression increase, 1) correspondence between tRNA abundance and amino acid composition increases, and 2) average protein cost decreases (figs. 2 and 3). These results suggest that both cost selection and translational selection operate in T. castaneum genes, increasing in strength with increasing levels of gene expression. However, amino acid composition is primarily determined by S/C score. Selection for translational efficiency then adjusts tRNA pools to match amino acid composition. This leads to a negative correlation between tRNA abundance and S/C score. As a consequence, highly expressed proteins where the use of less complex amino acids is favored will show tighter correspondence between tRNA abundance and amino acid composition. Thus, the increase in correspondence between tRNA and amino acid composition with expression levels is a consequence of underlying selection on the amino acid composition that minimizes the use of complex amino acids.
Interestingly, the negative correlation between tRNA gene copy number and S/C score is not limited to T. castaneum. We compiled the numbers of tRNA genes for 10 species from different phylogenetic groups, from bacteria to humans (supplementary table S1, Supplementary Material online). Despite dramatic differences in numbers of tRNA genes among species, they are positively correlated (supplementary table S2, Supplementary Material online). As a result, for every species examined, there is a significant negative correlation between tRNA gene number and the S/C score (table 5). The amino acid composition is also relatively constant among species (Grantham et al. 1980; Doolittle 1981; Barrai et al. 1995). For example, the correlation between amino acid composition in yeast (data from Akashi 2003) and T. castaneum is very high (rS = 0.930, P < 0.0001). Consequently, the negative correlation between S/C score and amino acid composition is also prevalent, if not universal (Dufton 1997). These two pervasive correlations predict that amino acid composition and tRNA gene numbers should be positively correlated in all these species. Furthermore, if expression imposes strong enough cost selection to favor the use of less complex amino acids in highly expressed genes, all these species should show increased correspondence between tRNA abundance and amino acid composition with increasing levels of gene expression. These observations demonstrate the phylogenetically widespread coupling of cost selection when measured using S/C score with translational selection. Why should that be? To this end, we note the connection between the work of Dufton (1997) and, Drummond and Wilke (2008). Dufton (1997) observes negative correlation between amino acid composition and S/C score and argues that protein amino acid composition reflects selection to minimize both, the cost of amino acid biosynthesis and “conformational disruption” caused by large, chemically complex amino acid side chains. Drummond and Wilke (2008) identify selection against protein misfolding (translational robustness) as an underlying force that generates major patterns of sequence evolution that are conserved across different species, including correlations between gene expression, codon bias and protein evolution. It is then likely that S/C score reflects amino acid propensity to generate misfolded proteins. Given that misfolded proteins may impose a substantial fitness cost (Geiler-Samerotte et al. 2011), we suggest that selection against protein misfolding may explain the widespread correlations described above. The proposed adaptations that would reduce negative consequences of protein misfolding include 1) reduction of the proportion of mistranslated proteins, 2) reduction of the proportion of proteins that misfold if mistranslated, and 3) reduction of the proportion of proteins that misfold even if translated without mistakes (Drummond and Wilke 2008). The correspondence between tRNA abundance and amino acid composition increases the accuracy of translation, increasing the proportion of error-free proteins. The negative correlation between tRNA and S/C score results in increase in the proportion of properly folded proteins even if they are mistranslated as newly misincorporated amino acid is likely to be a small/simple amino acid, carried by a more abundant tRNA. Note also that if mistranslation occurs not because of incorrect codon–anticodon pairing, but because the tRNA is charged with a wrong amino acid, the mischarged amino acid is also likely to be a small/simple one (assuming that the pool of free amino acids is roughly proportional to that encoded in the genome). Finally, the proportion of error-free proteins that undergo spontaneous misfolding is reduced as a direct consequence of avoiding the use of large/complex amino acids altogether (negative correlation between S/C score and amino acid composition). Thus, a single force—selection against protein misfolding—can generate the correlations among amino acid composition, tRNA abundance, and S/C score present in a wide range of organisms. From this perspective, the S/C score is actually a proxy for propensity to induce misfolding and might represent the biochemical bases of the proposed misfolding hypothesis (Dufton 1997; Drummond and Wilke 2008).
Table 5.
Spearman’s Correlation between S/C Score and tRNA Gene Number in Different Species.
| Species | rS | P |
|---|---|---|
| Bacillus subtilis | −0.523 | 0.018 |
| Saccharomyces cerevisiae | −0.760 | 0.0001 |
| Caenorhabditis elegans | −0.674 | 0.001 |
| Tribolium castaneum | −0.670 | 0.001 |
| Drosophila melanogaster | −0.558 | 0.011 |
| Strongylocentrotus purpuratus | −0.481 | 0.032 |
| Danio rerio | −0.543 | 0.014 |
| Gallus gallus | −0.565 | 0.010 |
| Homo sapiens | −0.640 | 0.002 |
| Arabidopsis thaliana | −0.523 | 0.018 |
| Zea mays | −0.608 | 0.005 |
Gene Expression and Gene Structure
Using a subset of 8,689 genes with introns, we examined the association between gene expression and three gene features: CDS length, intron size, and intron density. With regard to CDS length, it is tempting to suggest that selection favors the reduction in protein size when expression levels become sufficiently high; but why CDS length increases across genes with low and medium expression remains unclear. The pattern is not dependent on intron presence or unique to T. castaneum, as the same pattern is observed for T. castaneum genes that lack introns (data not shown) and in other eukaryotes, such as: C. elegans, D. melanogaster, A. thaliana, and Homo sapiens (fig.1 in Carmel and Koonin 2009). As a consequence of the nonlinear relationship between expression levels and CDS length, even though the average cost of protein synthesis per amino acid decreases steadily across all expression levels (fig. 3) the total protein cost decreases only among highly expressed proteins (correlations between expression level and total protein cost for low: rS = 0.239, medium: rS = 0.196, high: rS = −0.116 expression classes; P < 0.0001 in all cases; correlation coefficients are based on total protein cost using S/C score, but similar correlations are obtained using high-energy bonds measure). These results indicate that if selection does act to minimize the cost of translation and transcription, it is only strong enough to influence CDS length among genes in the highest expression classes.
Unlike CDS length, patterns of variation in intron size and intron density among expression levels in T. castaneum differ from the trends observed in other eukaryotes (Comeron 2004; Fahey and Higgins 2007; Carmel and Koonin 2009). In our data set, total and average intron size increase among highly expressed genes, which is contrary to the expectation for selection to reduce transcriptional cost (Castillo-Davis et al. 2002) and suggests the presence of fitness benefits associated with larger intron sizes in highly expressed genes. Additionally, in contrast to other eukaryotes where intron density either increases or decreases with expression (Comeron 2004; Ren et al. 2006; Fahey and Higgins 2007; Lanier et al. 2008; Carmel and Koonin 2009), intron density in T. castaneum is largely independent of expression levels. The near constancy of intron density in differentially expressed genes indicates that there is no tendency for intron loss in highly expressed genes as would be expected in the presence of selection favoring reduction in the transcriptional cost and implies beneficial effects of intron presence that appear to be independent of expression levels in T. castaneum. Various advantages associated with intron presence have been proposed (Coghlan and Wolfe 2000; Fedorova and Fedorov 2003; Roy and Gilbert 2006; Niu 2007). They range from the immediate benefits of intron presence related to genome stability (Niu 2007) to long-term advantages associated with the increase in effectiveness of selection (Comeron and Kreitman 2000, 2002). Further investigation is required in order to understand which of these benefits maintain intron size and intron density in T. castaneum.
Supplementary Material
Supplementary tables S1 and S2 and figure S1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Data Accessibility
Data sets: DRYAD entry doi: 10.5061/dryad.r0t1q.
Acknowledgments
We thank Eric Watson and Heath Blackmon for useful discussions throughout this study and Esther Betrán and anonymous reviewers for helpful comments on the manuscript. This work was supported by funding from National Institutes of Health grant #2R01GM065414-05A1 to J.P.D.
References
- Agris PF, Vendeix FAP, Graham WD. tRNA’s wobble decoding of the genome: 40 years of modification. J Mol Biol. 2007;366(1):1–13. doi: 10.1016/j.jmb.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Akashi H. Inferring weak selection from patterns of polymorphism and divergence at “silent” sites in Drosophila DNA. Genetics. 1995;139(2):1067–1076. doi: 10.1093/genetics/139.2.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H. Gene expression and molecular evolution. Curr Opin Genetics Dev. 2001;11(6):660–666. doi: 10.1016/s0959-437x(00)00250-1. [DOI] [PubMed] [Google Scholar]
- Akashi H. Translational selection and yeast proteome evolution. Genetics. 2003;164(4):1291–1303. doi: 10.1093/genetics/164.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H, Gojobori T. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc Natl Acad Sci U S A. 2002;99(6):3695–3700. doi: 10.1073/pnas.062526999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson SG, Kurland CG. Codon preferences in free-living microorganisms. Microbiol Rev. 1990;54(2):198–210. doi: 10.1128/mr.54.2.198-210.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrai I, Volinia S, Scapoli C. The usage of oligopeptides in proteins correlates negatively with molecular weight. Int J Pept Protein Res. 1995;45(4):326–331. doi: 10.1111/j.1399-3011.1995.tb01045.x. [DOI] [PubMed] [Google Scholar]
- Beletskii A, Bhagwat AS. Transcription-induced mutations: increase in C to T mutations in the nontranscribed strand during transcription in Escherichia coli. Proc Natl Acad Sci U S A. 1996;93(24):13919–13924. doi: 10.1073/pnas.93.24.13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129(3):897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel L, Koonin EV. A universal nonmonotonic relationship between gene compactness and expression levels in multicellular eukaryotes. Genome Biol Evol. 2009;1:382–390. doi: 10.1093/gbe/evp038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Davis CI, Mekhedov SL, Hartl DL, Koonin EV, Kondrashov FA. Selection for short introns in highly expressed genes. Nat Genet. 2002;31(4):415–418. doi: 10.1038/ng940. [DOI] [PubMed] [Google Scholar]
- Chamary VJ, Parmley LJ, Hurst DL. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet. 2006;7:98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
- Coghlan A, Wolfe KH. Relationship of codon bias to mRNA concentration and protein length in Saccharomyces cerevisiae. Yeast. 2000;16(12):1131–1145. doi: 10.1002/1097-0061(20000915)16:12<1131::AID-YEA609>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Comeron JM. Selective and mutational patterns associated with gene expression in humans. Genetics. 2004;167(3):1293–1304. doi: 10.1534/genetics.104.026351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM. Weak selection and recent mutational changes influence polymorphic synonymous mutations in humans. Proc Natl Acad Sci U S A. 2006;103(18):6940–6945. doi: 10.1073/pnas.0510638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM, Kreitman M. The correlation between intron length and recombination in Drosophila: dynamic equilibrium between mutational and selective forces. Genetics. 2000;156(3):1175–1190. doi: 10.1093/genetics/156.3.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM, Kreitman M. Population, evolutionary and genomic consequences of interference selection. Genetics. 2002;161(1):389–410. doi: 10.1093/genetics/161.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick FHC. Codon—anticodon pairing: the wobble hypothesis. J Mol Biol. 1966;19(2):548–555. doi: 10.1016/s0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- Cutter AD, Wasmuth JD, Blaxter ML. The evolution of biased codon and amino acid usage in nematode genomes. Mol Biol Evol. 2006;23(12):2303–2315. doi: 10.1093/molbev/msl097. [DOI] [PubMed] [Google Scholar]
- Doolittle R. Similar amino acid sequences: chance or common ancestry? Science. 1981;214(4517):149–159. doi: 10.1126/science.7280687. [DOI] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134(2):341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufton MJ. Genetic code synonym quotas and amino acid complexity: cutting the cost of proteins? J Theor Biol. 1997;187(2):165–173. doi: 10.1006/jtbi.1997.0443. [DOI] [PubMed] [Google Scholar]
- Duret L. tRNA gene number and codon usage in the C. elegans genome are co-adapted for optimal translation of highly expressed genes. Trends Genet. 2000;16(7):287–289. doi: 10.1016/s0168-9525(00)02041-2. [DOI] [PubMed] [Google Scholar]
- Duret L. Evolution of synonymous codon usage in metazoans. Curr Opin Genetics Dev. 2002;12(6):640–649. doi: 10.1016/s0959-437x(02)00353-2. [DOI] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc Natl Acad Sci U S A. 1999;96(8):4482–4487. doi: 10.1073/pnas.96.8.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey M, Higgins D. Gene expression, intron density, and splice site strength in Drosophila and Caenorhabditis. J Mol Evol. 2007;65(3):349–357. doi: 10.1007/s00239-007-9015-y. [DOI] [PubMed] [Google Scholar]
- Fedorova L, Fedorov A. Introns in gene evolution. Genetica. 2003;118(2):123–131. [PubMed] [Google Scholar]
- Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond DA. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci U S A. 2011;108(2):680–685. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh W-K, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425(6959):737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Grantham R, Gautier C, Gouy M. Codon frequencies in 119 individual genes confirm corsistent choices of degenerate bases according to genome type. Nucleic Acids Res. 1980;8(9):1893–1912. doi: 10.1093/nar/8.9.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heizer EM, Raiford DW, Raymer ML, Doom TE, Miller RV, Krane DE. Amino acid cost and codon-usage biases in 6 prokaryotic genomes: a whole-genome analysis. Mol Biol Evol. 2006;23(9):1670–1680. doi: 10.1093/molbev/msl029. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. 2008;42(1):287–299. doi: 10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J Mol Biol. 1981;151(3):389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 1985;2(1):13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- Ingvarsson P. Molecular evolution of synonymous codon usage in Populus. BMC Evol Biol. 2008;8(1):307. doi: 10.1186/1471-2148-8-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvarsson PK. Gene expression and protein length influence codon usage and rates of sequence evolution in Populus tremula. Mol Biol Evol. 2007;24(3):836–844. doi: 10.1093/molbev/msl212. [DOI] [PubMed] [Google Scholar]
- Jansen R, Gerstein M. Analysis of the yeast transcriptome with structural and functional categories: characterizing highly expressed proteins. Nucleic Acids Res. 2000;28(6):1481–1488. doi: 10.1093/nar/28.6.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahali B, Basak S, Ghosh TC. Reinvestigating the codon and amino acid usage of S. cerevisiae genome: A new insight from protein secondary structure analysis. Biochem Biophys Res Commun. 2007;354(3):693–699. doi: 10.1016/j.bbrc.2007.01.038. [DOI] [PubMed] [Google Scholar]
- Kanaya S, Yamada Y, Kinouchi M, Kudo Y, Ikemura T. Codon usage and tRNA genes in eukaryotes: correlation of codon usage diversity with translation efficiency and with CG-dinucleotide usage as assessed by multivariate analysis. J Mol Evol. 2001;53(4):290–298. doi: 10.1007/s002390010219. [DOI] [PubMed] [Google Scholar]
- Kanaya S, Yamada Y, Kudo Y, Ikemura T. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene. 1999;238(1):143–155. doi: 10.1016/s0378-1119(99)00225-5. [DOI] [PubMed] [Google Scholar]
- Kim HS, Murphy T, Xia J, Caragea D, Park Y, Beeman RW, Lorenzen MD, Butcher S, Manak JR, Brown SJ. BeetleBase in 2010: revisions to provide comprehensive genomic information for Tribolium castaneum. Nucleic Acids Res. 2010;38:D437–D442. doi: 10.1093/nar/gkp807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Abdulovic AL, Gealy R, Lippert MJ, Jinks-Robertson S. Transcription-associated mutagenesis in yeast is directly proportional to the level of gene expression and influenced by the direction of DNA replication. DNA Repair. 2007;6(9):1285–1296. doi: 10.1016/j.dnarep.2007.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliman RM, Hey J. Reduced natural selection associated with low recombination in Drosophila melanogaster. Mol Biol Evol. 1993;10:1239–1258. doi: 10.1093/oxfordjournals.molbev.a040074. [DOI] [PubMed] [Google Scholar]
- Lanier W, Moustafa A, Bhattacharya D, Comeron JM. EST analysis of Ostreococcus lucimarinus, the most compact eukaryotic genome, shows an excess of introns in highly expressed genes. PloS One. 2008;3(5):e2171. doi: 10.1371/journal.pone.0002171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos B, Bettencourt BR, Meiklejohn CD, Hartl DL. Evolution of proteins and gene expression levels are coupled in Drosophila and are independently associated with mRNA abundance, protein length, and number of protein-protein interactions. Mol Biol Evol. 2005;22(5):1345–1354. doi: 10.1093/molbev/msi122. [DOI] [PubMed] [Google Scholar]
- Lloyd AT, Sharp PM. Codon usage in Aspergillus nidulans. Mol Gen Genet. 1991;230(1):288–294. doi: 10.1007/BF00290679. [DOI] [PubMed] [Google Scholar]
- Lobry JR, Gautier C. Hydrophobicity, expressivity and aromaticity are the major trends of amino-acid usage in 999 Escherichia coli chromosome-encoded genes. Nucleic Acids Res. 1994;22(15):3174–3180. doi: 10.1093/nar/22.15.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G. Biased gene conversion: implications for genome and sex evolution. Trends Genet. 2003;19(6):330–338. doi: 10.1016/S0168-9525(03)00116-1. [DOI] [PubMed] [Google Scholar]
- Moriyama EN, Powell JR. Codon usage bias and tRNA abundance in Drosophila. J Mol Evol. 1997;45(5):514–523. doi: 10.1007/pl00006256. [DOI] [PubMed] [Google Scholar]
- Niu D-K. Protecting exons from deleterious R-loops: a potential advantage of having introns. Biol Direct. 2007;2(1):11. doi: 10.1186/1745-6150-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden JF. Analysis of codon usage [PhD thesis]. [Nottingham (UK)]: University of Nottingham. 1999 [Google Scholar]
- Percudani R. Restricted wobble rules for eukaryotic genomes. Trends Genet. 2001;17(3):133–135. doi: 10.1016/s0168-9525(00)02208-3. [DOI] [PubMed] [Google Scholar]
- Percudani R, Ottonello S. Selection at the wobble position of codons read by the same tRNA in Saccharomyces cerevisiae. Mol Biol Evol. 1999;16(12):1752–1762. doi: 10.1093/oxfordjournals.molbev.a026087. [DOI] [PubMed] [Google Scholar]
- Percudani R, Pavesi A, Ottonello S. Transfer RNA gene redundancy and translational selection in Saccharomyces cerevisiae. J Mol Biol. 1997;268(2):322–330. doi: 10.1006/jmbi.1997.0942. [DOI] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011;12(1):32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JR, Moriyama EN. Evolution of codon usage bias in Drosophila. Proc Natl Acad Sci U S A. 1997;94(15):7784–7790. doi: 10.1073/pnas.94.15.7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince EG, Kirkland D, Demuth JP. Hyperexpression of the X-chromosome in both sexes results in extensive female-bias of X-linked genes in the flour beetle. Genome Biol Evol. 2010 doi: 10.1093/gbe/evq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Bergero R, Zeng K, Charlesworth D. Patterns of codon usage bias in Silene latifolia. Mol Biol Evol. 2011;28:771–780. doi: 10.1093/molbev/msq251. [DOI] [PubMed] [Google Scholar]
- Raiford D, Heizer E, Miller R, Akashi H, Raymer M, Krane D. Do amino acid biosynthetic costs constrain protein evolution in Saccharomyces cerevisiae? J Mol Evol. 2008;67(6):621–630. doi: 10.1007/s00239-008-9162-9. [DOI] [PubMed] [Google Scholar]
- Ren X-Y, Vorst O, Fiers MWEJ, Stiekema WJ, Nap J-P. In plants, highly expressed genes are the least compact. Trends Genet. 2006;22(10):528–532. doi: 10.1016/j.tig.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43(1):223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x. [DOI] [PubMed] [Google Scholar]
- Richards S, Gibbs RA, Weinstock GM, et al. The genome of the model beetle and pest Tribolium castaneum. Nature. 2008;452(7190):949–955. doi: 10.1038/nature06784. [DOI] [PubMed] [Google Scholar]
- Richmond RC. Non-Darwinian evolution: a critique. Nature. 1970;225(5237):1025–1028. doi: 10.1038/2251025a0. [DOI] [PubMed] [Google Scholar]
- Roy SW, Gilbert W. The evolution of spliceosomal introns: patterns, puzzles and progress. Nat Rev Genet. 2006;7(3):211–221. doi: 10.1038/nrg1807. [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Seligmann H. Cost-minimization of amino acid usage. J Mol Evol. 2003;56(2):151–161. doi: 10.1007/s00239-002-2388-z. [DOI] [PubMed] [Google Scholar]
- Shah P, Gilchrist MA. Effect of correlated tRNA abundances on translation errors and evolution of codon usage Bias. PLoS Genet. 2010;6(9):e1001128. doi: 10.1371/journal.pgen.1001128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Kumar S. Gene expression intensity shapes evolutionary rates of the proteins encoded by the vertebrate genome. Genetics. 2004;168(1):373–381. doi: 10.1534/genetics.104.028944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swire J. Selection on synthesis cost affects interprotein amino acid usage in all three domains of life. J Mol Evol. 2007;64(5):558–571. doi: 10.1007/s00239-006-0206-8. [DOI] [PubMed] [Google Scholar]
- Trotta E. The 3-base periodicity and codon usage of coding sequences are correlated with gene expression at the level of transcription elongation. PloS One. 2011;6:e21590. doi: 10.1371/journal.pone.0021590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urrutia AO, Hurst LD. The signature of selection mediated by expression on human genes. Genome Res. 2003;13(10):2260–2264. doi: 10.1101/gr.641103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. Energy constraints on the evolution of gene expression. Mol Biol Evol. 2005;22(6):1365–1374. doi: 10.1093/molbev/msi126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sets: DRYAD entry doi: 10.5061/dryad.r0t1q.