

Homoprotocatechuate 2,3-dioxygenase (FeHPCD) utilizes an active site FeII to activate O2 in a reaction cycle that ultimately results in aromatic ring cleavage. Here, the roles of the conserved active site residue Tyr257 are investigated by solving the X-ray crystal structures of the Tyr257-to-Phe variant (Y257F) in complex with the substrate homoprotocatechuate (HPCA) and the alternative substrate 4-nitrocatechol (4NC). These are compared with structures of the analogous wild type enzyme complexes. In addition, the oxy intermediate of the reaction cycle of Y257F-4NC + O2 is formed in crystallo and structurally characterized. It is shown that both substrates adopt a previously unrecognized, slightly non-planar, strained conformation affecting the geometries of all aromatic ring carbons when bound in the FeHPCD active site. This global deviation from planarity is not observed for the Y257F variant. In the Y257F-4NC-oxy complex, the O2 is bound side-on to the FeII, while the 4NC is chelated in two adjacent sites. The ring of the 4NC in this complex is planar, in contrast to the equivalent FeHPCD intermediate, which exhibits substantial local distortion of the substrate hydroxyl moiety (C2-O−) that is hydrogen bonded to Tyr257. We propose that Tyr257 induces the global and local distortions of the substrate ring in two different ways. First, van der Waals conflict between the Tyr257-OH substituent and the substrate C2 carbon is relieved by adopting the globally strained structure. Second, Tyr257 stabilizes the localized out-of-plane position of the C2-O− by forming a stronger hydrogen bond as the distortion increases. Both types of distortion favor transfer of one electron out of the substrate to form a reactive semiquinone radical. Then, the localized distortion at substrate C2 promotes formation of the key alkylperoxo intermediate of the cycle resulting from oxygen attack on the activated substrate at C2, which becomes sp3 hybridized. The inability of Y257F to promote the distorted substrate structure may explain the observed 100-fold decrease in the rates of the O2 activation and insertion steps of the reaction.
Homoprotocatechuate 2,3-dioxygenase (FeHPCD) from Brevibacterium fuscum catalyzes the fission of the O-O bond of O2 with incorporation of both atoms into the aromatic substrate (HPCA) resulting in ring cleavage as shown in Scheme 1.1–5 The mechanism shown in Scheme 1 begins with O2 and HPCA binding in adjacent coordination sites of the active site FeII.4,6–10 Electron transfer from HPCA to the O2 via the iron would give both substrates radical character, allowing facile recombination to form an alkylperoxo intermediate from which O-O bond fission and aromatic ring cleavage could ensue. Both the initial intermediate, in which each substrate has radical character, and the alkylperoxo intermediate resulting from oxygen attack have been structurally characterized by X-ray crystallography after a crystal of the enzyme soaked with the slow substrate 4-nitrocatechol (4NC) was exposed to low concentrations of O2. 3 While the chemical nature of the alkylperoxo species is apparent from the structure, the diradical character of the initial intermediate was surmised from the pronounced lack of planarity observed for the aromatic substrate. It was proposed that the non-planar ring results from a localized semiquinone radical (SQ•) on the ring C2 (See Scheme 1 for numbering system used here) where the attack by oxygen (at the level of O2•−) will occur. Accordingly, in the alkylperoxo intermediate, this carbon in observed to be fully sp3 hybridized and 4 coordinate.
Scheme 1.
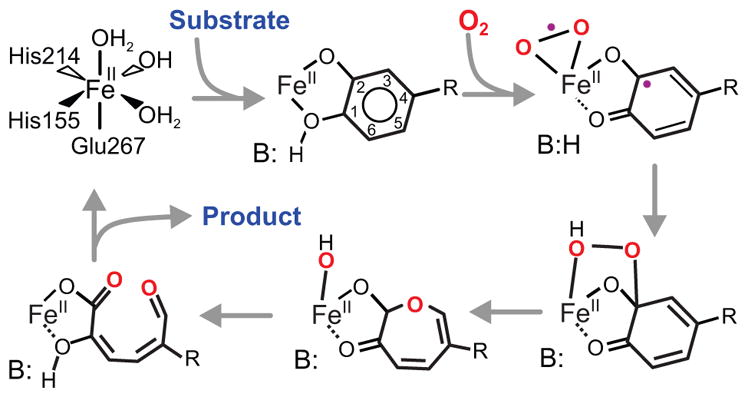
Proposed reaction mechanism for extradiol dioxygenases. In the case of FeHPCD, R is –CH2COO− (optimal substrate HPCA) or –NO2 (alternative substrate 4NC), B(H) is His200. The ring carbon numbering system shown is adopted for both substrates for ease of presentation.
The structural studies from our laboratory and others have shown that there are several second sphere active site residues that may contribute to catalysis (e.g. Figure 1).3,5,9–11 Extensive investigations of an active site His (His200 in the case of FeHPCD) have shown that it plays many roles including stabilization of the Fe-bound O2 via hydrogen bonding and charge interaction, acid-base catalysis of the alkylperoxo intermediate formation and subsequent oxygen insertion chemistry, and possibly, stabilization of the bound oxygen in the side-on orientation properly aligned to attack the aromatic substrate.3,12–15 Intermediates trapped from solution reactions of FeHPCD and its variants at His200 show that iron converts transiently to the FeIII state in the absence of H200, but it is always observed in the FeII state in its presence. 13,14 Thus, when His200 is present, one or two electrons appear to pass from the aromatic substrate to the oxygen with either no change in iron oxidation state or a transient change that persists for much less than the freezing time for rapid freeze quench (RFQ) experiments (~10 ms).14 This suggests that there is a considerable driving force for electron transfer that is not apparent from the redox potentials of the isolated aromatic substrate and O2.
Figure 1.
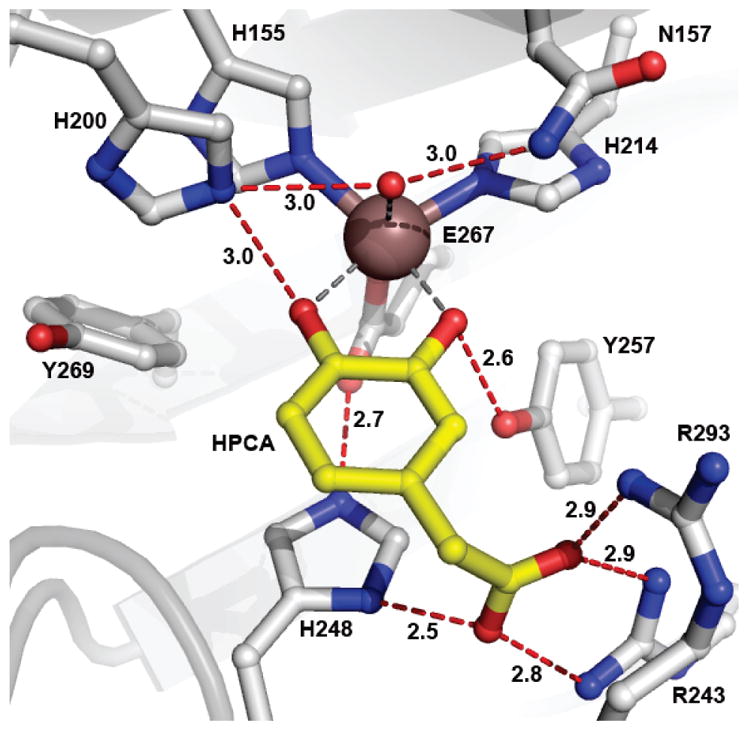
Active site environment of FeHPCD in complex with the optimal substrate HPCA (PDB 4GHG, subunit C). Atom color code: gray, carbon (enzyme); yellow, carbon (substrate); blue, nitrogen; red, oxygen; bronze, iron. Red dashed lines show hydrogen bonds (Å). Gray dashed lines indicate bonds or potential bonds to iron (Å). Additional distances are given in Table S3. The cartoon depicts the secondary structure elements.
Another second sphere amino acid, Tyr257, is hydrogen bonded to the deprotonated C2-O− of bound HPCA (Figure 1). The orientation of Tyr257 is such that this interaction might stabilize the non-planar structure of the substrate in the reaction cycle intermediate. In the accompanying report,16 it is shown that mutation of Tyr257 to Phe (Y257F) results in a significant decrease in the rate constants for many steps with reduction approaching 100-fold in the oxygen binding and insertion steps despite the presence of the acid-base catalyst His200. The results suggest that the putative HPCA SQ•-FeII-O2•− is either not formed or is unreactive, leading to formation of an observed HPCA quinone-FeII-(H)peroxo species that then slowly yields the correct ring cleaved product.
Here, structural studies of the Y257F variant, its complexes with HPCA and 4NC, and the intermediate that results from exposure of the Y257F-4NC complex to O2 are reported. The results give insight into the structural basis for the observed decreases in rate constants and the change in the chemical nature of the reactive intermediate as well as the strategy used by aromatic dioxygenases to simultaneously maintain high rates and high fidelity in ring cleaving reactions.
EXPERIMENTAL PROCEDURES
Reagents and Enzymes
All chemicals were purchased from Sigma-Aldrich and were used without purification except for HPCA and 4NC, which were recrystallized from water at 4 °C to remove minor contaminants. Recombinant B. fuscum FeHPCD and the Y257F variant were expressed and purified as previously described.1,3,12,16.
X-ray Crystallography
Crystals of FeHPCD and Y257F were grown by the hanging-drop method at 20 °C in either 13% PEG6000, 0.1 M calcium chloride, 0.1 M Tris-HCl pH 6.5 – 7.4 or 18% PEG8000, 0.1 M calcium acetate, 0.1 M sodium cacodylate pH 6.5. For X-ray data collection at 100K, crystals were cryo-cooled in liquid nitrogen following a brief transfer into cryoprotectant solution containing either 20–25% PEG400 or 20% glycerol in the mother liquor solution. Crystal morphology, cell dimensions, crystal packing or structure are not affected by the choice of buffer or calcium salt used in our earlier or present studies of the FeHPCD or its variants. 3,11
For anaerobic complex preparation, all mother liquor solutions, enzyme crystals (FeHPCD and Y257F), and aromatic substrates (4NC and HPCA stocks) were equilibrated in the anaerobic glovebox atmosphere (Belle Technology) for at least 18 hours prior to mixing. The soaking reaction was initiated by anaerobic addition of 10 mM 4NC or HPCA to enzyme crystals. After 0.5 – 3 hour incubation, crystal complexes were rapidly transferred into mother liquor solution containing 25% PEG400 prior to cryo-cooling in liquid nitrogen inside the anaerobic glovebox. For aerobic complex preparation, crystals of Y257F-4NC complex, formed inside the glovebox, were exposed to atmospheric conditions prior to cryo-cooling in liquid nitrogen.
X-ray diffraction data, collected at 100 K under the stream of liquid nitrogen, were processed using XDS17,18 or HKL2000 19 packages. The coordinates of the FeHPCD (PDB 3OJT) were used as an initial model in rigid body refinement followed by cycles of restrained refinement with Refmac5 20 as part of the CCP4 program suite 21 and model building using Coot 22. TLS was used in the final round of restrained refinement, with a single subunit defined as a group. Link restraints to the metal were removed from the refinement to avoid bias in the refined metal-ligand distances. NCS restraints were not used during refinement, and the 4 subunits of the single enzyme molecule present in the asymmetric unit were refined independently. Ligand refinement protocols were essentially the same as that described previously 3, with specific details given in the Supplementary Information section. X-ray data processing and refinement statistics are summarized in Tables S1 and S2. All structure figures were produced using PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC.
RESULTS
Past structural studies have shown that the aromatic substrate participates in several specific interactions when bound in the active site of FeHPCD. These include hydrogen bonding, electrostatic and steric interactions with the 3 completely conserved active site residues His200, His248 and Tyr257. The X-ray crystal structures of anaerobic complexes of FeHPCD with HPCA (1.50 Å, PDB 4GHG) and 4NC (1.55 Å, PDB 4GHH) reported here illustrate these interactions in detail (Figures 1 and S1A). One new aspect that is apparent in these higher resolution structures of the FeHPCD complexes is the strained conformation of the bound substrates, 4NC and HPCA, as evidenced by the observed electron density (see below). The structures also emphasize the favorable stabilizing interaction with the Tyr257, as indicated by hydrogen bonding distances of 2.5 – 2.6 Å between substrate C2-O− and hydroxyl moiety of Tyr257 in complexes with HPCA and 4NC. When the Phe substitution eliminates this interaction with substrate, structural and functional consequences of several types occur and these are described here.
Structure of Y257F Variant
The resting state structure of the Y257F variant was solved at 1.55 Å resolution (Figure 2A). Structural comparison of the Y257F variant (PDB 4GHC) with FeHPCD (PDB 3OJT) shows no changes in the global or local conformation as indicated by RMSD values of 0.46 Å for superposition of all atoms within entire tetramer. As illustrated in Figure 2B, the positions of key active site residues, including the aromatic ring of residue-257, are unaltered by substitution of Tyr for Phe in the Y257F variant. Therefore, it is likely that the observed changes in kinetics for Y257F variant, 16 can be attributed to the loss of interactions between substrate and Tyr257.
Figure 2.
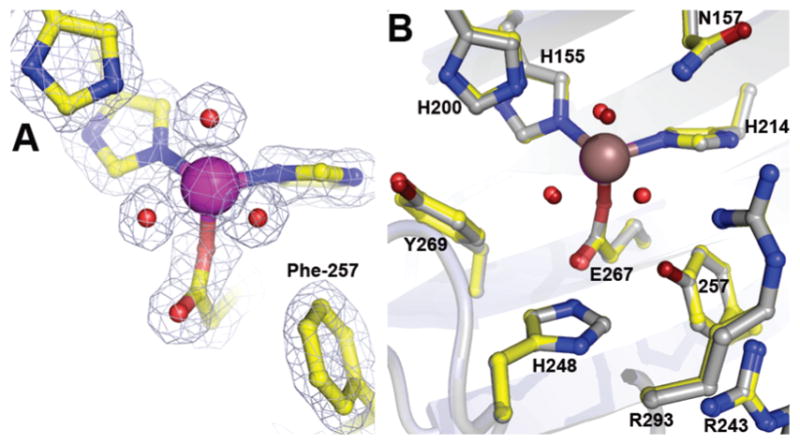
Structure of the Y257F variant in the resting state (A) and comparison of the active site environments (B) for the Y257F variant (PDB 4GHC) with that of FeHPCD (PDB 3OJT). The blue 2Fobs Fcalc electron density map is contoured at 1.6 σ. Atom color code: gray, carbon (FeHPCD); yellow, carbon (Y257F); blue, nitrogen; red, oxygen (Y257F); dark red, oxygen (FeHPCD); purple, iron (Y257F); bronze, iron (FeHPCD). Cartoons depict secondary structure elements for FeHPCD (gray) and Y257F variant (light blue).
Structure of Y257F-HPCA Complex
In order to evaluate the structural consequences of Tyr257 substitution on binding of substrate, the X-ray crystal structure of anaerobic Y257F-HPCA complex was solved at 1.85 Å resolution (PDB 4GHD). As illustrated in Figure 3, Phe at position 257 does not alter the mode of substrate binding, metal coordination distances, hydrogen-bonding interactions or structural integrity of the residues in the active site. Structures of the Y257F variant in the resting state and in complex with HPCA reveal small shifts of 0.2 – 0.6 Å in positions of H248 side chain and carbonyl moiety of R293 (Figure S2A). However, these small shifts result from optimization of the ionized substituent of HPCA in the anion-binding pocket formed by H248/R243/R293 rather than mutation of Y257F, because exactly the same shifts accompany binding of HPCA to FeHPCD (c.f. Figure S2A and B). The positions of residues in the active site and the aromatic substrate in the FeHPCD-HPCA and Y257F-HPCA complex structures are essentially the same, as illustrated in Figure 3B. This includes the asymmetry of the substrate coordination of the iron, which shows that only one of the two catecholic hydroxyls is deprotonated (Table S3). However, the slight deviation from planarity characteristic of the substrate ring in the FeHPCD-HPCA complex is not observed in the Y257F-HPCA complex (Figure 3B and 4).
Figure 3.
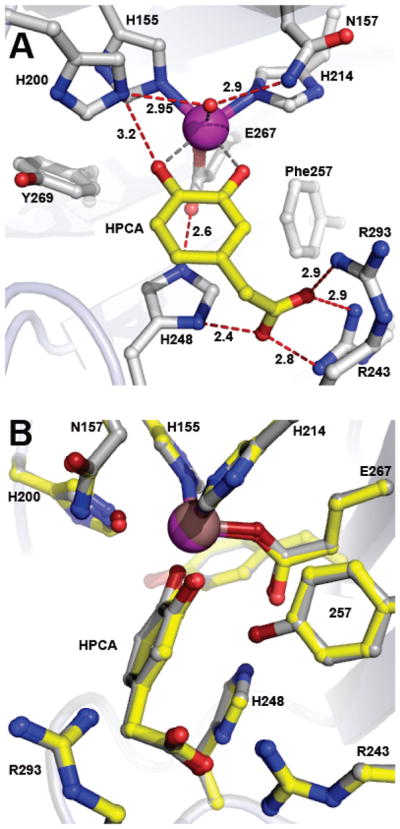
Structure of optimal substrate HPCA bound in the active site of the Y257F variant (PDB 4GHD). (A) Interactions between bound HPCA and residues in the active site (subunit C). Red dashed lines show hydrogen bonds (Å). Gray dashed lines indicate bonds or potential bonds to iron (Å). Additional distances are given in Table S3. Atom color code: gray, carbon (enzyme); yellow, carbon (HPCA); blue, nitrogen; red, oxygen; purple, iron (Y257F). (B) Comparison of active site environments in the FeHPCD-HPCA (PDB 4GHG) and Y257F-HPCA (PDB 4GHD) complexes. Atom color code: gray, carbon (FeHPCD); yellow, carbon (Y257F); blue, nitrogen; dark red, oxygen (FeHPCD); red, oxygen (Y257F); bronze, iron (FeHPCD); purple, iron (Y257F). Additional rotated view for structure overlay in panel B is shown in Figure S3A.
Figure 4.

Front and side views of the optimal substrate HPCA bound to iron in the active sites of the Y257F variant (A, PDB 4GHD) and FeHPCD (B, PDB 4GHG) enzymes. Side view comparison of metal-bound HPCA in the FeHPCD and Y257F enzymes is shown in panel C. The blue 2Fobs–Fcalc electron density map is contoured at 1.2 σ. The green ligand-omit Fobs–Fcalc difference map was calculated without HPCA in the models, and it is contoured at 7.0 σ (Y257F) or 6.0 σ (FeHPCD). Atom color code: gray, carbon (enzyme); yellow, carbon (HPCA in the Y257F complex); dark gray, carbon (HPCA in the FeHPCD complex); blue, nitrogen; dark red, oxygen (FeHPCD); red, oxygen (Y257F); purple, iron (Y257F); bronze, iron (FeHPCD).
Structure of Y257F-4NC Complex
A high-resolution X-ray crystal structure of Y257F variant in complex with alternative substrate 4NC was solved at 1.60 Å resolution (PDB 4GHE). The structure of the Y257F-4NC complex observed in subunit C is shown in Figure 5. As is the case for FeHPCD, the binding positions of HPCA and 4NC in the active site of Y257F are nearly superimposable and there are negligible changes in the positions of the active site residues (Figure S4). Due to the differences in bulk and charge of the ring substituents of HPCA and 4NC, slight shifts relative to the resting state in the positions of H248 and main chain carbonyl of R293 are observed (Figure S2, S4). Again, these shifts are the same as those observed when HPCA binds to FeHPCD. Spectroscopic studies show that the 4NC is fully deprotonated in the Y257F-4NC complex in solution (data not shown), and the similar iron coordination distances observed for the 4NC hydroxyls suggest that this is also the case in the crystal (Figure 5A, Table S3).
Figure 5.
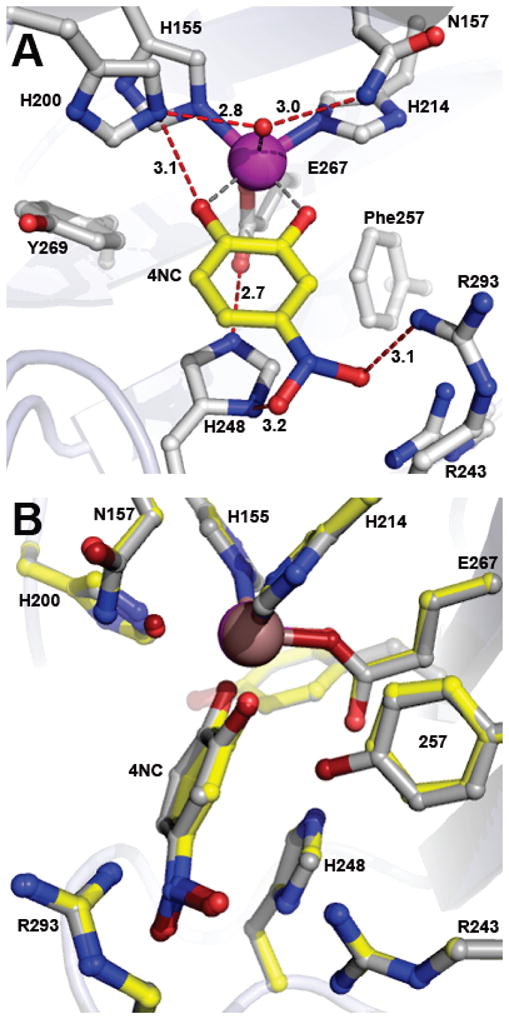
Structure of alternative substrate 4NC bound in the active site of the Y257F variant (PDB 4GHE). (A) Interactions between bound 4NC and residues in the active site (subunit C). Red dashed lines show hydrogen bonds (Å). Gray dashed lines indicate bonds or potential bonds to iron (Å). Additional distances are given in Table S3. Atom color code: gray, carbon (enzyme); yellow, carbon (4NC); blue, nitrogen; red, oxygen; purple, iron (Y257F). (B) Comparison of active site environments in the FeHPCD-4NC (PDB 4GHH) and Y257F-4NC (PDB 4GHE) complexes. Atom color code: gray, carbon (FeHPCD); yellow, carbon (Y257F); blue, nitrogen; dark red, oxygen (FeHPCD); red, oxygen (Y257F); bronze, iron (FeHPCD); purple, iron (Y257F). Additional rotated view for structure overlay in panel B is shown in Figure S3B.
It is also of interest to note that although binding of 4NC does not elicit any conformational changes within active site environment of either FeHPCD or the Y257F variant, some deviations from ideal geometry of 4NC are evidenced from the observed electron density. Specifically, distortion of the –NO2 moiety relative to the plane of the ring is observed (Figure 6), which in the context of active site environment, can be explained by steric and electrostatic effects. The anion-binding pocket formed by residues H248/R243/R293 is likely to polarize the nitro substituent, inducing asymmetry in the normally delocalized moiety. The out-of-plane bending of the substituent eliminates steric clashes. Taking this into account, the refined model of 4NC bound in the active site of Y257F variant describes the observed electron density well (Figure 6A), without clashes (Figure 5A), and the conformation of –NO2 moiety is similar to that observed in the FeHPCD-4NC complex structure (Figure 6C). With the exception of the nitro substituent, the ring of 4NC substrate in the Y257F complex is nearly planar, in comparison to that in the FeHPCD-4NC complex (Figure 5B and 6C).
Figure 6.

Front and side views of the alternative substrate 4NC bound to iron in the active sites of Y257F variant (A, PDB 4GHE) and FeHPCD (B, PDB 4GHH) enzymes. The blue 2Fobs–Fcalc electron density map is contoured at 1 σ. The green ligand-omit Fobs–Fcalc difference map was calculated without 4NC in the models and it is contoured at 6.0 σ. (FeHPCD). (C) Side view comparison of metal-bound 4NC in the active sites of FeHPCD and Y257F enzymes. (D) Side view comparison of metal-bound 4NC in the active site of FeHPCD with model of 4NC refined using idealized geometry restraints (same orientation as in panel C). Atom color code: gray, carbon (enzyme); yellow, carbon (4NC in the Y257F complex); dark gray, carbon (4NC in the FeHPCD complex); green, carbon (4NC model with idealized geometry, panel D); blue, nitrogen; dark red, oxygen (FeHPCD); red, oxygen (Y257F); purple, iron (Y257F); bronze, iron (FeHPCD).
Structure of Y257F-4NC-O2 Complex
Several experimental strategies were employed to examine the reactivity of Y257F-4NC complex in crystallo using varied concentrations of O2. In our first attempt, the procedure employed previously to form intermediates with the FeHPCD crystals was used. The preformed anaerobic Y257F-4NC complex in the crystal was exposed to very low O2 concentrations (~50 ppm), but no intermediate was observed even after prolonged incubation times. Then, higher O2 concentrations were used (1 atm of air, ~ 250 μM dissolved O2, 22 °C). In the case of the FeHPCD-4NC crystals, these conditions could not be used because a mixture of intermediates, present simultaneously in each subunit, could not be resolved.3 However, in the case of the Y257-4NC crystals, a single new species with additional electron density in the O2 binding site was observed in two of the 4 subunits in the asymmetric unit. The X-ray crystal structure of the Y257F-4NC-O2 complex trapped under these in crystallo reaction conditions was solved at 1.67 Å resolution (PDB 4GHF). The occupancy of the O2 binding site in the subunits containing the oxy-complex was estimated to be 50 %, somewhat lower than the occupancy of the equivalent complex formed in the FeHPCD enzyme (> 80%). Additional experimental strategies such as reaction at 4 °C (increased solubility of O2) or using pressure cell (4DX Systems) to briefly expose Y257F-4NC crystal to 0.5 – 2 bar of O2 prior to cryo-cooling in liquid nitrogen did not improve the observed occupancy of bound O2 in the active site.
The occurrence of different intermediates in specific subunits in the crystal is uniformly observed for this homotetrameric enzyme and is thought to derive from differential crystal packing forces.3,11 In the case of Y257F-4NC, the intermediates observed in subunits of the air-exposed crystal included the Y257F-4NC-oxy intermediate in subunits C and D and the substrate complex in subunit A, whereas ligand occupancy in subunit B was insufficient for refinement. The Y257F-4NC-oxy complex structures show that dioxygen is bound in a side-on orientation (Figure 7 and S5). This binding orientation is also observed in the structure of the FeHPCD-4NC-oxy complex, but the Fe-O bond lengths are longer for the Y257F complex, consistent with weaker binding affinity. The binding position of aromatic substrate in the active site of the air-exposed crystal is similar to that observed for the anaerobic Y257F-4NC and FeHPCD-4NC complex structures (Figure S6). However, in contrast to the structure of the FeHPCD-4NC-oxy complex, the aromatic ring of 4NC remains undistorted in the Y257F complex (Figure 7B).
Figure 7.
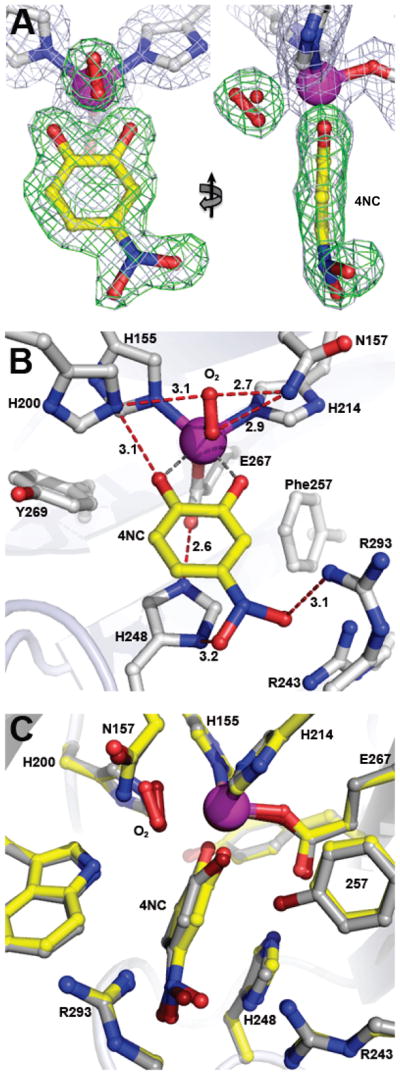
Intermediate observed in the active sites of Y257F variant reacted with 4NC in the presence of atmospheric concentrations of O2 (PDB 4GHF). (A) Front and side views for complex observed in subunit C, with a 50:50 mixture of dioxygen and solvent refined in the O2-binding site. The blue 2Fobs–Fcalc electron density map is contoured at 1 σ. The green ligand-omit Fobs–Fcalc difference map was calculated in the absence of all substrates in the final model and it is contoured at 6.5 σ. Atom color code: gray, carbon (enzyme); yellow, carbon (4NC); blue, nitrogen; red, oxygen; purple, iron. (B) Interactions between bound 4NC, dioxygen (50%) and residues in the active site (subunit C). Solvent present at partial occupancy in the O2-binding site (e.g. panel A) is omitted for clarity. Red dashed lines show hydrogen bonds (Å). Gray dashed lines indicate bonds or potential bonds to iron (Å). Additional distances are given in Table S3. Atom color code: same as in panel A. (C) Comparison of the active site environments in the FeHPCD-[4NC-semiquinone]-O2•− (PDB 2IGA, subunit C) and Y257F-4NC-oxy (PDB 4GHF) complexes. Atom color code: gray, carbon (FeHPCD); yellow, carbon (Y257F); blue, nitrogen; dark red, oxygen (FeHPCD); red, oxygen (Y257F); bronze, iron (FeHPCD); purple, iron (Y257F). Additional views and comparisons are shown in Figures S5 and S6.
DISCUSSION
One of the most unusual and unexpected results that emerged from the study of the 4NC oxygenation reaction catalyzed by FeHPCD in crystallo was the observation of an effectively stable oxy-complex. 3 The current study shows that an oxy complex can also be stabilized in the crystal of the Y257F-4NC. As in the case of the oxy complex of the FeHPCD enzyme, the oxygen ligand binds side-on, but the complexes differ in at least three other respects. First, the plane of the ring of 4NC is significantly distorted in the FeHPCD complex, but nearly planar in that of Y257F. Second, it requires much higher O2 concentrations to form the intermediate in the crystal of Y257F. Third, the alkylperoxo and product complexes are represented in other enzyme subunits within the asymmetric unit of the FeHPCD crystal, but this is not the case for the Y257F crystal. The bearing of these results on the oxygen activation and insertion mechanism of extradiol dioxygenases is discussed here.
Two Types of Aromatic Ring Distortion
A fully aromatic ring is normally observed to be planar in high-resolution crystal structures. This result is expected based on the large resonance stabilization energy that follows from the sp2 hybridized carbons of the ring and the resulting π-interactions. Nevertheless, our current and past studies have revealed two distinct types of ring distortions in substrate and oxy complexes of FeHPCD (Figures 5B, 6C–D, 7B). 3 The first involves a uniform bending of the ring, which affects the relative positions of all of the ring carbons. This global distortion is observed in the anaerobic substrate complexes with both HPCA and 4NC. The second is a localized and more significant distortion that moves specifically the C2-O− below the plane of the ring and is observed in the oxy complex of 4NC (it may also occur in the HPCA complex, but this species has not been stabilized in structural studies). Importantly, both types of ring distortions are lost for the equivalent complexes made using the Y257F variant (Figures 3–7). The distortion in the FeHPCD-substrate complex may arise from three subtle effects: (i) The plane defined by substrate binding sites on the iron and in the distal anion-binding pocket is rotated from that defined by the iron coordination geometry. Specifically, the anion-binding pocket for the substituent at the 4-position of substrate, defined by residues H248/R243/R293, is oriented below the equatorial plane of the metal coordination defined by the binding sites for substrate H155 and H214 and is optimal for longer anionic substituent of the HPCA substrate. (ii) The stabilization of the shorter polar substituents will draw electron density from the substrate, perturbing its aromaticity. This perturbation is directly detected in the asymmetric electron density distribution of bound 4NC. 3 (iii) Tyr257 is in van der Waals contact with the C2 carbon of the substrate, thereby preventing the ring from remaining planar when the substrate is bound between the anchoring points in the active site. In effect, Tyr257 acts as a fulcrum on which the ring is bent (Figure 8A).
Figure 8.

Representative steric interactions between ring carbon atoms of 4NC-derived intermediates and 2nd sphere residue in position 257 in the FeHPCD and Y257F enzymes. (A) The models and van der Waals interactions (surface) for the FeHPCD enzyme: 4NC complex (PDB 4GHH, subunit C), [4NC-semiquinone]-O2•− (PDB 2IGA, subunit C) and alkylperoxo intermediate (PDB 2IGA, subunit D). (B) The models and van der Waals interactions (surface) for the Y257F variant: 4NC complex (PDB 4GHE, subunit C) and 4NC-oxy (PDB 4GHF, subunit C). Atom color code: gray, carbon; blue, nitrogen; red, oxygen. (C) Overlay of 4NC-derived intermediate structures, illustrating the gradient of localized distortion of the 4NC ring at C2. Atom color code: light gray, carbon (planar 4NC from PDB 4GHE, subunit C); gray, carbon (strained 4NC from PDB 4GHH, subunit C); dark gray, carbon (4NC-semiquinone from PDB 2IGA, subunit C); yellow, carbon (alkylperoxo intermediate from PDB 2IGA, subunit D); blue, nitrogen; dark red, oxygen (alkylperoxo intermediate); red, oxygen.
For the Y257F variant, the van der Waals conflict is removed and a planar ring is observed (Figure 8B). The deviation from planarity may have a mechanistic purpose in that the transfer of an electron from the substrate will make the ring easier to distort. Conversely, this first type of distortion of the ring will promote electron transfer to the oxygen as it binds leading to the second type of ring distortion.
Distortion of the Aromatic Ring of 4NC in the Oxy Complex
The substantial and localized ring distortion observed for 4NC in the FeHPCD-4NC-SQ-superoxo complex is likely to be due to electron transfer out of the aromatic ring to the side-on bound oxygen seen in the crystal structure. The localization of the resultant radical at C2 would likely come about due to the strong bond formed with the iron by the C2-O− and its interactions with Y257. Indeed, one explanation for the nearly planar 4NC ring seen in the Y257F-4NC-oxy complex is that the semiquinone radical maybe delocalized over all of the ring carbons because the hydrogen bond between Tyr257 and the 4NC C2-O− is necessary to stabilize one specific oxygen atom below the plane of the ring.
Alternative explanations for the planar ring structure in the Y257F-4NC-oxy complex are that either no electron transfer has occurred or that 2 electrons have been transferred to form a 4NC-quinone-FeII-(H)peroxo species. Structural studies of the 4NC-quinone complex of FeHPCD have been conducted and show the expected flat ring (unpublished observation). The distribution of electron density in the bound quinone was shown to be readily distinguishable from that of bound 4NC, indicating that the ligand structures reported here for Y257F are not consistent with that of the 4NC quinone. Based on the structural data alone, we cannot distinguish between the remaining possibilities that the substrate remains aromatic or is oxidized to the semiquinone in the Y257F-4NC-oxy complex with full delocalization of the radical. In the accompanying study of the reaction in solution, it is shown that the oxy intermediate formed is likely to contain fully aromatic 4NC bound to FeII along with a weakly bound O2. 16 If this is the intermediate seen in the crystal, then the Fe-O2 bond might be expected to be weak, and this is consistent with the long Fe-O2 bonds observed here.
It is also possible that migration of density to the nitro group contributes to the distortion. However, it is shown here that the 4NC ring is nearly planar in the Y257F-4NC complex, suggesting that the inductive effect of the nitro substituent on ring planarity is minor.
Role of Ring Distortion in Promoting Oxygen Attack
The overall range of ring distortions of both types for the enzymes and substrate complexes described here is quite dramatic as summarized in Figure 8C. The current results suggest that these global and local distortions of the substrate aromatic ring caused by the interactions with Tyr257 are important for efficient catalysis. Four outcomes attributable to the lack of substrate ring distortion in Y257F might be anticipated. First, the lack of a driving force for electron transfer from substrate to O2 would result in a weaker Fe-O2 bond. Accordingly, exposure to O2 at atmospheric concentrations is necessary to form the oxy complex in crystals of Y257F-4NC even at partial occupancy as opposed to the trace O2 concentrations required to form the intermediate at full occupancy in the FeHPCD-4NC crystal. Second, the absence of radical character in both the substrate and bound O2 would decrease their mutual reactivity. Third, failure to localize the radical character of the substrate would decrease its reactivity still further. Fourth, the formation of an sp3 hybridized C2 carbon following the oxygen attack would push the oxygen bound to C2 closer to Y257. Consequent formation of a stronger hydrogen bond would promote the reaction by stabilizing the resulting alkylperoxo intermediate. The Phe257 residue of Y257F variant could carry out none of these stabilizing roles, and as a result, the reaction between bound O2 and the substrate would be slowed. We speculate in the accompanying report that the normal reaction between the insipient substrate and O2 radicals does not occur, giving time for two electrons to be transferred from the substrate to the bound O2. 16 The resulting yellow quinone-FeII-(hydro)peroxo intermediate was shown to react at least two orders of magnitude slower than the (unobserved) intermediate formed in FeHPCD. The formation of the 4NC quinone would not be affected as strongly by the Y257F mutation because the quinone is planar and steric interaction with residue 257 is absent.
Role of His248
In some Type II extradiol dioxygenases such as Sphingomonas paucimobilis SYK-6 protocatechuate 4,5-dioxygenase (LigAB) or E. coli 2,3-dihydroxyphenylpropionate1,2-dioxygenase (MhpB), a histidine is present in place of the tyrosine conserved in Type I extradiol dioxygenases, such as Tyr257 in FeHPCD.23 It has been proposed that this second sphere His (His115 in MhpB or His127 in LigAB) can act as a base catalyst to promote ionization of the substrate during binding, with its basicity increased by interaction with nearby acidic side chain (e.g. Asp114 in MhpB or Glu86 in LigAB).15,24 In the active site of FeHPCD, the only acid-base residue in the vicinity of Tyr257 is His248, another residue conserved in Type I extradiol dioxygenases. However, structural analysis shows that His248 is unlikely to hydrogen bond with Tyr257, and we show here that no change occurs in the relative positions of these residues when the potential hydrogen bonding interactions are removed in the Y257F variant and its ligand complexes. Consequently, the absence of any direct charge or hydrogen bonding interactions between Tyr257 and nearby residues strongly suggests that it does not serve as an active site base. In contrast, another potential role for His248 is suggested by the structural analysis reported here, specifically stabilization of the orientation of Glu267, one of the conserved metal ligands. In fact, each of the first coordination sphere ligands (His155, His214, Glu267) has an acid-base hydrogen-bonding partner from the 2nd sphere (Asp154, His213, His248) as shown in Figure S7. Whereas His155 and His214 appear to be rather limited in their orientational flexibility due to steric effects, Glu267 could potentially adopt a much larger conformational range in the absence of His248 interaction. Perhaps the reason His248 is one of only 9 conserved residues (and one of the 3 second sphere residues) is that it must serve to maintain the structural and functional integrity of the 2-His-1-carboxylate triad in Type I extradiol dioxygenases. In comparison, conservation of His residue in position equivalent to that of His248 in FeHPCD is not observed in Type II enzymes such as LigAB, probably because the positions of the Glu residue within 2-His-1-carboxylate motif in Type I and II extradiol dioxygenases are different relative to the plane of the bound substrate. His248 in FeHPCD also forms a part of the anion-binding pocket that stabilizes the charged side chain substituent of the optimal substrate, HPCA. However, this function is a unique feature of FeHPCD rather than a common attribute of large and diverse family of enzymes that utilize the 2-His-1-carboxylate motif. This would argue in favor of the primary role of His248 in stabilization of coordination geometry (Glu267) of the catalytic triad.
Conclusion
The reactions of FeHPCD in crystallo have provided important insights into how this enzyme, and enzymes in general, use multiple elements of their structure in a coordinated fashion to promote efficient and specific catalysis. The studies described here suggest that steric interactions and a specific hydrogen bond and between Tyr257 and the substrate stabilize potential alternative structures of the substrate that can only be fully realized by loss of an electron to the only acceptor available, O2. The downstream effect of this chain of events is to activate both the substrate and O2 for reaction at only one substrate carbon. For FeHPCD in solution this phase of the reaction cycle is apparently complete well within the dead time of the stopped flow device. For Y257F in solution, the putative diradical species is not stabilized, so that there is a change in mechanism such that a reaction between a peroxo adduct and a 4NC quinone occurs to yield the same alkylperoxo intermediate. This is a much more subtle effect than those we have reported for variants at the His200 position, but it supports the idea that subtle changes in the active site are effective at not only determining the specificity of the reaction, but also optimizing the rate to increase flux and lessen the chance for adventitious chemistry.
Supplementary Material
Acknowledgments
We acknowledge Soleil Synchrotron (beamline Proxima I, St Aubin, France), Swiss Light Source (beamline PXII, PSI, Villigen, Switzerland) and Advanced Photon Source (SBC, beamline 19BM, Argonne, USA) for access to synchrotron radiation facilities. We thank Andrew W. Thompson, Beatriz Guimaraes, Martin Fuchs and Randy Alkire for technical assistance with X-ray data collection. We are grateful for resources from the University of Minnesota Supercomputing Institute.
Funding
This work is supported by Biological Sciences Research Council Grant BB/H001905/1 (to E.G.K) and National Institutes of Health Grants GM 24689 (to J.D.L.).
ABBREVIATIONS
- FeHPCD
recombinant homoprotocatechuate 2,3-dioxygenase from Brevibacterium fuscum
- Y257F
Tyr257Phe variant of FeHPCD
- HPCA
homoprotocatechuate or 3,4-dihydroxyphenylacetate or 4-carboxymethyl catechol
- 4NC
4-nitrocatechol
- SQ•
semiquinone radical
- O2•−
superoxide anion radical
Footnotes
Accession Codes
The coordinates and structure factor files have been deposited in the Protein Data Bank (PDB) [www.rcsb.org/pdb] with accession codes: 4GHG, FeHPCD-HPCA; 4GHH, FeHPCD-4NC; 4GHC, Y257F; 4GHD, Y257F-HPCA; 4GHE,Y257F-4NC; and 4GHF, Y257F-4NC + O2.
SUPPORTING INFORMATION AVAILABLE
Additional experimental procedures, Tables S1-S3, and Figures S1-S7. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Miller MA, Lipscomb JD. Homoprotocatechuate 2,3-dioxygenase from Brevibacterium fuscum - A dioxygenase with catalase activity. J Biol Chem. 1996;271:5524–5535. doi: 10.1074/jbc.271.10.5524. [DOI] [PubMed] [Google Scholar]
- 2.Groce SL, Miller-Rodeberg MA, Lipscomb JD. Single-turnover kinetics of homoprotocatechuate 2,3-dioxygenase. Biochemistry. 2004;43:15141–15153. doi: 10.1021/bi048690x. [DOI] [PubMed] [Google Scholar]
- 3.Kovaleva EG, Lipscomb JD. Crystal structures of Fe2+ dioxygenase superoxo, alkylperoxo, and bound product intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lipscomb JD. Mechanism of extradiol aromatic ring-cleaving dioxygenases. Curr Opin Struct Biol. 2008;18:644–649. doi: 10.1016/j.sbi.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vetting MW, Wackett LP, Que L, Jr, Lipscomb JD, Ohlendorf DH. Crystallographic comparison of manganese- and iron-dependent homoprotocatechuate 2,3-dioxygenases. J Bacteriol. 2004;186:1945–1958. doi: 10.1128/JB.186.7.1945-1958.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arciero DM, Lipscomb JD. Binding of 17O-labeled substrate and inhibitors to protocatechuate 4,5-dioxygenase-nitrosyl complex. Evidence for direct substrate binding to the active site Fe2+ of extradiol dioxygenases. J Biol Chem. 1986;261:2170–2178. [PubMed] [Google Scholar]
- 7.Bugg TDH. Non-heme iron-dependent dioxygenases: Mechanism and structure. In: de Visser SP, Kumar D, editors. Iron-containing enzymes: Versatile catalysts of hydroxylation reactions in nature. The Royal Society of Chemistry; Cambridge, UK: 2011. pp. 42–66. [Google Scholar]
- 8.Vaillancourt FH, Bolin JT, Eltis LD. The ins and outs of ring-cleaving dioxygenases. Crit Rev Biochem Mol Biol. 2006;41:241–267. doi: 10.1080/10409230600817422. [DOI] [PubMed] [Google Scholar]
- 9.Han S, Eltis LD, Timmis KN, Muchmore SW, Bolin JT. Crystal structure of the biphenyl-cleaving extradiol dioxygenase from a PCB-degrading pseudomonad. Science. 1995;270:976–980. doi: 10.1126/science.270.5238.976. [DOI] [PubMed] [Google Scholar]
- 10.Senda T, Sugiyama K, Narita H, Yamamoto T, Kimbara K, Fukuda M, Sato M, Yano K, Mitsui Y. Three-dimensional structures of free form and two substrate complexes of an extradiol ring-cleavage type dioxygenase, the BphC enzyme from Pseudomonas sp. strain KKS102. J Mol Biol. 1996;255:735–752. doi: 10.1006/jmbi.1996.0060. [DOI] [PubMed] [Google Scholar]
- 11.Kovaleva EG, Lipscomb JD. Intermediate in the O-O bond cleavage reaction of an extradiol dioxygenase. Biochemistry. 2008;47:11168–11170. doi: 10.1021/bi801459q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groce SL, Lipscomb JD. Aromatic ring cleavage by homoprotocatechuate 2,3-dioxygenase: Role of His200 in the kinetics of interconversion of reaction cycle intermediates. Biochemistry. 2005;44:7175–7188. doi: 10.1021/bi050180v. [DOI] [PubMed] [Google Scholar]
- 13.Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Münck E, Lipscomb JD. Trapping and spectroscopic characterization of an FeIII-superoxo intermediate from a nonheme mononuclear iron-containing enzyme. Proc Natl Acad Sci U S A. 2010;107:16788–16793. doi: 10.1073/pnas.1010015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mbughuni MM, Chakrabarti M, Hayden JA, Meier KK, Dalluge JJ, Hendrich MP, Münck E, Lipscomb JD. Oxy-intermediates of homoprotocatechuate 2,3-dioxygenase: Facile electron transfer between substrates. Biochemistry. 2011;50:10262–10274. doi: 10.1021/bi201436n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendel S, Arndt A, Bugg TDH. Acid-base catalysis in the extradiol catechol dioxygenase reaction mechanism: Site-directed mutagenesis of His-115 and His-179 in Escherichia coli 2,3-dihydroxyphenylpropionate 1,2-dioxygenase (MhpB) Biochemistry. 2004;43:13390–13396. doi: 10.1021/bi048518t. [DOI] [PubMed] [Google Scholar]
- 16.Mbughuni MM, Meier KK, Münck E, Lipscomb JD. Substrate-mediated oxygen activation by homoprotocatechuate 2,3-dioxygenase: Intermediates formed by a tyrosine 257 variant. Biochemistry. 2012;51:XXXX–XXXX. doi: 10.1021/bi301114x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Cryst. 1993;26:795–800. [Google Scholar]
- 18.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 21.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR. Overview of the CCP4 suite and current developments. Acta Crystallogr D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;E60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 23.Sugimoto K, Senda T, Aoshima H, Masai E, Fukuda M, Mitsui Y. Crystal structure of an aromatic ring opening dioxygenase LigAB, a protocatechuate 4,5-dioxygenase, under aerobic conditions. Structure (London) 1999;7:953–965. doi: 10.1016/s0969-2126(99)80122-1. [DOI] [PubMed] [Google Scholar]
- 24.Bugg TDH, Ramaswamy S. Non-heme iron-dependent dioxygenases: Unravelling catalytic mechanisms for complex enzymatic oxidations. Curr Opin Chem Biol. 2008;12:134–140. doi: 10.1016/j.cbpa.2007.12.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.