

Abstract
MDM2 oncogenic protein is the principal cellular antagonist of the p53 tumor suppresser gene. p53 activity needs exquisite control to elicit appropriate responses to differential cellular stress conditions. p53 becomes stabilized and active upon various types of stresses. However, too much p53 is not beneficial to cells and causes lethality. At the steady state, p53 activity needs to be leashed for cell survival. Early studies suggested that the MDM2 oncoprotein negatively regulates p53 activity through the induction of p53 protein degradation. MDM2 serves as an E3 ubiquitin ligase of p53; it catalyzes polyubiquitination and subsequently induces proteasome degradation to downregulate p53 protein level. However, the mechanism by which MDM2 represses p53 is not a single mode. Emerging evidence reveals another cellular location of MDM2-p53 interaction. MDM2 is recruited to chromatin, specifically the p53 responsive promoter regions, in a p53 dependent manner. MDM2 is proposed to directly inhibit p53 transactivity at chromatin. This article provides an overview of the mechanism by which p53 is repressed by MDM2 in both ubiquitination dependent and ubiquitination independent pathways.
Keywords: MDM2, ubiquitination dependent, ubiquitination independent, antirepression model
The Story of p53
The discovery of p53 as a tumor suppressor gene
p53 has been the most prominent and intensively studied tumor suppressor gene over the last 30 years of cancer research. In 1979, David Lane and Arnold Levine simultaneously discovered a 53,000 Da protein in the immunoprecipitated complex of the simian virus 40 (SV-40) T antigen protein.1,2 This protein was later named p53 and designated as an oncogene. By using the p53 cDNA clone, together with the Ras oncogene, 3 groups successfully either immortalized or fully transformed cells.3-5 This first cloned p53 cDNA was actually a dominant negative allele, with a mutation at codon 135 (valine to alanine), which led “p53” to act as an oncogene. The Levine group subsequently showed that the wild-type p53 protein can actively inhibit oncogene transformation,6 and the Vogelstein lab found that the p53 gene frequently mutated in human colon carcinomas,7 consistent with native p53 acting as a tumor suppressor gene. Following this pioneering work, many more lines of evidence pointed to the tumor suppressor function of p53 in human carcinogenesis. For example, the p53 mutations were found in very diversified human tumor types,8 and the Li-Fraumeni syndrome of multiple cancers at an early age was discovered to be due to inherited p53 mutations.9
p53 is a sequence specific transcription factor
The sequences of the p53 locus from more than 16,000 tumor samples have been collected and analyzed. Strikingly, 97% of the mutations fall into the DNA binding domain of p53.10,11 This sequence specific DNA binding domain plays an undoubtedly crucial role in the tumor suppressor function of p53. Besides the evolutionarily conserved DNA binding domain between amino acid 108 and 298, p53 also contains a transactivation domain in the N-terminus that is extensively modified at the post-translational level. Therefore, p53 is able to bind and transactivate its target genes. The consensus sequence for p53 binding has 2 copies of the inverted pentameric sequence PuPuPuC (A/T)(T/A)GPyPyPy separated by a 0 to 13 base-pair long intervening fragment. There are 300 to 1,600 predicted binding sites for p53 throughout the human genome, according to microarray and computational analysis.12,13 So far, about one hundred genes have been shown to contain p53 responsive elements and have been experimentally demonstrated to be transcriptional targets of p53. As a transcription factor that regulates a broad range of target genes falling into different functional groups, p53 is able to coordinate diverse cellular responses to a variety of cell stress factors.
p53 responds to a broad range of cellular stresses
Sitting at the nexus of a complicated network, p53 senses and integrates diverse signals and converts them into highly coordinated gene expression patterns. The transactivation activity of p53 is kept silent or extremely low in normal cells and is activated when cells are exposed to stresses like DNA damage, oncogene activation, and hypoxia. Once activated, depending on the stress type and the microenvironment, p53 selectively turns on its transcriptional target genes that function in cell cycle arrest, DNA damage repair, or apoptosis to generate different cellular adaptive responses. It is not clear how the information is integrated to selectively target p53 to different sets of target gene promoters. p53 chooses to either pause cell growth to allow time for DNA repair or kill cells bearing nonrepairable lesions. Since the active p53 can inhibit cell growth or even kill cells, a stringent regulatory mechanism is required to prevent the errant activation of p53.
p53 function is controlled by post-translational modifications
p53 modifications, including phosphorylation, ubiquitination, acetylation, methylation, sumoylation, and neddylation, make up a very complex epigenetic code that intricately modulates p53 functions. Among these modifications, ubiquitination, phosphorylation, and acetylation are the most extensively studied modifications on the p53 protein. They are involved in the regulation of all 3 steps required for p53 activation: (1) p53 stabilization, (2) DNA binding, and (3) transcriptional activation. Ubiquitination largely contributes to the control of p53 stability, and this modification is mainly mediated by oncoprotein MDM2.
p53-MDM2 Network
p53 function is highly controlled by protein stability
It is well accepted that p53 function is tightly controlled by its protein stability. p53 has very short half-life in normal cells, whereas its half-life is dramatically prolonged in human tumor cells.14 Other evidence that proved the importance of the regulation of p53 stability lies in the fact that the quickly increased p53 protein level is the priming step for full activation of p53 when cells are facing diverse cellular stresses like DNA damage and hypoxia conditions. There is long-lasting interest in the search for a gene capable of degrading p53. Biochemical studies identified a 90-kDa protein in p53 immunoprecipitated complex, and it was identified as MDM2.
MDM2 is the primary negative regulator of p53
MDM2, a gene isolated from a highly amplified chromosome locus in a murine tumor cell line 3T3-DM, was designated with great tumorigenic properties.15 Later, the tumorigenic potential of MDM2 was closely linked to its repressive function of p53 tumor suppressor gene. First, physical association was detected between p53 and MDM2 proteins through immunoprecipitation studies.16 Second, overexpression of the MDM2 protein confers tumorigenic properties on murine tumor cells, and cells carrying an inactive p53 mutant exhibit similar transforming potentials.15 Moreover, MDM2 gene amplification, frequently detected in human tumors, was only coupled with wild-type p53 but not the inactive p53 mutant.17 These observations led to the hypothesis that overexpression of MDM2 might serve as the molecular mechanism by which the cell can inactivate p53 to transform a normal cell into a cancer cell. This hypothesis was then validated by the Levine group. They showed that MDM2 indeed inhibits p53 transactivity by forming a tight complex with p53.18,19 Mouse genetic work strengthens the prominence of MDM2 as the repressive regulator of p53. MDM2 deficient mice showed very early embryonic lethality, due to the overactive p53 whereas the concomitant deletion of both alleles of MDM2 and p53 rescued the embryonic lethality of MDM2 knockout mice.20,21 Taken together, these observations support the notion that MDM2 is the primary negative regulator of p53.
Ubiquitination Dependent Function of MDM2
MDM2 is an E3 ubiquitin ligase toward p53
Biochemical studies were carried out to explore the underlying molecular mechanism of MDM2 as a repressive regulator of p53. Since the change in p53 protein level seems to be closely related to the p53 function, the initial effort was to test whether MDM2 influenced the protein degradation of p53. The transient transfection experiment showed that MDM2 greatly reduced p53 protein level by inducing proteasome degradation of p53.22 In contrast, a MDM2 mutant, defective in p53 interaction, was not effective in the induction of p53 protein degradation.22 Therefore, MDM2 causes a dramatic reduction of p53 protein level through proteasome mediated degradation, and this function of MDM2 relies on the association between p53 and MDM2.
The protein degradation of p53 maintains a high rate under steady state. p53 protein is almost nondetectable in normal cells because of a very short half-life, ranging from 5 to 30 minutes in various cell types.23,24 The degradation of p53 is ubiquitination dependent and mediated by the proteasome, as is the degradation of more than 70% of cellular proteins. Ubiquitination requires the sequential actions of 3 enzymes.25 E1 activating enzyme forms a thioester between the C-terminal glycine of ubiquitin and its own active site cysteine.25 Ubiquitin is then transferred to the active site cysteine of an E2 conjugating enzyme.25 An E3 ubiquitin ligase facilitates the transfer of ubiquitin to the protein substrate, resulting in rapid proteasome degradation of the protein substrate.25 The E3 ubiquitin ligase, unlike E1 and E2, is specific to the protein substrate. E3s cooperate with E1 and E2 to catalyze the conjugation of polyubiquitin chains onto protein substrates so the ubiquitin conjugated substrate can be recognized by the 26 S proteasome for proteolysis. E3s provide substrate selectivity through a specific self-containing substrate recognition domain or via other cofactors in the E3 ubiquitin complex.
The analysis of amino acid sequence identified a RING (Really Interesting New Gene) finger domain in the carboxyl terminal end of MDM2, and this domain was characterized with E3 ubiquitin ligase activity. When incubated with E1, the ubiquitin activating enzyme, and E2, the ubiquitin conjugating enzyme, MDM2 was able to catalyze ubiquitin-protein adduct on both itself and p53.26 The ubiquitinated p53 then translocates to 26S proteasome for proteolysis. The RING finger domain in the carboxyl terminus of MDM2 contains cysteine-rich consensus sequences that form 2 interleaved zinc-binding sites with a total of 8 cysteines and histidines comprising the sites for zinc coordination.27,28 The structure of RING finger domain brings together the E2 active site and the acceptor lysines on the substrate.29 Mutation of putative zinc coordination sites in key cysteine or histidine residues abrogates the intrinsic E3 ubiquitin ligase activity of MDM2. In accordance, the same mutations abolish the capability of MDM2 to function as ubiquitination enzyme of p53.28 The combination of both in vivo and in vitro biochemical evidence shows that MDM2 encloses E3 ubiquitin ligase activity targeting both p53 protein and itself for polyubiquitination and subsequent proteasome degradation.27 The ubiquitination activity of MDM2 is dependent on the C-terminal RING finger domain, and this RING finger domain confers substrate specificity with unclear mechanism.27
MDM2 was discovered as the principal physiological E3 ubiquitin ligase of p53, but it was not the first identified E3 ubiquitin ligase specific for p53. E6-AP (E6-Associated Protein) was the first discovered E3 ubiquitin ligase of p53. E6-AP targets p53 within a complex of high-risk human papillomavirus E6 proteins.30,31 Since E6-AP negatively regulates p53 activity, high-risk HPV infected cells are released from cell cycle arrest allowing viral genome replication.31 E6-AP contains a HECT (Homolog of E6-AP C Terminus) domain involved in catalyzing ubiquitination.31 E6-AP, as the first known E3 ubiquitin ligase of p53, is outshone by MDM2 because mouse genetic studies fail to offer encouraging data. The genetic ablation of E6-AP in mice caused only modest increases of cytoplasmic p53 protein level, and this phenotype is restricted to neurons.32 In addition to E6-AP, Arf-BP1 (Arf Binding Protein 1), COP1, Pirh2, and synoviolin have been characterized as E3 ubiquitin ligases of p53 in various contexts.33-36 Their physiological significance, however, remains to be determined. The pivotal and indispensable role of MDM2 as the key negative regulator of p53 is underscored by mouse genetic studies.37
MDM2 is required but might not be sufficient for p53 ubiquitination and degradation. The ubiquitination of p53 seems not to be a simple 1-step event, as once thought, and appears to entail other players. The distinction between monoubiquitination and polyubiquitination of p53 adds additional complexity to its regulation.38 MDM2 possess E3 ligase activity and prefers to catalyze monoubiquitination of p53 when the amount of MDM2 protein is limited in cells.38 The monoubiquitin conjugated p53 specie has preference to undergo nuclear export. In turn, p53 is polyubiquitinated and degraded by the proteasome in the absence of stress.38 Recently, CBP and p300 have been proposed to mediate this polyubiquitination step of p53, and they are defined as E4 ubiquitin ligase. CBP and p300, harboring a conserved Zn2+-binding Cys-His-rich region within the first 595 aa,39,40 seemingly bear E4 ubiquitin ligase activity through this putative atypical ligase domain.41,42 CBP and p300 require the priming step of monoubiquitination of p53 driven by MDM2 and then act as a ubiquitin chain extending factor to add ubiquitin monomers to form polyubiquitin chains on p53.41,42 In summary, MDM2 coordinates with CBP and p300, named as E4 ubiquitin ligase, to catalyze polyubiquitination of p53 and induce proteasome mediated degradation of p53 protein.
Furthermore, ubiquitination is a reversible process. HAUSP, the first recognized deubiquitinating enzyme with substrate specificity, chops off the ubiquitin moiety from ubiquitin conjugated p53 to induce p53 stabilization. Deubiquitinating enzyme provides another layer of regulation of p53 ubiquitination mediated by MDM2 and antagonizes the degradation function of MDM2 toward p53.43
The relationship between MDM2 and p53 is not unidirectional. Despite the negative regulation of p53 by MDM2, p53 positively regulates MDM2 by acting as the transcription factor of the mdm2 gene. Therefore, the 2-way relationship between MDM2 and p53 forms an autoregulatory negative feedback loop. Keeping the balance between 2 proteins maintains a low cellular level of p53 and limits the duration and potency of the p53 response upon stresses. This core feedback loop illustrates the simplest version of the relationship between MDM2 and p53, which is embedded inside an intricate network composed of additional molecules and linkages to MDM2-p53 pathway. Other proteins converge onto the MDM2-p53 axis and provide additional layers of MDM2 regulation to fine-tune p53 activity.
MDM2 ligase activity is regulated by post-translational modifications
MDM2 function is tightly controlled by post-translational protein modifications. Among all kinds of modifications, phosphorylation is the most prevalent one occurring on the MDM2 protein. MDM2 was initially discovered as a 90-kDa phosphonucleus protein.18 MDM2 is composed of 491 amino acids, and 20% are either serine or threonine residues.44 Phosphorylation happens on multiple sites in MDM2, and the phosphorylated residues cluster into 2 functional domains: (1) the N-terminal domain that interacts with p53 and inhibits p53 transactivity and (2) the highly disordered acidic domain in the central part of MDM2 that serves as the docking site for many binding partners.44 Phosphorylation of MDM2 modulates MDM2 function in p53 ubiquitination in many ways. Phosphorylation of MDM2 could directly affect E3 ligase activity of MDM2, interfere with the physical association between MDM2 and p53, or restrain MDM2 in certain subcellular compartments to protect p53 from MDM2-driven degradation.
The DNA-PK (DNA-activated protein kinase) phosphorylates MDM2 in the p53 binding domain at Serine17, and the phosphorylation on this site has been reported to have significant impact on the ability of MDM2 to ubiquitinate p53. The analysis of MDM2 Ser17Ala mutant supports a model that DNA-PK induces phosphorylation of Mdm2 on Ser17, which in turn renders the association of MDM2 with p53 to ensure p53 activation.45
ATM (ataxia telangiectasia-mutated), another phosphatidylinositol 3-kinase (PI3-K) family member, phosphorylates both p53 and MDM2, contributing to p53 induction upon genotoxic stress. ATM is able to phosphorylate MDM2 at Ser395, close to the RING finger E3 ligase domain.46 The phosphorylation on Ser395 of MDM2 does not affect the binding affinity with p53 but instead makes MDM2 less capable of promoting nuclear export of p53 into cytoplasm. Cytoplasm is the major compartment of p53 degradation; therefore, p53 degradation is slowed down and p53 protein accumulates.47 The reverse reaction of phosphorylation on MDM2 Ser395 is catalyzed by the Wip1 phosphatases, also known as PPM1D.48 The downregulation of MDM2 Ser395 phosphorylation is predicted to inhibit p53 function, and the data on Wip1 studies suggest that this is the case. Wip1 mediated dephosphorylation results in stabilization of MDM2, enhances Mdm2-p53 binding affinity, and subsequently augments p53 ubiquitination.48 Thus, Wip1 antagonizes ATM to facilitate MDM2 directed p53 degradation.48 The inhibitory role of Wip1 in the control of p53 function implies an oncogenic role of Wip1, and Wip1 indeed is amplified and overexpressed in a number of human cancers.48
Tyr394, an adjacent site of Ser395 on MDM2, was shown as a c-Ab1 dependent phosphorylation site in response to DNA damage.49 In the nucleus, c-Ab1 stimulation induces apoptosis partly through its control of the MDM2-p53 pathway. C-Ab1 induced MDM2 phosphorylation protects p53 from degradation by weakening the interaction between MDM2 and p53.50 In contrast, the substitution of Tyrosine 394 by the phosphorylation dead Phenylalanine promotes p53 degradation mediated by MDM2.49 Altogether, c-Ab1 neutralizes the inhibitory effect of MDM2 toward p53 activity and provokes p53 induced apoptosis.
The central acidic domain of MDM2 is highly disordered and requires protein modifications to adopt fixed structures to favor binding with other proteins. Multiple phosphorylation sites have been identified in this domain. Glycogen synthase kinase-3β (GSK-3β), a serine/threonine kinase, targets Serine240 and Serine254 of MDM2 for phosphorylation.51 Unlike other protein kinases mentioned above, GSK-3β mediated phosphorylation enhances MDM2 activity for efficient degradation of p53.51 The inhibitor of GSK-3β rescued p53 from degradation in an Mdm2-dependent manner.51 In a search for the mechanism of increased degradation ability of MDM2, both the binding affinity of p53-MDM2 complex and the subcellular location of MDM2 were examined, and neither of them was altered upon GSK-3β phosphorylation.51 Later, a novel mechanism was proposed and verified. MDM2 interacts with multiple proteasome subunits, and the GSK-3β phosphorylated MDM2 exhibited stronger binding affinity to make p53 more accessible to the proteasome for degradation.52 At steady state, GSK-3β is active and assists mdm2 in efficiently degrading p53 by maintaining the phosphorylation level of MDM2 central domain. GSK-3β becomes inactive upon stress, such as ionizing radiation, and results in hypophosphorylation of the central domain of Mdm2.52 p53 degradation is halted and p53 stability is increased. GSK-3β is not the only kinase targeting MDM2 acidic domain. Casein kinase (CK1) phosphorylates another cluster of serine residues ranging from 240 to 250 amino acid on MDM2.53 CK1 mediated phosphorylation of MDM2 attenuates MDM2 mediated p53 proteolysis. Inactivation of CK1 results in accumulation of MDM2 and decreased p53 activity.54 Taken together, CK1 and GSK-3β exert opposing effects on p53 activity by modulating the phosphorylation status of MDM2 central domain.
The PI-3K/Akt signaling pathway plays a critical role in tumorigenesis by promoting cell proliferation and cell survival. MDM2 was identified as a bona fide substrate of Akt. The phosphorylation of MDM2 was detected on serine residue 166 and 186 once Akt was stimulated by cell survival signals.55 Serine 166 and 186 are located in the vicinity of MDM2 NLS (nucleus localization sequence). In turn, the phosphorylation of MDM2 at these 2 sites promotes the nucleus entrance of MDM2; enhances the interaction with p300, an E4 ubiquitin ligase of p53; and inhibits its interaction with p19ARF, an inhibitor of MDM2 E3 ligase activity.44 This heterogeneous complex favors proteasome degradation of p53 and results in reduced p53 protein level. Therefore, cells with Akt activation can circumvent p53 to gain tumorigenicity. Collectively, upon cell survival signals, Akt induces phosphorylation of MDM2 and enhances the MDM2 mediated degradation of p53. The activation of Akt endows cells with growth and survival advantages at least in part by repressing p53 activity.
MDM2 itself is subjected to ubiquitination and degradation, just like its ligase substrate p53. In fact, MDM2 also has a short half-life.56 The intrinsic E3 ligase activity of MDM2 drives its own ubiquitination.57 However, data from the RING mutant knock in mice do not support the notion that the endogenous Mdm2 regulates its own stability by autoubiquitination.58 Only recently has a putative candidate of E3 ubiquitin ligase of MDM2 been proposed. Surprisingly, it is the classic histone acetyltransferase PCAF (p300-CBP-associated factor) involved in the acetylation of diverse transcription factors including p53.59 PCAF bears a zinc-finger like domain that has been characterized as an atypical E3 ligase domain.59 The inactivation of PCAF abrogates the ubiquitination of MDM2, thus stabilizing MDM2 protein and weakening p53 function.59 PCAF brings additional complexity to the already elaborate p53 network by its 2-fold regulatory roles in modulating p53 activity: direct acetylation of p53 and indirect control of p53 ubiquitination level through MDM2. SUMO, the small ubiquitin-like modifier, tags to proteins to control multiple cellular functions like protein stability, nucleus localization, and protein trafficking. Sumoylation happens on lysine 182 on MDM2 and has been proposed to control the switch of substrate specificity of MDM2.60,61 Further validation of this hypothesis will be necessary due to very limited information of MDM2 sumoylation.
Other MDM2 Interacting Proteins Acting in the p53 Pathway
It is widely accepted that MDM2 mediated ubiquitination is the primary mechanism to induce p53 degradation. The post-translational modifications, mainly phosphorylation, fine-tune this regulatory function of MDM2. In addition, a large array of MDM2 interacting proteins converge onto the MDM2-p53 axis to compose additional layers of regulation of MDM2. These interacting proteins can both positively and negatively modify the degradation capability of MDM2 toward p53.
Negative Regulators of MDM2
ARF (Alternative reading frame), 1 of the 3 proteins encoded by INK4a/ARF/INK4b locus, is proven a tumor suppressor gene, and mice depleted of both alleles of ARF are prone to spontaneous cancers.62 The tumor suppressor function of ARF is largely ascribed to its regulation of the p53-MDM2 pathway in response to oncogenic signals like Ras, MYC, and E1A.63 Upon oncogenic stress, ARF expression is dramatically increased. In turn, ARF forms a complex with MDM2 and exhibits an inhibitory effect on MDM2 mediated p53 ubiquitination. p53 therefore is derepressed from MDM2 inhibition and impedes the aberrant cell growth induced by oncogene activation. More than 1 mechanism has been presented by which ARF inhibits MDM2 activity. Elegant biochemical studies established a model showing that ARF sequesters MDM2 by forming an ARF-MDM2 complex in the nucleolus to prevent p53 from degradation.64 Alternatively, ARF could directly impair the E3 ubiquitin ligase activity of MDM2, presumably by altering the appropriate conformation of MDM2 functional domain, and block p53 degradation.65 Besides MDM2, ARF also stimulates p53 activity by modulating other E3 ubiquitin ligases of p53 like ARF-BP1.33 Although the mechanism by which ARF acts as a stimulator of p53 needs further exploration, MDM2 continues to be the primary mediator in ARF regulated p53 activation.
Ribosome biogenesis is highly related to tumor progression since vigorous ribosome production is requisite to fulfill the needs of uncontrolled cancer cell growth. For example, mutagenesis screening in zebrafish identified several ribosome proteins involved in tumorigenesis.66 The p53-MDM2 pathway, as the major defensive line of tumorigenesis, has been linked to ribosome proteins (RPs). The extensive studies of RPs offer multiple candidates as MDM2 partners.67 L5, L11, and L23, the large subunit RPs in 60S ribosome, are among these candidates. These 3 RPs interact with MDM2, block the ubiquitin ligase activity of MDM2, and lead to p53 accumulation. On the basis of these observations, one working model was proposed to connect ribosome stress, one kind of outcome caused by aberrant ribosome biogenesis, and the MDM2-p53 pathway. Ribosome stress may generate the free form of RPs to disperse in the nucleus, in turn activating p53 to induce cell cycle arrest and allow for the quality control of impaired ribosome.67 L5, L11, and L23 are not the only MDM2 interacting PRs. A broader range of RPs have been identified to intervene with the MDM2-p53 pathway, and these RPs can either upregulate or downregulate p53 activity. Our immature understanding of this extremely elaborate RP-MDM2-p53 network will need to be advanced by further investigation.
One inhibitory factor of cell growth is known as 14-3-3 sigma; its inactivation via hypermethylation has been well documented in breast cancer samples.68 The prospective tumor suppression properties of 14-3-3 sigma have been connected to its regulation of the p53-MDM2 pathway. 14-3-3 sigma activates p53 mainly through the adverse effect on MDM2 ligase activity and the blockage of nuclear export of p53.69 14-3-3 sigma functions as an anchor to sequester MDM2 in the cytoplasm and to retain p53 in the nucleus.69 The final outcome of 14-3-3 sigma activation is decreased p53 ubiquitination and increased p53 protein level. 14-3-3 sigma was initially described as an induced protein of p53 in response to DNA damage signal. Together with the reciprocal activation of 14-3-3 sigma toward p53, a positive regulatory loop is constructed to link 14-3-3 sigma and p53, and the loop is tied up by MDM2. That 14-3-3 sigma elicits positive regulation of p53 by inhibiting MDM2 upon stress might explain the loss of 14-3-3 sigma in several type of carcinomas.
Positive Regulators of MDM2
Yin Yang 1 (YY1) is a multifunctional transcription factor and an essential gene for development.70 YY1 targets the MDM2-p53 pathway through a transcription independent mechanism. YY1 forms a ternary complex with MDM2-p53 and strengthens the association between MDM2 and p53.70 YY1 induces polyubiquitination of p53 by facilitating MDM2 ligase activity.70 In addition, YY1 interferes with the ARF-MDM2 interface, and MDM2 is released from the sequestration of ARF.70 YY1 is a positive cofactor for MDM2 E3 ubiquitin ligase and enhances the ubiquitination and degradation of p53. KAP1, a nuclear co-repressor gene, is another positive regulator of MDM2 and causes p53 reduction.71 KAP1 was identified as a novel MDM2 binding partner and functions in the same mode as YY1.71 KAP1 is proposed to be involved in tumor progression due to its communication with the MDM2-p53 pathway.
HAUSP also serves as a positive regulator of MDM2; however, it leads to a complicated outcome in the regulation of the p53-MDM2 pathway. The deubiquitinating enzyme HAUSP functions as a double-edged sword in regulating p53 stability.72 HAUSP has dual and direct roles in deubiquitinating and stabilizing both MDM2 and p53 proteins. The overall effect on p53 will be determined by the homeostatic level of HAUSP under certain conditions.72 The complete loss of HAUSP leads to p53 induced apoptosis due to the elevated p53 protein level since MDM2 deubiquitination and degradation are completely abolished. In contrast, the partial impairment of HAUSP exhibits a greater stabilization effect toward MDM2 over p53, and p53 becomes destabilized.72
MDM2 has a structurally related binding partner MDMX, also called MDM4. Like MDM2, MDMX interacts with the N-terminal transactivation domain of p53 to crowd out co-transcriptional factors of p53 like p300. Unlike MDM2, the C-terminal RING finger domain in MDMX does not perform E3 ubiquitin ligase activity against p53. MDMX could stimulate MDM2 activity to degrade p53. Transfection of MDMX stabilized MDM2 protein by inhibiting its self-ubiquitination.73 MDMX may optimize E3 ligase activity of MDM2 by forming heterodimer through their RING finger domains when the ratio of MDMX: MDM2 is low in cells.74
Acting as a positive regulator of MDM2 is only one side of the story of MDMX. MDMX is another essential repression factor of p53 and is as important as MDM2. MDMX was discovered in 1996 and did not raise much attention in relation to p53 until mice studies established an essential and nonredundant role of MDMX as a p53 negative regulator. Similar to MDM2, MDMX knockout mice is embryonically lethal and can be rescued by p53 inactivation.75 The lack of intrinsic E3 ligase activity of MDMX raises a long-standing question: How does MDMX repress p53 activity? The assumption is that a p53 degradation-independent mechanism is involved in the molecular basis of MDMX repressive function, as discussed in more detail in the following session.
Here we summarize the classic model of MDM2 as a repressor of p53 (Fig. 1). MDM2 associates with p53, and the RING finger E3 ligase domain in MDM2 conjugates polyubiquitin chains onto p53 for proteasome degradation; therefore, p53 is kept at a low level in normal cells. Upon stress, a series of post-translational modifications occurring on MDM2 and many other MDM2 interacting proteins, directly inhibit the E3 ligase function of MDM2, or restrain MDM2 in certain subcellular locations to release p53 from the degradation mediated by MDM2. MDM2 loses the ability to ubiquitinate p53, and p53 becomes stabilized in response to stresses. In this classic model, MDM2 directed p53 ubiquitination and degradation play the central role. However, emerging evidence suggests dual roles of MDM2 as a repressor of p53 activity. Ubiquitination dependent and ubiquitination independent mechanisms are jointly present to control p53 activity.
Figure 1.

Ubiquitination dependent function of MDM2: the classic model of MDM2 as a repressor of p53. In the nucleus, MDM2 associates with p53 and conjugates monoubiquitin onto p53. MDM2 facilitates p53 translocation from nucleus into cytoplasm, where MDM2, together with other co-factor, presumably E4 ubiquitin ligase of p53, catalyzes polyubiquitin chains onto p53 to induce proteasome degradation. Simultaneously, p53 ubiquitination and degradation take place in the nucleus. Upon cellular stresses, a series of post-translational modifications occur on MDM2. The modified MDM2 dissociates from p53, and p53 is prevented from MDM2 mediated ubiquitination and degradation. p53 therefore becomes stabilized. P = phosphorylation; Ub = ubiquitin; Poly-Ub = polyubiquitination.
Ubiquitination Independent Function of MDM2
The evidence of a ubiquitin independent function of MDM2 existed even before MDM2 was defined as an E3 ubiquitin ligase. In 1993, researchers from the Vogelstein laboratory reported their observations that MDM2 overexpression could conceal the activation domain of p53 without changing the homeostasis of p53.19 More direct proof comes from another independent study. A truncated fragment of MDM2, containing the N-terminal 130 amino acids but not the C-terminal RING finger ligase domain, is sufficient to inhibit p53-mediated transcriptional activation.76 These observations suggest that MDM2 uses both ubiquitination dependent and independent mechanisms to repress p53 transactivity.
It is challenging to study the ubiquitination independent mechanism of MDM2 repression of p53. This is because the potent E3 ligase domain of MDM2 easily induces up- or downregulation of the p53 protein level and shadows the effect of this potential degradation independent mechanism when MDM2 gene is being manipulated under experimental conditions. Thus, an in vitro and reconstitutive system was used as the starting point to gain insight into this nontraditional mechanism through which much more advanced understanding has been achieved.
By using a reconstituted in vitro transcription system, the additional inhibitory effect of MDM2 other than degradation was revealed to lie in a central region in MDM2 encompassing amino acids 50-222.77 By using a fusion protein of this fragment of MDM2 sticky to a DNA-binding domain to allow p53 independent promoter recruitment, it was clarified that this inhibitory domain of MDM2 directly represses basal transcription machinery.77 The inhibitory domain of MDM2 binds to TFIIE, a subunit in the basal transcription complex, and probably stops the formation of preinitiation complex to synthesize mRNA.77 This is a complementary mechanism for the blockage of p53 from the basal transcription machinery due to the masking of the p53 transactivation domain by MDM2 binding.77
Not only the essential transcription factors but also the p53 specific transcription co-activators, CBP and p300, are shielded from p53 by MDM2.78 CBP and its homolog, p300, are required to stimulate the transactivity of p53. p53 interacts with both MDM2 and CBP/p300 through its N-terminal transactivation domain.78 MDM2 compromises the interaction between p53 and CBP/p300 to hinder the further activation of p53 transcription activity directed by these co-activators. Taken together, on the p53 responsive promoters, MDM2 restrains p53 function by disengaging p53 from its transcription machinery, an event not dependent on MDM2 ubiquitin ligase activity.
Notably, MDM2 was demonstrated to interfere with the process of p53 acetylation, a protein modification indispensable for p53 transactivation.79 Both in vivo and in vitro data established a role of MDM2 in preventing p300 stimulated p53 acetylation in a trimeric p53-MDM2-p300 complex.80 Moreover, MDM2 lessens p53 acetylation signal by promoting the deacetylation process. MDM2 recruits HDAC1 containing complex in a p53 independent manner.81 HDAC1 is a histone deacetylase enzyme and is able to deacetylate p53. In turn, p53 deacetylation was promoted in the presence of the MDM2-HDAC1 complex. Since the acetyl group is removed from lysine residue on p53, ubiquitination is now upregulated on the same lysines. p53 protein degradation is accelerated, resulting in reduced p53 protein level and activity. The MDM2-HDAC1 complex inhibits p53 activity through the acceleration of p53 protein degradation, the consequence of a reduced p53 acetylation level.81 Overall, MDM2 does not favor p53 acetylation reaction, which is a prerequisite condition for p53 transactivation.
This ubiquitination independent repressive function of MDM2 relies on its direct interference with the p53 transcription apparatus. It is thus reasonable to hypothesize that MDM2 localizes to p53 responsive elements of its target genes. Under physiological conditions, chromatin immunoprecipitation studies identified MDM2 tethered to the p53 responsive p21 promoter region.82 MDM2 conjugates ubiquitin monomer to histone protein H2B in the vicinity of the p53 binding site and constitutes a transcription repressive atmosphere in the local region on chromatin.82 The MDM2 mediated histone ubiquitination comprises a transcription repressive function in the context of p53 responsive genes. This function of MDM2 might be expanded to a p53 independent context as well.
The idea of MDM2 recruitment to the p53 responsive promoters has been gradually accepted as more supporting evidence has become available. It has been demonstrated that endogenous MDM2 protein localizes to p21 and MDM2 promoters at the steady state, and MDM2 localization to these DNA fragments is p53 dependent.83 In comparison, MDM2 decreases the association with p53 on the chromatin once p53 is activated.83 This phenomenon suggests that the binding of MDM2 to chromatin sustains a dormant status of p53.
An Antirepression Model of p53 Activation Including MDM2 and MDMX
Questions will be raised following the discussion of MDM2 recruitment onto the chromatin region. What is the functional consequence of the chromatin associated MDM2? How does this portion of MDM2 contribute to p53 activation? Studies from the Gu laboratory established the link between p53 acetylation and the motion of MDM2 recruitment to the chromatin. A refined antirepression model was proposed to delineate the role of MDM2 recruitment in the process of p53 activation84 (Fig. 2). MDMX, a MDM2-similar protein and another proven key repressor of p53, was also involved in this refined model. In the studies from this laboratory, MDM2 and MDMX were examined and proven present on the promoter of p53 responsive genes, p21, MDM2, and PIG3.79 p53 was kept in a preset repressed state when staying in the same complex with MDM2 and MDMX on chromatin. When p53 acetylation was fully activated, the recruitments of MDM2 and MDMX on those p53 target promoters were abrogated due to decreased association between acetylated form of p53 and MDM2/MDMX.79 p53 was released from the originally repressed status, and this step was designated as an antirepression step in this modified p53 activation model. Conversely, an acetylation dead mutant of p53 showed no effect in stripping off MDM2 or MDMX from the tested p53 target promoters, and p53 remained in the inactive form.79 The repressed status of p53 implies that p53 is intrinsically active at the steady state and that the inactivation of p53 is purely due to the binding of MDM2 or MDMX on p53 associated chromatin. In fact, the functional defects of inactive p53 mutant defective in acetylation can be rescued by removing MDM2 and MDMX from cells.79 This is the most critical piece of evidence to prove the existence of a preset repression state of p53 and the necessity of an antirepression step in this refined p53 activation model.
Figure 2.
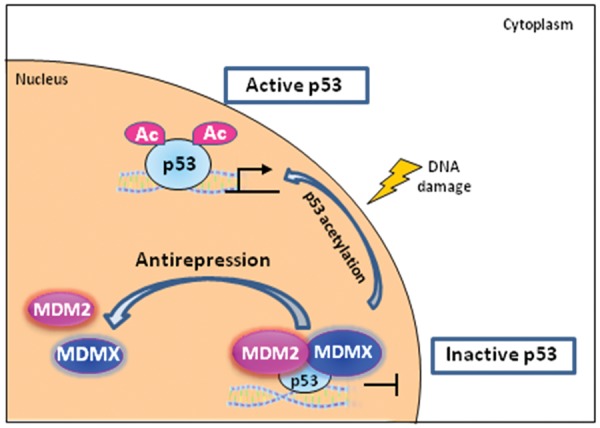
Ubiquitination independent function of MDM2: the antirepression model of p53 activation. Under homeostasis, p53 is kept in a preset repressed state when staying in the same complex with MDM2 and MDMX on chromatin. When p53 acetylation is fully activated, the recruitments of MDM2 and MDMX on those p53 target promoters will be abrogated due to the decreased association between the acetylated form of p53 and MDM2/MDMX. p53 is then released from the originally repressed status, and this step is designated as antirepression step in this modified p53 activation model. Ac = acetylation.
Concluding Remarks
p53, MDM2, and MDMX studies have plentiful and valuable information that can be only partially covered in this review due to space limitations. We focus on the regulatory mechanism of MDM2 in the repression of p53 activity. Both ubiquitination dependent and ubiquitination independent mechanisms of MDM2 mutually exist and are equally important. In particular, the ubiquitination independent mechanism is only at the initial stage of being unveiled. Many questions await elucidation. How many and to what extent are p53 transcription targets repressed by MDM2 and MDMX on chromatin? Does MDM2 or MDMX have promoter preference or tissue specificity? How does the chromatin associated portion of MDM2 or MDMX respond to various stress conditions? Additional exploration is expected to advance our understanding of this mechanism. Finally, the implications of current knowledge and future investigation for cancer biology and therapy are potentially significant. As MDM2 is a bona fide, clinically relevant drug target for cancer therapy, better understanding of its working mechanism will offer more effective and less toxic cancer therapy design.
Acknowledgments
We thank Chao Dai, a member of the Gu laboratory, for her critical comments.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA131439 and P01 CA080058 to W. Gu.. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1. Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261-3 [DOI] [PubMed] [Google Scholar]
- 2. Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43-52 [DOI] [PubMed] [Google Scholar]
- 3. Eliyahu D, Raz A, Gruss P, Givol D, Oren M. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature. 1984;312:646-9 [DOI] [PubMed] [Google Scholar]
- 4. Jenkins JR, Rudge K, Currie GA. Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature. 1984;312:651-4 [DOI] [PubMed] [Google Scholar]
- 5. Parada LF, Land H, Weinberg RA, Wolf D, Rotter V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature. 1984;312:649-51 [DOI] [PubMed] [Google Scholar]
- 6. Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083-93 [DOI] [PubMed] [Google Scholar]
- 7. Nigro JM, Baker SJ, Preisinger AC, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342:705-8 [DOI] [PubMed] [Google Scholar]
- 8. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323-31 [DOI] [PubMed] [Google Scholar]
- 9. Iwakuma T, Lozano G, Flores ER. Li-Fraumeni syndrome: a p53 family affair. Cell Cycle. 2005;4:865-7 [DOI] [PubMed] [Google Scholar]
- 10. Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81-137 [DOI] [PubMed] [Google Scholar]
- 11. Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19:607-14 [DOI] [PubMed] [Google Scholar]
- 12. Cawley S, Bekiranov S, Ng HH, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499-509 [DOI] [PubMed] [Google Scholar]
- 13. Hoh J, Jin S, Parrado T, Edington J, Levine AJ, Ott J. The p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci U S A. 2002;99:8467-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rotter V. p53, a transformation-related cellular-encoded protein, can be used as a biochemical marker for the detection of primary mouse tumor cells. Proc Natl Acad Sci U S A. 1983;80: 2613-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fakharzadeh SS, Trusko SP, George DL. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J. 1991;10:1565-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hinds PW, Finlay CA, Frey AB, Levine AJ. Immunological evidence for the association of p53 with a heat shock protein, hsc70, in p53-plus-ras-transformed cell lines. Mol Cell Biol. 1987;7:2863-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80-3 [DOI] [PubMed] [Google Scholar]
- 18. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237-45 [DOI] [PubMed] [Google Scholar]
- 19. Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857-60 [DOI] [PubMed] [Google Scholar]
- 20. Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203-6 [DOI] [PubMed] [Google Scholar]
- 21. Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378: 206-8 [DOI] [PubMed] [Google Scholar]
- 22. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296-9 [DOI] [PubMed] [Google Scholar]
- 23. Rogel A, Popliker M, Webb CG, Oren M. p53 cellular tumor antigen: analysis of mRNA levels in normal adult tissues, embryos, and tumors. Mol Cell Biol. 1985;5:2851-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davidoff AM, Humphrey PA, Iglehart JD, Marks JR. Genetic basis for p53 overexpression in human breast cancer. Proc Natl Acad Sci U S A. 1991;88:5006-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503-33 [DOI] [PubMed] [Google Scholar]
- 26. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25-7 [DOI] [PubMed] [Google Scholar]
- 27. Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275:8945-51 [DOI] [PubMed] [Google Scholar]
- 28. Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci U S A. 1999;96:11364-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009; 78:399-434 [DOI] [PubMed] [Google Scholar]
- 30. Huibregtse JM, Scheffner M, Howley PM. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol Cell Biol. 1993;13:775-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495-505 [DOI] [PubMed] [Google Scholar]
- 32. Jiang YH, Armstrong D, Albrecht U, et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799-811 [DOI] [PubMed] [Google Scholar]
- 33. Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071-83 [DOI] [PubMed] [Google Scholar]
- 34. Dornan D, Bheddah S, Newton K, et al. COP1, the negative regulator of p53, is overexpressed in breast and ovarian adenocarcinomas. Cancer Res. 2004;64:7226-30 [DOI] [PubMed] [Google Scholar]
- 35. Leng RP, Lin Y, Ma W, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779-91 [DOI] [PubMed] [Google Scholar]
- 36. Yamasaki S, Yagishita N, Sasaki T, et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase “Synoviolin.” EMBO J. 2007;26:113-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Rozieres S, Maya R, Oren M, Lozano G. The loss of mdm2 induces p53-mediated apoptosis. Oncogene. 2000;19:1691-7 [DOI] [PubMed] [Google Scholar]
- 38. Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972-5 [DOI] [PubMed] [Google Scholar]
- 39. De Guzman RN, Goto NK, Dyson HJ, Wright PE. Structural basis for cooperative transcription factor binding to the CBP coactivator. J Mol Biol. 2006;355:1005-13 [DOI] [PubMed] [Google Scholar]
- 40. Freedman SJ, Sun ZY, Poy F, et al. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci U S A. 2002;99:5367-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grossman SR, Deato ME, Brignone C, et al. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science. 2003;300:342-4 [DOI] [PubMed] [Google Scholar]
- 42. Shi D, Pop MS, Kulikov R, Love IM, Kung AL, Grossman SR. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc Natl Acad Sci U S A. 2009;106:16275-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li M, Chen D, Shiloh A, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648-53 [DOI] [PubMed] [Google Scholar]
- 44. Meek DW, Knippschild U. Posttranslational modification of MDM2. Mol Cancer Res. 2003;1:1017-26 [PubMed] [Google Scholar]
- 45. Mayo LD, Turchi JJ, Berberich SJ. Mdm-2 phosphorylation by DNA-dependent protein kinase prevents interaction with p53. Cancer Res. 1997;57:5013-6 [PubMed] [Google Scholar]
- 46. Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, Shkedy D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc Natl Acad Sci U S A. 1999;96:14973-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maya R, Balass M, Kim ST, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lu X, Nguyen TA, Zhang X, Donehower LA. The Wip1 phosphatase and Mdm2: cracking the “Wip” on p53 stability. Cell Cycle. 2008;7:164-8 [DOI] [PubMed] [Google Scholar]
- 49. Goldberg Z, Vogt Sionov R, Berger M, et al. Tyrosine phosphorylation of Mdm2 by c-Abl: implications for p53 regulation. EMBO J. 2002;21:3715-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sionov RV, Coen S, Goldberg Z, et al. c-Abl regulates p53 levels under normal and stress conditions by preventing its nuclear export and ubiquitination. Mol Cell Biol. 2001;21:5869-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kulikov R, Boehme KA, Blattner C. Glycogen synthase kinase 3-dependent phosphorylation of Mdm2 regulates p53 abundance. Mol Cell Biol. 2005;25:7170-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kulikov R, Letienne J, Kaur M, Grossman SR, Arts J, Blattner C. Mdm2 facilitates the association of p53 with the proteasome. Proc Natl Acad Sci U S A. 2010;107:10038-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Winter M, Milne D, Dias S, et al. Protein kinase CK1delta phosphorylates key sites in the acidic domain of murine double-minute clone 2 protein (MDM2) that regulate p53 turnover. Biochemistry. 2004;43:16356-64 [DOI] [PubMed] [Google Scholar]
- 54. Inuzuka H, Tseng A, Gao D, et al. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(beta-TRCP) ubiquitin ligase. Cancer Cell. 2010;18:147-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598-603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chang YC, Lee YS, Tejima T, et al. mdm2 and bax, downstream mediators of the p53 response, are degraded by the ubiquitin-proteasome pathway. Cell Growth Differ. 1998;9:79-84 [PubMed] [Google Scholar]
- 57. Stommel JM, Wahl GM. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J. 2004;23:1547-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Clegg HV, Itahana K, Zhang Y. Unlocking the Mdm2-p53 loop: ubiquitin is the key. Cell Cycle. 2008;7:287-92 [DOI] [PubMed] [Google Scholar]
- 59. Linares LK, Kiernan R, Triboulet R, et al. Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nat Cell Biol. 2007;9:331-8 [DOI] [PubMed] [Google Scholar]
- 60. Buschmann T, Fuchs SY, Lee CG, Pan ZQ, Ronai Z. SUMO-1 modification of Mdm2 prevents its self-ubiquitination and increases Mdm2 ability to ubiquitinate p53. Cell. 2000;101:753-62 [DOI] [PubMed] [Google Scholar]
- 61. Miyauchi Y, Yogosawa S, Honda R, Nishida T, Yasuda H. Sumoylation of Mdm2 by protein inhibitor of activated STAT (PIAS) and RanBP2 enzymes. J Biol Chem. 2002;277:50131-6 [DOI] [PubMed] [Google Scholar]
- 62. Sharpless NE, Ramsey MR, Balasubramanian P, Castrillon DH, DePinho RA. The differential impact of p16(INK4a) or p19(ARF) deficiency on cell growth and tumorigenesis. Oncogene. 2004;23:379-85 [DOI] [PubMed] [Google Scholar]
- 63. Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265-75 [DOI] [PubMed] [Google Scholar]
- 64. Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20-6 [DOI] [PubMed] [Google Scholar]
- 65. Llanos S, Clark PA, Rowe J, Peters G. Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus. Nat Cell Biol. 2001;3:445-52 [DOI] [PubMed] [Google Scholar]
- 66. Amsterdam A, Sadler KC, Lai K, et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004;2:E139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Deisenroth C, Zhang Y. Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene. 2010;29:4253-60 [DOI] [PubMed] [Google Scholar]
- 68. Lee MH, Lozano G. Regulation of the p53-MDM2 pathway by 14-3-3 sigma and other proteins. Semin Cancer Biol. 2006;16:225-34 [DOI] [PubMed] [Google Scholar]
- 69. Yang HY, Wen YY, Chen CH, Lozano G, Lee MH. 14-3-3 sigma positively regulates p53 and suppresses tumor growth. Mol Cell Biol. 2003;23:7096-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sui G, Affar el B, Shi Y, et al. Yin Yang 1 is a negative regulator of p53. Cell. 2004;117:859-72 [DOI] [PubMed] [Google Scholar]
- 71. Wang C, Ivanov A, Chen L, et al. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005;24:3279-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell. 2004;13:879-86 [DOI] [PubMed] [Google Scholar]
- 73. Stad R, Little NA, Xirodimas DP, et al. Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep. 2001;2:1029-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Poyurovsky MV, Priest C, Kentsis A, et al. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 2007;26:90-101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Marine JC. MDM2 and MDMX in cancer and development. Curr Top Dev Biol. 2011;94:45-75 [DOI] [PubMed] [Google Scholar]
- 76. Leng P, Brown DR, Shivakumar CV, Deb S, Deb SP. N-terminal 130 amino acids of MDM2 are sufficient to inhibit p53-mediated transcriptional activation. Oncogene. 1995;10:1275-82 [PubMed] [Google Scholar]
- 77. Thut CJ, Goodrich JA, Tjian R. Repression of p53-mediated transcription by MDM2: a dual mechanism. Genes Dev. 1997;11:1974-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wadgaonkar R, Collins T. Murine double minute (MDM2) blocks p53-coactivator interaction, a new mechanism for inhibition of p53-dependent gene expression. J Biol Chem. 1999;274:13760-7 [DOI] [PubMed] [Google Scholar]
- 79. Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kobet E, Zeng X, Zhu Y, Keller D, Lu H. MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc Natl Acad Sci U S A. 2000;97:12547-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ito A, Kawaguchi Y, Lai CH, et al. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Minsky N, Oren M. The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol Cell. 2004;16:631-9 [DOI] [PubMed] [Google Scholar]
- 83. White DE, Talbott KE, Arva NC, Bargonetti J. Mouse double minute 2 associates with chromatin in the presence of p53 and is released to facilitate activation of transcription. Cancer Res. 2006;66:3463-70 [DOI] [PubMed] [Google Scholar]
- 84. Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609-22 [DOI] [PMC free article] [PubMed] [Google Scholar]