

Abstract
Adenovirus-mediated gene transfer into a tumor mass can be improved by combining it with conditionally-replicating adenovirus (CRAd) when both vectors co-infect the same cancer cell. We investigated the efficiency of enhancing transgene expression and effectiveness of cancer killing of two advenoviruses (Ads), one expressing E2F-1 (AdE2F-1) and another expressing a truncated form of E2F-1 that lacks the transactivation domain (AdE2Ftr), when combined with oncolytic Adhz60. We found that AdE2F-1 with Adhz60 actually decreased E2F-1 expression and viral replication through a mechanism apparently involving repression of the cyclin-E promoter and decreased expression of early and late structural proteins necessary for viral replication. In contrast, AdE2Ftr with Adhz60 resulted in increased E2Ftr expression, AdE2Ftr replication, and cancer cell death both in vitro and in vivo. These results indicate that AdE2Ftr coupled with a CRAd enhances AdE2Ftr-mediated cancer cell death.
Keywords: Lung, cancer, virotherapy, E2Ftr, apoptosis, in vivo
Introduction
Inefficient adenovirus-mediated gene transfer to the tumor mass has been a primary concern in the practical application of gene therapy. However, new strategies are improving gene transfer and potential therapeutic efficacy. One promising strategy to increase the therapeutic gene transfer of replication-defective adenovirus (Ad) is to combine treatment with a conditionally replicating adenovirus (CRAd).
Previous studies have demonstrated that several cancer cell lines co-transduced with the replication-enabling plasmid expressing E1A- and ElA-deleted adenovirus induced the production and amplification of E1A-deleted adenovirus and its transgene (Dion et al., 1996; Goldsmith et al., 1994). This strategy of trans-complementation has been widely documented in other cell types and using multiple different viruses. Recently, the combination of CRAd with Adp27 was shown to enhance the therapeutic index of Adp27, resulting in stronger tumor suppression than treatment with either virus alone (Lee et al., 2006). Another study showed that transduction of CRAd and an adenovirus expressing herpes simplex virus thymidine kinase gene (AdHSTK) increased the transduction efficiency of HSTK and increased its sensitivity to ganciclovir more efficiently than AdHSTK alone and resulted in stronger growth suppression of established lung cancer xenografts (Oh et al., 2010). Park et al. developed a potent novel genetic immunotherapy against Lewis lung carcinoma by combining a CRAd (Δ24RGD) with a replication-defective adenovirus expressing interferon-beta (AdIFN-β) (Park et al., 2010).
We and others have previously shown that adenoviral vectors expressing the transcription factor E2F-1 (AdE2F-1) efficiently induces apoptosis in cancer cells in vitro and in vivo (Dong et al., 1999; Dong et al., 2002; Elliott et al., 2001; Itoshima et al., 2000; Vorburger et al., 2005). E2F-1 was first described as a transcription factor that binds to two sites on the adenovirus E2 promoter (Kovesdi et al., 1986a, b). This binding of E2F-1 to the E2 promoter is important for expression of viral E2, which is directly involved in viral DNA replication (Huang and Hearing, 1989). Therefore, we evaluated whether AdE2F-1 mediated-tumor suppression could be enhanced by combining treatment with CRAd, and similarly whether E2F-1 could enhance CRAd replication in cancer cells.
In this study, we evaluated whether transgene expression of two replication-defective adenoviruses can be increased by co-infection with E1B-deleted CRAd (Adhz60) in lung cancer cells. We studied either Ad expressing E2F-1 (AdE2F-1) or a truncated form of E2F-1 that lacks the transactivation domain (E2Ftr) (AdE2Ftr) (Bell et al., 2006; Fan and Bertino, 1997). We chose to study E2Ftr to determine the role it plays in the transactivation domain of E2F-1 in adenovirus replication. We also investigated the potential application of this combination strategy to enhance AdE2F-1-mediated tumor suppression in vivo. To our surprise, we found that Adhz60 was unable to support replication of AdE2F-1 and resulted in decreased ectopic expression of E2F-1 and key proteins required for Adhz60 replication (e.g., cyclin E, E1A, adenoviral late proteins [ALP]). This effect was associated with repression of the cyclin E promoter and was independent of known E2F-1 repressors, such as pRB family members. We found that AdE2F-1 coupled with Adhz60 resulted in inefficient cancer cell killing. In contrast, Adhz60 enabled the replication of AdE2Ftr, resulting in increased E2Ftr expression and, importantly, significantly improved the destruction of lung cancer cells in vitro and in vivo.
Results
E2Ftr expression but not E2F-1 expression increased by E1B-deleted CRAd
According to previous studies, CRAd can support the replication of replication-defective adenoviruses when co-transduced in the same cancer cells, resulting in increased transgene expression from the replication-defective adenovirus (Lee et al., 2006; Lee et al., 2004). To determine whether the E1B-deleted CRAd (Adhz60) would support replication of AdE2F-1 or AdE2Ftr to increase their transgene expression, we transduced A549 lung cancer cells with AdE2F-1, AdE2Ftr, or AdLacZ (MOI of 25), together with increasing doses of Adhz60. After 48h, expression of E2F-1 or E2Ftr was analyzed by Western blot. Contrary to our expectation, Adhz60 did not increase E2F-1 levels, but did increase E2Ftr expression (Fig. 1A). As expected, neither E2F-1 nor E2Ftr was detected in cells co-transduced with control AdLacZ + Adhz60. Although E2F-1 expression was not increased, Adhz60 did enable replication of AdE2Ftr, which may represent a more desirable outcome, in terms of therapeutic potential, as E2Ftr lacks the oncogenic potential of E2F-1.
Fig. 1. Ectopic expression of E2Ftr but not E2F-1 increases viral replication protein expression in combination with CRAd.
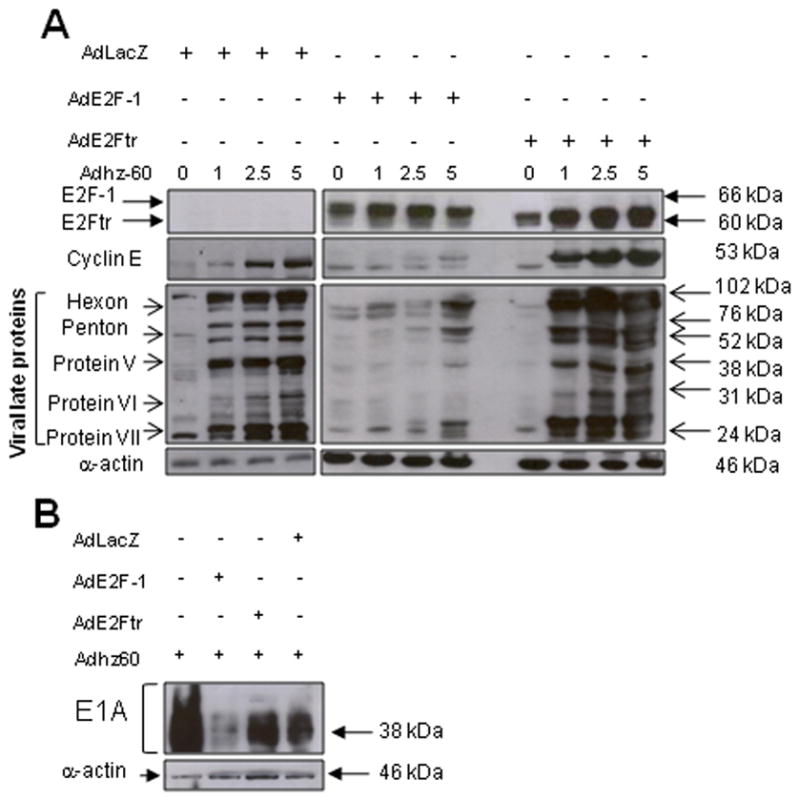
(A) A549 cells were infected with AdE2F-1, AdE2Ftr, or AdLacZ (multiplicity of infecton [MOI] of 25) together with increasing doses of Adhz60. (B) A549 cells were cotransduced with Adhz60 (MOI of 2.5) and AdE2F-1, AdE2Ftr, or AdLacZ (MOI of 25). After 48h, expression of E2F-1, E2Ftr, cyclin E, adenovirus late proteins, E1A or α-actin (loading control) were detected in cell lysates by Western blot. Images are representative of three separate experiments.
E2F-1 expression is associated with downregulation of cyclin E, adenovirus late protein (ALP), and E1A expression
In order to determine why E2F-1 levels did not increase in the presence of Adhz60, we further analyzed expression of viral proteins important in replication. Previously, we found that cyclin E expression was required for an efficient adenovirus replication (Zheng et al., 2008), and that cyclin E was a target gene of E2F-1 (Geng et al., 1996). We found that cyclin E was downregulated in the presence of overexpressed E2F-1, whereas cyclin E levels were unaffected by E2Ftr. In fact, cyclin E levels increased in an Adhz60 dose-dependent manner in cells co-infected with AdE2Ftr + Adhz60, with a similar effect observed with co-infection of AdLacZ + Adhz60 (Fig. 1A). This result suggests that ectopic expression of E2F-1, but not E2Ftr, is associated with downregulation of Adhz60 induced-cyclin E.
We also assessed whether decreased cyclin E levels could affect expression of ALP, which are required for virion formation and release. We found that downregulation of cyclin E in the presence of E2F-1 is associated with decreased ALP levels (Fig. 1A). In contrast, co-infection with AdE2Ftr + Adhz60 or AdLacZ + Adhz60 increased ALP expression in an Adhz60 dose-dependent fashion (Fig. 1A). These data suggest that decreased cyclin E levels in the presence of E2F-1 are associated with reduced levels of ALP.
Since there is a relationship between E2F-1 and decreased ALP levels, we next asked whether E2F-1 could also affect early events in adenovirus replication. A549 cells were infected with Adhz60 (MOI of 2.5) alone or in combination with AdE2F-1, AdE2Ftr, or AdLacZ (MOI of 25). After 48h, expression of E1A (a key protein for viral replication) was analyzed. We have found that E2F-1 also was associated with decreased expression of E1A, whereas neither E2Ftr nor LacZ affected expression of E1A (Fig. 1B). Collectively, these data suggest that there is an association between E2F-1 and a decrease of cyclin E, E1A, and ALP proteins.
Relationship between E2F-1 and decrease of adenovirus E1A and late proteins is reproducible in a panel of cancer cells
Next, we wanted to confirm that the association between E2F-1 and cyclin E downregulation was not limited to A549 cells. We therefore tested the efficacy of Adhz60-induced cyclin E expression in a panel of cancer cells. Lung cancer cells H1299 and H460, or osteosarcoma cells Saos2, were infected with Adhz60 (MOI of 2.5), as described above. Forty-eight hours later, an immunoblot revealed that infection with Adhz60 induced expression of cyclin E in all three cell lines (Fig. 2A). In order to determine the effect of ectopic expression of E2F-1 in these different cancer cell-types, Adhz60 was infected with AdE2F-1, AdE2Ftr, or AdLacZ. Expression of cyclin E, E1A, and ALP was analyzed at 48h. As expected, there was a relationship between E2F-1 and decreased expression of cyclin E, E1A, and ALP proteins in all cell lines tested (Fig. 2B). In contrast, neither E2Ftr nor LacZ was associated with decreased expression of cyclin E, E1A, or ALP (Fig. 2B). These data suggest that E2F-1 expression is associated with decreased expression of cyclin E, E1A, and ALP in a panel of cancer cells, but E2Ftr expression allows abundant expression of these viral replication proteins.
Fig. 2. Ectopic expression of E2F-1 but not E2Ftr represses viral replication protein expression in multiple cancer cell lines.
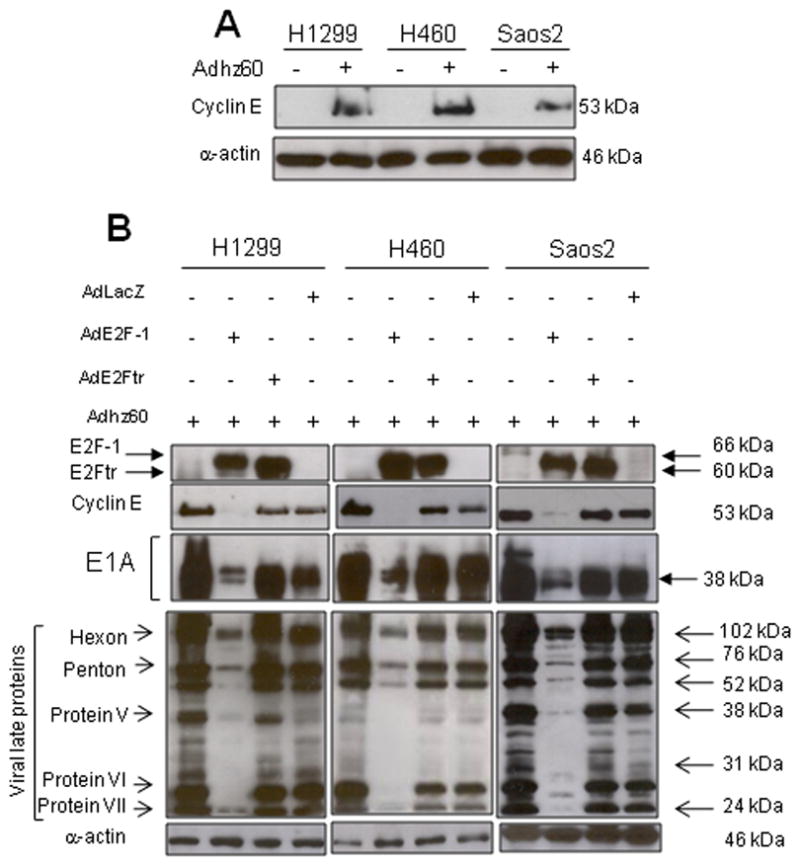
(A) H1299, H460, or Saos2 cells were infected with Adhz60 (MOI of 2.5). After 48h, expression of cyclin E was analyzed in cell lysates by Western blot. (B) H1299, H460, or Saos2 cells were cotransduced with Adhz60 (MOI of 2.5) and AdE2F-1, AdE2Ftr, or AdLacz (MOI of 25). After 48h, expression of adenovirus early and late proteins were detected by Western blot. Expression of α-actin was also detected as a loading control. Images are representative of results from three separate experiments.
Collectively, our data suggested that the combination of AdE2F-1 + Adhz60 would not improve therapeutic efficacy, at least not in tumors with a similar phenotype to cancer cell lines tested in this study. This is because there is a close relationship between E2F-1 expression with decreased expression of cyclin E and poor production of adenoviral early and late genes. However, we reasoned that the combination of Adhz60 with AdE2Ftr may have enhanced therapeutic effects, especially as E2Ftr is a known stronger inducer of apoptosis than E2F-1 (Gomez-Gutierrez et al., 2010a) and does not interfere with Adhz60 replication.
Decreased Adhz60-induced cyclin E promoter activation is associated with E2F-1
Because we showed a close relationship between E2F-1 and decreased expression of cyclin E in the presence of Adhz60, we assessed whether ectopic expression of E2F-1 might be associated with repression of cyclin E promoter. A549 cells were transfected with a plasmid expressing luciferase under regulation of the cyclin E promoter. This was followed by infection with AdE2F-1, AdE2Ftr (MOI of 25), Adhz60 (MOI of 2.5) singly, or either AdE2F-1 or AdE2Ftr in combination with Adhz60. After 48h, luciferase activity was analyzed. We found that AdE2F-1 alone induced significant luciferase activity in comparison with untreated or AdE2Ftr-infected cells. As expected, Adhz60 alone, even at a low MOI, induced the greatest luciferase activity, indicating activation of the cyclin E promoter. However, when Adhz60 was combined with AdE2F-1, luciferase activity declined significantly, but was unaffected by combination of Adhz60 with AdE2Ftr (P < 0.05; Fig. 3A). This result suggests that E2F-1 alone induces cyclin E promoter activity, but in combination with Adhz60, it may act to repress activity of adenovirus E2 promoter, resulting in potential inhibition of Adhz60 replication and lower cyclin E expression. These results confirm a mechanism of action of E2F-1 that may make it a less attractive therapeutic option compared with E2Ftr.
Fig. 3. Regulation of cyclin E promoter activity by E2F-1.
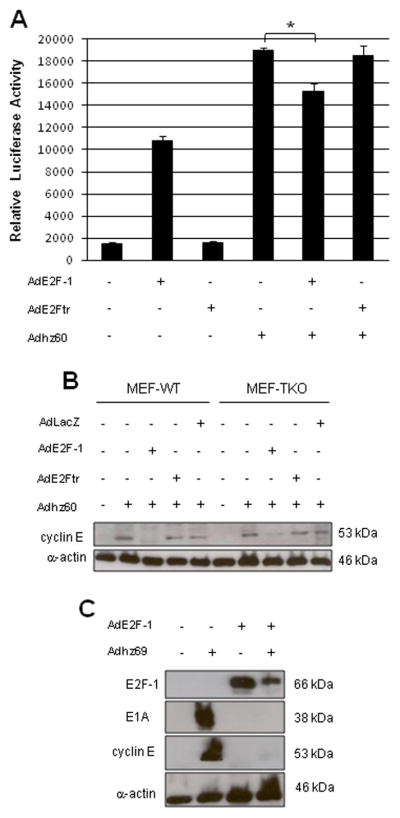
(A) A549 cells were transfected with cyclin E promoter reporter plasmid followed by no infection (mock), or infection with AdE2F-1, AdE2Ftr (MOI of 25), Adhz60 (MOI of 2.5) alone, or either AdE2F-1 or AdE2Ftr in combination with Adhz60. Luciferase activity was determined at 48h. Results represent the mean of three independent experiments ± standard deviation (SD; error bars) (*P < 0.05). (B) WT or triple knockout (TKO) MEF cells were infected with Adhz60 (MOI of 2.5) alone, or in the following combinations: AdE2F-1 + Adhz60, AdE2Ftr + Adhz60, or AdLacZ + Adhz60. After 48h, expression of cyclin E was analyzed by Western blot. (C) A549 cells were infected with Adhz69 (MOI of 2.5), AdE2F-1 (MOI of 25) alone or in combination. After 48h, expression of E2F-1, cyclin E, E1A, or α-actin were analyzed by Western blot. Images are representative of results from three separate experiments.
pRB family members are not involved in downregulation of cyclin E in presence of E2F-1
Members of the pRB family exert their cell cycle regulatory functions by binding and sequestering E2F family of transcription factors (Dyson, 1998). A combination of pRB and E2F-1 form transcriptional repressor complexes on promoters of E2F-regulated genes (Yamasaki et al., 1998). Therefore, pRB family members may inhibit E2F-1 activity, resulting in repression of E2F-1 target genes. To test this hypothesis, we used a mouse embryonic fibroblast (MEF) cell line deficient in pRB, p107, and p130 (three pRB family members). This was designated as triple knockout (TKO) cells. WT MEF and TKO MEF were infected as above. At 48h, expression of cyclin E was analyzed. Interestingly, we observed that there was an association between E2F-1 expression and decreased cyclin E levels in both WT and TKO MEF, whereas neither E2Ftr nor LacZ affected cyclin E levels in either experimental group (Fig. 3B). These results suggest that E2F-1 is associated with inhibition of Adhz60 induced-cyclin E, and that pRB family members may not be involved.
Increased expression of E1A does not prevent decrease of cyclin E and E1A in association with E2F-1
Since we observed that expression of E2F-1 driven by adenovirus was associated with downregulation of key proteins for CRAd replication, we next asked whether expression of higher levels of E1A, driven by a strong cytomegalovirus (CMV) promoter, could overcome the downregulation of cyclin E and E1A in association with E2F-1. To do this, we used either A549 cells that were not infected (mock), or were infected with Adhz69 (MOI of 2.5), a CRAd that expresses E1A under the regulation of the CMV promoter (Rao et al., 2004; Zheng et al., 2005), AdE2F-1 (MOI of 25), or AdE2F-1 + Adhz69. After 48h, an immunoblot revealed that E2F-1 expression decreased in cells infected with Adhz69 + AdE2F-1, as well as decreased expression of cyclin E and E1A (Fig. 3C). This result suggests that, despite E1A expression being driven by a strong CMV promoter, E2F-1 was still associated with downregulation of key proteins for CRAd replication.
Combination of AdE2Ftr with Adhz60 increases AdE2Ftr-mediated cell killing effect
We next evaluated the cell killing potential of combining replication-defective adenoviral vectors with Adhz60. To study this, A549 cells were mock-infected or infected with AdE2F-1, AdE2Ftr, or AdLacZ alone, or in combination with Adhz60. We then determined cell growth inhibition by MTT assay, assessed apoptosis by Annexin V staining, and analyzed adenovirus yield. MTT assay revealed minimal growth inhibition using AdE2F-1, AdE2Ftr, or AdLacZ alone. There was, however, a significant reduction in the number of viable cells with AdE2Ftr + Adhz60 (37% viability) compared with AdE2Ftr or Adhz60 alone (85% or 70% of viability, respectively) (P < 0.05) (Fig. 4A). The combination of AdE2F-1 with Adhz60 was less effective at cell killing (74% viability); in fact, the combination of AdLacZ + Adhz60 induced greater reduction of viable cells (60%) than AdE2F-1 + Adhz60 treatment. This can be explained by replication of both AdLacZ and Adhz60, which results in higher virus production and oncolysis, whereas E2F-1 inhibits virus replication and oncolysis.
Fig. 4. Effect of combination therapy of AdE2Ftr with Adhz60 on cell killing activity.

A549 cells were not infected (mock) or infected with Adhz60 (MOI of 2.5), AdE2F-1, AdE2Ftr or AdLacZ (MOI of 25) alone, or in the following combinations: AdE2F-1 + Adhz60, AdE2Ftr + Adhz60, or AdLacZ + Adhz60. At 72 h postinfection, the following assays were performed: (A) Cell growth inhibition measured by MTT assay. (B) Percentage of apoptotic cells was determined by annexin V-PE staining and analyzed as describe in material and methods. (C) Adenovirus yield in A549 cells at 72h after infection. Results represent the mean of three independent experiments ± standard deviation (SD; error bars) (*P < 0.05).
Previously, we showed that AdE2Ftr induces apoptosis by activation of caspase in a panel of cancer cells (Gomez-Gutierrez et al., 2010a). We therefore confirmed the level of apoptosis by annexin V staining in experimental groups. We found that AdE2Ftr or Adhz60 alone induced 8.6% or 18% apoptosis, respectively (Fig. 4B). The combination of AdE2Ftr + Adhz60 induced significantly more apoptosis (43%) compared with either virus alone (P < 0.05), whereas the combination of AdLacZ + Adhz60 induced only 22% apoptosis (Fig. 4B).
The effect of E2F-1 in the release of infectious virus was measured. Cell culture medium (supernatant) was collected. Then, virus titers were determined by standard infection unit measure. We found that titers from cells infected with Adhz60 alone reached 1×106 infection units (IFU)/ml. When Adhz60 was infected in combination with AdE2F-1, virus titers decreased 10 fold (1×105 IFU/ml), whereas titers were similar to the Adhz60 alone levels in cells with combined Adhz60 and AdE2Ftr infection (Fig. 4C). These data show that oncolytic adenovirus Adhz60 enables replication of AdE2Ftr, which results in enhanced E2Ftr expression and increased cell-killing effects compared with AdE2Ftr infection alone.
Adhz60 enhances E2Ftr expression and killing effect in a panel of cancer cells
We next tested whether Adhz60 would enable replication of AdE2Ftr and enhance E2Ftr expression in a panel of cancer cells. H1299, H460, or Saos2 cell lines were infected with AdE2Ftr (MOI of 25) alone or in combination with Adhz60 (MOI of 2.5). E2Ftr expression was analyzed by a Western blot at 48h. We found that Adhz60 significantly enhanced expression of E2Ftr in all three cancer cell lines tested (Fig. 5A). Next, caspase activity was assessed; a colorimetric assay for caspase-9 activity revealed that the combination of AdE2Ftr + Adhz60 induced greater caspase-9 activity in comparison with AdE2Ftr alone (P < 0.05) (Fig. 5B). Apoptosis was further validated by annexin V staining, which revealed that Adhz60 increased AdE2Ftr-mediated apoptosis (Fig. 5C). These data suggest that oncolytic adenovirus Adhz60 enables AdE2Ftr replication, increases E2Ftr expression, and induces a higher percentage of apoptosis in a panel of cancer cells in comparison with AdE2Ftr alone.
Fig. 5. Combination therapy effectiveness of AdE2Ftr with Adhz60 on a panel of cancer cells.

(A) H1299, H460, or Saos2 cells were infected with AdE2Ftr (MOI of 25) in the absence or presence of Adhz60 (MOI of 2.5). After 48h, expression of E2Ftr, or α-actin were analyzed by Western blot. (B) After 48 h of infections above mentioned, the OD405 values of cell lysates were measured as caspase-9 activity. The results were expressed as the fold change in treated cells over that of the control cells. Values represent the mean of three independent experiments ± standard deviation (SD; error bars) (*P < 0.05). (C) Percentage of apoptosis determined by annexin V-PE staining and analyzed as describe in material and methods. Results represent the mean of three independent experiments ± standard deviation (SD; error bars) (*P < 0.05).
Combination therapy of AdE2Ftr with Adhz60 induces strong tumor suppression in vivo
The effectiveness of adenoviral combination therapy was assessed in vivo using mice bearing ectopic subcutaneous A549 lung tumors. Balb/c nude mice were injected subcutaneously with 5×106 A549 cells. After 6 days, mice with palpable tumors were randomized and directly injected intratumoraly with AdE2Ftr + AdLacZ (control), AdLacZ + Adhz60, or AdE2Ftr + Adhz60 (1×108 plaque forming units [pfu] per virus, per mouse) at 0, 2, and 5 days (5 mice per group). In this experiment, we did not include AdE2F-1, because our in vitro data showed that the combination of AdE2F-1 with Adhz60 is not feasible. Tumor volume was measured in two dimensions with calipers at 5-day intervals after initial adenoviral treatment. There was a reduction in tumor volume of about 30% in mice treated with AdLacZ + Adhz60 in comparison with control mice injected with AdE2Ftr + AdLacZ (Fig. 6A). However, there was about a 70% decrease in tumor volume in mice treated with the combination of AdE2Ftr + Adhz60 compared with control (P < 0.05; Fig. 6A). Twenty-five days after the last adenovirus injection, the tumors were excised and tumor size between groups was compared visually. Tumors from mice injected with AdE2Ftr + AdLacZ were identifiably larger than tumors from mice injected with AdE2Ftr + Adhz60 (Fig. 6B). Additionally, tumors were weighed. Tumors from AdE2Ftr + AdLacZ treatment weighed 1.5 ± 1.1g and 0.45 ± 0.6g from AdLacZ + Adhz60 treatment, but AdE2Ftr + Adhz60-treated tumors weighed significantly less at 0.09 ± 0.25g compared with control groups (P < 0.05) (Fig. 6C). These results clearly indicate the improved efficacy of combination therapy of AdE2Ftr with Adhz60 compared with AdE2Ftr + AdLacZ.
Fig. 6. In vivo anti-tumor effect of combination adenoviral therapy.

Six days after subcutaneous administration of 5×106 A549 cells, mice with palpable tumors were randomized to receive a local injection of AdE2Ftr + AdLacZ, AdLacZ + Adhz60, or AdE2Ftr + Adhz60 on days 0, 2, and 5 (vertical arrows). The total virus dose was 3 injections at 1×108 pfu viral particles. (A) Tumor volume was plotted against time (n=5 per group) (*P<0.05) (B) On day 25, after last adenovirus treatment tumors were excised and photographed to show relative tumor size between groups. (C) Tumors were also weighed on day 30. The results are expressed as the mean from each treatment group ± standard error (SE; error bars) (P < 0.05). (D) Immunohistochemistry of lung cancer tumors at 25 days after last combination therapy treatment. An mAb anti-human E2F-1 was used to detect E2Ftr expression and an pAb anti-human cleaved Caspase-3 (Asp175)(5A1E) was used to confirm apoptosis. Photographs were taken with ×20 magnification and analyzed with NIS-Elements BR 3.0 software (Nikon Instruments, Melville, NY).
Tumors were harvested at 25 days after the last adenovirus injection and subjected to histopathological analysis. Immunohistochemical analysis revealed a strong E2Ftr expression (~ 65% of positive cells) in tumors of mice injected with AdE2Ftr + Adhz60, which was associated with a significant percentage of cleaved caspase-3-positive cells (~70%) (Fig. 6D). In contrast, E2Ftr and cleaved caspase-3 were not detected in tumors treated with AdE2Ftr + AdLacZ, whereas there was a modest percentage (~37%) of cleaved caspase-3-positive cells in tumors treated with AdLacZ + Adhz60. These results indicate that Adhz60 enhanced and prolonged expression of E2Ftr, which resulted in strong activation of caspase-3 in lung tumor mass (Fig. 6D).
Discussion
Researchers have long been hampered in their gene transfer attempts at the successful application of a replication-defective adenovirus into the tumor mass. This means that several adenoviral gene treatments are required to achieve significant tumor mass reduction, which may result in increased adverse effects. In recent years, combining replication-defective adenovirus with CRAd has been used to overcome these difficulties with promising experimental results reported (Lee et al., 2006; Lee et al., 2004; Oh et al., 2010; Park et al., 2010). We proposed that AdE2F-1-mediated tumor suppression (Dong et al., 1999; Dong et al., 2002; Elliott et al., 2001; Itoshima et al., 2000; Vorburger et al., 2005) could be enhanced by combining treatment with CRAd, in that CRAd supplies E1A to enable AdE2F-1 replication, and AdE2F-1 provides the transcription factor to transactivate the CRAd E2 promoter.
Our study was conducted to investigate the potential application of the combinational therapy of AdE2F-1 or AdE2Ftr with Adhz60 (E1B-deleted). Contrary to our expectations, we found that Adhz60 was unable to support AdE2F-1 replication, as E2F-1 expression did not increase. We suggested that a possible responsible mechanism involves repression of the cyclin E promoter by ectopic expression of E2F-1, thereby decreasing expression of cyclin E, as well as both early and late adenoviral proteins, which play a key role in adenoviral replication (Zheng et al., 2008). This effect was reproducible in four separate cancer cell line types (A549, H1299, H460, Saos2). In contrast, E2Ftr, which lacks the transactivation domain of E2F-1, did not affect the expression of these key replication proteins.
E2F-1 has two contrasting functions: It can induce apoptosis (Dong et al., 2002; Itoshima et al., 2000; Vorburger et al., 2005), or it can induce progression of the cell cycle (Johnson et al., 1993; Muller and Helin, 2000). It appears, in some circumstances, that E2F-1 acts as a tumor suppressor gene by downregulation of anti-apoptotic genes (Dong et al., 1999; Phillips et al., 1999; Yang et al., 1999; Yang et al., 2000). In other circumstances, it acts as an oncogene by inducing S-phase entry and cell transformation (Arata et al., 2000; Xu et al., 1995; Yang and Sladek, 1995). We do not know the mechanism by which E2F-1 sometimes could act as an oncogene and in others as a tumor suppressor gene. In addition, binding of E2F-1 to the E2 promoter is important for expression of viral E2, which is directly involved in viral DNA replication (Huang and Hearing, 1989). However, exogenous overexpression of E2F-1 appears to have a contradictory effect. Thus, in our case, E2F-1 acts as a tumor suppressor gene, and at high levels, may form homodimers or heterodimers with other transcription factors, such as DP-1, and then binds and represses activation of the adenovirus E2 promoter, thereby in inhibition of CRAd replication.
We previously reported that the construction of adenovirus expressing E2Ftr under regulation of CMV promoter was unsuccessful, because it was likely that E2Ftr may competitively bind to the viral E2 promoter and inhibit Ad vector amplification (Gomez-Gutierrez et al., 2010b). However, in our present study, we found that E2Ftr did not interfere with the CRAd replication process. These current results further suggest that E2F-1 transactivation domain may play an important role in the negative regulation of CRAd E2 promoter. This also suggests that when research is planned to arm a CRAd with a therapeutic gene, especially a transcription factor, an assay of transcomplementation should be performed in order to determine whether such transcription factor (therapeutic gene) does not interfere with the CRAd replication.
The ideal approach to improve the efficacy of CRAds is to arm these vectors with therapeutic genes. However, the limited space in the Ad backbone makes that capacity for packaging transgenes into CRAds difficult to carry out. To overcome this concern, the application of replication-defective viruses carrying therapeutic genes in combination with CRAd vectors expressing E1A in trans has been proposed (Oh et al., 2010; Park et al., 2010).
In the current study, we show that adenoviral combination therapy of AdE2Ftr with Adhz60 enhanced E2Ftr expression in four cancer cell types, but moreover, the AdE2Ftr-mediated cancer cell death was enhanced by co-infection with Adhz60 in vitro and in vivo. For example, previously, we gave mice four doses of AdE2Ftr at a higher concentration of 1×109 pfu per treatment (Gomez-Gutierrez et al., 2010a). In the current study, we gave mice only three doses of AdE2Ftr + Adhz60 at a lower concentration of 1×108 pfu for each treatment. Comparing our results across experiments suggests that, even though we reduced the total amount of adenovirus 6-fold, we achieved a comparable anti-tumor effect.
Although co-delivery of two separate vectors provides suboptimal therapy, this study was highly useful to determine whether E2F-1 or E2Ftr is the better option to eventualy arm an oncolytic adenovirus with a tumor supressor gene, having thus in a single vector, the properties of oncolysis and apoptosis. We acknowledge the potential for adverse effects with these therapies, such as immune reactivity or pre-existing immunity targeting adenoviruses in humans, and that these effects can render repeated doses of adenovirus gene therapy vectors less effective. Therefore, we suggest that it is possible to overcome this concern by the application of a reduced dose of oncolytic adenovirus armed with E2Ftr, which may be as effective as multiple other treatments. Thus, lung cancer tumors could be effectively destroyed through oncolysis and apoptosis to achieve therapeutic results. The potential for augmentation of this gene therapy effect by chemotherapeutic agents remains to be elucidated as well. The results of the present studies clearly indicate that combination adenoviral therapy of AdE2Ftr with CRAd is a promising approach to treat lung cancer.
Materials and methods
Cell lines and culture conditions
The human lung cancer cell lines (A549, H1299, H460); osteosarcoma cell line Saos2; wild-type (WT) and pRB, p107, and p130 triple-knockout (TKO) mouse embryonic fibroblasts (MEF); and human embryonic kidney cell line (HEK-293) were purchased from the American Type Culture Collection (Rockville, MD). A549, H1299, HEK-293, and TKO MEFs were cultured in Dulbecco’s Modified Eagle Medium (DMEM). H460 cells were cultured in RPMI. Saos2 cells were cultured in McCoy’s medium. All media were supplemented with 10% heat-inactivated fetal bovine serum (FBS) and penicillin/streptomycin (100 U/mL). Cell culture reagents were obtained from Invitrogen (Carlsbad, CA). Cells were cultured in a 5% CO2 incubator at 37°C, and the medium was changed every 2 or 3 days.
Adenoviral vectors and plasmids
All three replication-defective recombinant adenoviral vectors used in this report have deletion of the viral E1 gene. The Ad5CMV-E2F-1 (AdE2F-1) and Ad5CMV-LacZ (AdLacZ) vectors contained the transgenes E2F-1 and nuclear-localized β-galactosidase, respectively, under the control of the CMV promoter, as previously described (Dong et al., 2002). AdTet-E2Ftr3 (AdE2Ftr) expressing E2F-1 lacking the transactivation domain was constructed previously by our laboratory (Gomez-Gutierrez et al., 2010b). Two CRAds were used, as previously reported by us (Rao et al., 2004; Zheng et al., 2005): Adhz60 with deletion of E1B gene and Adhz69 expressing E1A under regulation of CMV promoter. The plasmid containing 2.2 kb of the human cyclin E promoter driving the cDNA of firefly luciferase was kindly provided by Dr. Greg McCarty (Johns Hopkins University) (Loeb et al., 2002) and was used to assess the induction of cyclin E promoter.
Western blot analysis
Cells were harvested and lysed in a radioimmunoprecipitation assay (RIPA) buffer, as described previously (Gomez-Gutierrez et al., 2006). Cell lysates were centrifuged and protein concentration was determined by Detergent Compatible (DC) Protein Assay (Bio-Rad, Hercules, CA). Equal amounts of cellular protein were electrophoresed in 10% sodium dodecylsulfate polyacrylamide gels and transferred to Hybond polyvinylidene difluoride membranes (Amersham, Arlington Heights, IL). The membranes were first incubated with the following primary antibodies (Abs): mouse-antihuman-E2F-1 monoclonal (m)Ab, rabbit-antihuman cyclin E (p)Ab (Santa Cruz Biotech, Santa Cruz, CA), rabbit- antihuman adenovirus proteins virion (Abcam, ab6982, San Francisco, CA), mouse anti-adenovirus type 5 E1A (BD Pharmingen, San Diego CA), rabbit-antihuman-α-actin polyclonal (Sigma-Aldrich). Next, the membranes were incubated with antimouse immunoglobulin (Ig) or anti-rabbit Ig, peroxidase-linked, species-specific whole Abs (Amersham, Piscataway, NJ). Electrochemiluminescent reagents were used to detect the signals, according to the manufacturer’s instructions (Amersham).
Luciferase-promoter assay
A549 cells were plated in a 12-well plate at 1×105 cells/well and cultured in medium containing 10% FBS. Within 24h, cells were co-transfected with a cyclin E promoter construct and a phRL-CMV plasmid (containing the cDNA encoding Renilla luciferase) by using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA). Cells were not infected or infected with AdE2F-1, AdE2Ftr (both MOI of 25), Adhz60 (MOI of 2.5), Ad-E2F-1+Adhz60, or Ad-E2Ftr+Adhz60. After 48h, lysates were collected and assayed for luciferase activity using the Dual Luciferase Reporter Assay System (Progema, Madison, WI), as reported previously (Hao et al., 2007). Luciferase activity from untreated control cells was used to determine background signal. All luciferase values were normalized to the Renilla luciferase readout values and expressed as relative luciferase activity.
Cell viability assay
Cell proliferation was assessed three days after infection by measuring the conversion of the tetrazolium salt (MTT) to formazan, according to the manufacturer’s instructions (Sigma-Aldrich, St Louis, MO), as reported previously (Gomez-Gutierrez et al., 2010a). The supernatant from each plate was collected for measurement of absorbance at a wavelength of 570 nm. The results were expressed as the percentage of live cells.
Annexin V staining
Annexin V staining was performed, as described previously (Gomez-Gutierrez et al., 2006) and according to the manufacturer’s instructions (Annexin V-PE Apoptosis Kit, Pharmingen, San Diego CA). Cells were analyzed by fluorescence-activated cell sorter scan (FACScan) flow cytometer (Becton Dickinson, Franklin Lakes, NJ) with FlowJo software (Tree Star Inc., Ashland, OR).
Adenovirus titering
1×105 cells were plated in a 12-well plate. The next day, cells were infected with Adhz60 (MOI of 2.5) alone or in combination with Ad-E2F-1, Ad-E2Ftr, or Ad-LacZ (MOI of 25). Three days after infection, supernatants were collected. Adenovirus release was titered by using a TCID50 end-point dilution method in 96-well plates. In brief, 96-well plates were seeded with 3×103 HEK-293 cells and were infected after 48h with a 3-fold serially diluted virus. Infected cells were incubated for 7 days, after which wells with a cytopathic effect were scored visually, and infective unit (IFU) was calculated as described in Sandig et al., 2000.
Caspase-9 activity assay
1×106 cells were not infected (control) or infected with AdE2Ftr at (MOI of 25) in absence or presence of Adhz60 at MOI of 2.5, 48h postinfection. Caspase-9 activity was analyzed by caspase-9 colorimetric assay kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer’s instructions and as described previously (Hao et al., 2007). No cell lysates and no substrates were used as blanks. The results were expressed as the fold increase in treated cells over that of the control cells.
Lung cancer xenograft study
Tumors were formed by subcutaneous (s.c.) injection of 5×106 A549 lung cancer cells into bilateral flanks of athymic BALB/c nu/nu male mice (6–8 weeks-of-age; Charles River Laboratories, Wilmington, MA). Six days later, mice with palpable tumors were randomized, and tumors were directly injected with 1×108 pfu of each of: AdLacZ + AdE2Ftr (control), AdLacZ + Adhz60, or AdE2Ftr +Adhz60 (n=5 per group). The purified virus was diluted in a total volume of 100 μl of 0.9% of sterile normal saline. Injections were repeated at days 2 and 5 after initial infection. Tumors were measured every 3 days, and tumor volume was determined by externally measuring in 2 dimensions with a caliper. Volume (V) was determined by the equation V = (L × W 2)/2, where L = length, W = width of the tumor. Animal experiments were performed in accordance with institutional guidelines and approved by the University of Louisville Institutional Animal Care and Use Committee.
Immunohistochemistry
Tumors were excised at 25 days after last adenovirus injection, fixed in 10% formalin, embedded in paraffin blocks, and processed for histologic analysis and detection of E2Ftr expression and cleaved caspase-3. A mouse-antihuman-E2F-1 mAb (KH95) sc-125 (Santa Cruz Biotechnology) at a dilution of 1:200 was used to detect E2Ftr. A rabbit-antihuman cleaved caspase-3 (Asp175)(5A1E) mAb (Cell Signaling, Danvers, MA) at a dilution of 1:200 was used to detect cleaved caspase-3. The slides were then washed with PBS and incubated with the standard ultra-sensitive ABC peroxidase staining kit (PIERCE, Rockford, IL) and detected with diaminobenzidine tetrahydrochloride (DAB) solution containing 0.006% H2O2. Hematoxylin was used as a counterstain. Tissue sections stained without primary antibodies were used as negative controls. Photographs were taken with ×20 magnification and analyzed with NIS-Elements BR 3.0 software (Nikon Instruments Inc., Melville, NY).
Statistical analysis
The results of the in vitro assays and tumor growth in mice from three treatment groups were analyzed by the unpaired Student’s t test using a 1-way ordinary parametric analysis of variance. P < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by Lung Cancer Research Foundation (JGGG), and awards R01CA129975 (HSZ) from the National Cancer Institute and GMB081410 (KMM & HSZ) from the Kentucky Lung Cancer Research Program. The authors thank Melanie Scott and Margaret Abby for editing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arata Y, Fujita M, Ohtani K, Kijima S, Kato JY. Cdk2-dependent and -independent pathways in E2F-mediated S phase induction. The Journal of biological chemistry. 2000;275:6337–6345. doi: 10.1074/jbc.275.9.6337. [DOI] [PubMed] [Google Scholar]
- Bell LA, O’Prey J, Ryan KM. DNA-binding independent cell death from a minimal proapoptotic region of E2F-1. Oncogene. 2006;25:5656–5663. doi: 10.1038/sj.onc.1209580. [DOI] [PubMed] [Google Scholar]
- Dion LD, Goldsmith KT, Garver RI., Jr Quantitative and in vivo activity of adenoviral-producing cells made by cotransduction of a replication-defective adenovirus and a replication-enabling plasmid. Cancer Gene Ther. 1996;3:230–237. [PubMed] [Google Scholar]
- Dong YB, Yang HL, Elliott MJ, Liu TJ, Stilwell A, Atienza C, Jr, McMasters KM. Adenovirus-mediated E2F-1 gene transfer efficiently induces apoptosis in melanoma cells. Cancer. 1999;86:2021–2033. doi: 10.1002/(sici)1097-0142(19991115)86:10<2021::aid-cncr20>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Dong YB, Yang HL, Elliott MJ, McMasters KM. Adenovirus-mediated E2F-1 gene transfer sensitizes melanoma cells to apoptosis induced by topoisomerase II inhibitors. Cancer Res. 2002;62:1776–1783. [PubMed] [Google Scholar]
- Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- Elliott MJ, Dong YB, Yang H, McMasters KM. E2F-1 up-regulates c-Myc and p14(ARF) and induces apoptosis in colon cancer cells. Clin Cancer Res. 2001;7:3590–3597. [PubMed] [Google Scholar]
- Fan J, Bertino JR. Functional roles of E2F in cell cycle regulation. Oncogene. 1997;14:1191–1200. doi: 10.1038/sj.onc.1200940. [DOI] [PubMed] [Google Scholar]
- Geng Y, Eaton EN, Picon M, Roberts JM, Lundberg AS, Gifford A, Sardet C, Weinberg RA. Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene. 1996;12:1173–1180. [PubMed] [Google Scholar]
- Goldsmith KT, Curiel DT, Engler JA, Garver RI., Jr Trans complementation of an E1A-deleted adenovirus with codelivered E1A sequences to make recombinant adenoviral producer cells. Human gene therapy. 1994;5:1341–1348. doi: 10.1089/hum.1994.5.11-1341. [DOI] [PubMed] [Google Scholar]
- Gomez-Gutierrez JG, Garcia-Garcia A, Hao H, Rao XM, Montes de Oca-Luna R, Zhou HS, McMasters KM. Adenovirus-mediated expression of truncated E2F-1 suppresses tumor growth in vitro and in vivo. Cancer. 2010a;116:4420–4432. doi: 10.1002/cncr.25322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gutierrez JG, Rao XM, Garcia-Garcia A, Hao H, McMasters KM, Zhou HS. Developing adenoviral vectors encoding therapeutic genes toxic to host cells: comparing binary and single-inducible vectors expressing truncated E2F-1. Virology. 2010b;397:337–345. doi: 10.1016/j.virol.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gutierrez JG, Souza V, Hao HY, Montes de Oca-Luna R, Dong YB, Zhou HS, McMasters KM. Adenovirus-mediated gene transfer of FKHRL1 triple mutant efficiently induces apoptosis in melanoma cells. Cancer Biol Ther. 2006;5:875–883. doi: 10.4161/cbt.5.7.2911. [DOI] [PubMed] [Google Scholar]
- Hao H, Dong Y, Bowling MT, Gomez-Gutierrez JG, Zhou HS, McMasters KM. E2F-1 induces melanoma cell apoptosis via PUMA up-regulation and Bax translocation. BMC Cancer. 2007;7:24. doi: 10.1186/1471-2407-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MM, Hearing P. The adenovirus early region 4 open reading frame 6/7 protein regulates the DNA binding activity of the cellular transcription factor, E2F, through a direct complex. Genes Dev. 1989;3:1699–1710. doi: 10.1101/gad.3.11.1699. [DOI] [PubMed] [Google Scholar]
- Itoshima T, Fujiwara T, Waku T, Shao J, Kataoka M, Yarbrough WG, Liu TJ, Roth JA, Tanaka N, Kodama M. Induction of apoptosis in human esophageal cancer cells by sequential transfer of the wild-type p53 and E2F-1 genes: involvement of p53 accumulation via ARF-mediated MDM2 down-regulation. Clin Cancer Res. 2000;6:2851–2859. [PubMed] [Google Scholar]
- Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- Kovesdi I, Reichel R, Nevins JR. E1A transcription induction: enhanced binding of a factor to upstream promoter sequences. Science. 1986a;231:719–722. doi: 10.1126/science.2935935. [DOI] [PubMed] [Google Scholar]
- Kovesdi I, Reichel R, Nevins JR. Identification of a cellular transcription factor involved in E1A trans-activation. Cell. 1986b;45:219–228. doi: 10.1016/0092-8674(86)90386-7. [DOI] [PubMed] [Google Scholar]
- Lee CT, Lee YJ, Kwon SY, Lee J, Kim KI, Park KH, Kang JH, Yoo CG, Kim YW, Han SK, Chung JK, Shim YS, Curiel DT, Carbone DP. In vivo imaging of adenovirus transduction and enhanced therapeutic efficacy of combination therapy with conditionally replicating adenovirus and adenovirus-p27. Cancer Res. 2006;66:372–377. doi: 10.1158/0008-5472.CAN-05-1515. [DOI] [PubMed] [Google Scholar]
- Lee CT, Park KH, Yanagisawa K, Adachi Y, Ohm JE, Nadaf S, Dikov MM, Curiel DT, Carbone DP. Combination therapy with conditionally replicating adenovirus and replication defective adenovirus. Cancer Res. 2004;64:6660–6665. doi: 10.1158/0008-5472.CAN-04-1200. [DOI] [PubMed] [Google Scholar]
- Loeb DM, Korz D, Katsnelson M, Burwell EA, Friedman AD, Sukumar S. Cyclin E is a target of WT1 transcriptional repression. The Journal of biological chemistry. 2002;277:19627–19632. doi: 10.1074/jbc.M201336200. [DOI] [PubMed] [Google Scholar]
- Muller H, Helin K. The E2F transcription factors: key regulators of cell proliferation. Biochim Biophys Acta. 2000;1470:M1–12. doi: 10.1016/s0304-419x(99)00030-x. [DOI] [PubMed] [Google Scholar]
- Oh JY, Park MY, Kim DR, Lee JH, Shim SH, Chung JH, Yoon HI, Sung MW, Kim YS, Lee CT. Combination gene therapy of lung cancer with conditionally replicating adenovirus and adenovirus-herpes simplex virus thymidine kinase. Int J Mol Med. 2010;25:369–376. [PubMed] [Google Scholar]
- Park MY, Kim DR, Jung HW, Yoon HI, Lee JH, Lee CT. Genetic immunotherapy of lung cancer using conditionally replicating adenovirus and adenovirus-interferon-beta. Cancer Gene Ther. 2010;17:356–364. doi: 10.1038/cgt.2009.78. [DOI] [PubMed] [Google Scholar]
- Phillips AC, Ernst MK, Bates S, Rice NR, Vousden KH. E2F-1 potentiates cell death by blocking antiapoptotic signaling pathways. Mol Cell. 1999;4:771–781. doi: 10.1016/s1097-2765(00)80387-1. [DOI] [PubMed] [Google Scholar]
- Rao XM, Tseng MT, Zheng X, Dong Y, Jamshidi-Parsian A, Thompson TC, Brenner MK, McMasters KM, Zhou HS. E1A-induced apoptosis does not prevent replication of adenoviruses with deletion of E1b in majority of infected cancer cells. Cancer Gene Ther. 2004;11:585–593. doi: 10.1038/sj.cgt.7700739. [DOI] [PubMed] [Google Scholar]
- Sandig V, Youil R, Bett AJ, Franlin LL, Oshima M, Maione D, Wang F, Metzker ML, Savino R, Caskey CT. Optimization of the helper-dependent adenovirus system for production and potency in vivo. Proc Natl Acad Sci U S A. 2000;97:1002–1007. doi: 10.1073/pnas.97.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorburger SA, Hetrakul N, Xia W, Wilson-Heiner M, Mirza N, Pollock RE, Feig B, Swisher SG, Hunt KK. Gene therapy with E2F-1 up-regulates the protein kinase PKR and inhibits growth of leiomyosarcoma in vivo. Mol Cancer Ther. 2005;4:1710–1716. doi: 10.1158/1535-7163.MCT-05-0036. [DOI] [PubMed] [Google Scholar]
- Xu G, Livingston DM, Krek W. Multiple members of the E2F transcription factor family are the products of oncogenes. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:1357–1361. doi: 10.1073/pnas.92.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki L, Bronson R, Williams BO, Dyson NJ, Harlow E, Jacks T. Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1(+/−)mice. Nat Genet. 1998;18:360–364. doi: 10.1038/ng0498-360. [DOI] [PubMed] [Google Scholar]
- Yang HL, Dong YB, Elliott MJ, Liu TJ, Atienza C, Jr, Stilwell A, McMasters KM. Adenovirus-mediated E2F-1 gene transfer inhibits MDM2 expression and efficiently induces apoptosis in MDM2-overexpressing tumor cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 1999;5:2242–2250. [PubMed] [Google Scholar]
- Yang HL, Dong YB, Elliott MJ, Liu TJ, McMasters KM. Caspase activation and changes in Bcl-2 family member protein expression associated with E2F-1-mediated apoptosis in human esophageal cancer cells. Clin Cancer Res. 2000;6:1579–1589. [PubMed] [Google Scholar]
- Yang XH, Sladek TL. Overexpression of the E2F-1 transcription factor gene mediates cell transformation. Gene Expr. 1995;4:195–204. [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Rao XM, Gomez-Gutierrez JG, Hao H, McMasters KM, Zhou HS. Adenovirus E1B55K region is required to enhance cyclin E expression for efficient viral DNA replication. J Virol. 2008;82:3415–3427. doi: 10.1128/JVI.01708-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Rao XM, Snodgrass C, Wang M, Dong Y, McMasters KM, Zhou HS. Adenoviral E1a expression levels affect virus-selective replication in human cancer cells. Cancer biology & therapy. 2005;4:1255–1262. doi: 10.4161/cbt.4.11.2137. [DOI] [PubMed] [Google Scholar]