

Abstract
Classically, p53 is considered to be an overarching tumor suppressor gene, important in its role as a transcription factor for a number of genes critical for cell cycle arrest, apoptosis, and senescence. More recently, the scope of p53 function has been further broadened, with evidence emerging that supports essential roles for p53 in reproduction and metabolism. The homologous proteins Mdm2 and MdmX function as the primary negative regulators of p53 stability and activity. Canonically, Mdm2 is thought to regulate p53 through 2 mechanisms: 1) through directly binding the p53 transactivation domain, suppressing p53 activity, and 2) through functioning as an E3 ubiquitin ligase capable of ubiquitinating p53, targeting it for nuclear export and degradation. MdmX similarly functions to bind the p53 transactivation domain; however, it is not characterized to harbor any intrinsic E3 ubiquitin ligase activity. Despite extensive study, the advent of a number of mouse models has brought to light the necessity of studying the p53 pathway at physiological levels and emphasized the major differences that can exist between in vitro and in vivo analysis. While many questions remain, a focus on the use of in vivo models in p53 study is providing a clearer view of how this pathway is regulated, with a newfound emphasis on the role of the Mdm2:MdmX heterodimer, and with that a better understanding of how this pathway could be better manipulated for therapeutic gains.
Keywords: Mdm2, MdmX, p53, E3 ubiquitin ligase, mouse models
It has been over 30 years since p53 was first identified, and over the course of those 30 years, our knowledge of the breadth and importance of p53 function has continued to grow. Due to its role as a transcription factor for a number of downstream target genes important in cell cycle arrest, apoptosis, and senescence, as well as the fact that p53 is deleted, mutated, or misregulated in a majority of cancers, p53 is now well characterized as a key tumor suppressor gene.1-5 In more recent years, the importance of p53 has been further expanded as it is now implicated to have key roles in reproduction and metabolism in addition to its role as a tumor suppressor.6-8 With such a range of functions, as well as the strong correlation between disease and aberrant p53 expression and function, a thorough understanding of p53 regulation is critical. Mdm2 was identified as a p53-interacting protein in the early 1990s and is now generally accepted to function as the primary negative regulator of p53 via its ability to act as an E3 ubiquitin ligase for p53 as well as its ability to bind to the p53 transactivation site and inhibit p53 activity.9-12 While additional E3s for p53, including Pirh2,13 COP1,14 and TOPORS,15 have been identified, Mdm2 appears to be irreplaceable in its role in controlling p53 stability. MdmX, a homolog of Mdm2, has also been revealed to be a key regulator of p53.16 Similarly to Mdm2, MdmX is capable of binding to p53 via its N-terminus, inhibiting p53 activity.16 Although the RING finger domains of Mdm2 and MdmX share a high level of homology, MdmX does not function as an E3 ubiquitin ligase for p53.16-18 While MdmX appears to play a unique and critical role in p53 regulation, it remains unclear exactly how and where MdmX is exerting this effect. Many of the strides that have been taken toward a better understanding of p53 regulation in vivo have been through the utilization of knock-out and knock-in mouse models. Despite the extensive study surrounding the function and regulation of p53, newly innovated mouse models continue to reveal that much remains unknown about how this critical gene and its protein product are regulated at physiological levels as well as the striking differences that can exist between in vitro and in vivo studies. Here, we discuss the important role that mouse models have played in our current understanding of in vivo p53 regulation and how this valuable tool can be used to further elucidate Mdm2:MdmX:p53 interactions as well as speculate further on how Mdm2 and MdmX are functioning to regulate p53 when expressed at endogenous levels.
Knock-Out Mouse Models: Setting the Stage
Mdm2 is well characterized in vitro to regulate both p53 stability and activity, and through the development of an Mdm2 knock-out mouse model, it was made clear just how necessary Mdm2 is in this capacity. Deletion of the Mdm2 gene results in early embryonic lethality, most likely due to a failure to inhibit p53 function, as the timing of lethality in mutants coincides with an increase in the cell cycle in wild-type animals.19 The lethality of Mdm2 –/– mice is completely rescued with concomitant deletion of p53, which further suggests that Mdm2 is necessary in a p53-dependent manner, specifically to downregulate p53 in embryogenesis.19,20 In addition to demonstrating the necessity of Mdm2 in proper p53 regulation, the lethality of Mdm2 –/– mice also served to demonstrate that too much, as well as too little, p53 activity can be detrimental. In a similar manner, knock-out of MdmX also results in p53-dependent early embryonic lethality.21 Despite the fact that MdmX is not historically thought to affect p53 stability as does Mdm2, deletion of MdmX results in activation of p53 and subsequent loss of cellular proliferation.21 The p53-dependent embryonic lethality of MdmX –/– mice implicates MdmX as a critical component in p53 regulation and suggests that, despite the similarities between Mdm2 and MdmX and how they interact with p53, these 2 proteins have nonredundant roles in p53 regulation in vivo. Haploinsufficiency yields divergent phenotypes for Mdm2 and MdmX, with MdmX +/– mice exhibiting increased radiosensitivity compared to Mdm2 +/– mice and gender-based differences in radiosensitivity existing in Mdm2 +/– but not MdmX +/– mice, further emphasizing the potential for differing mechanisms of p53 regulation between the 2 proteins.22 It has been hypothesized that Mdm2 and MdmX cannot compensate for one another because each protein serves a unique role in either the timing or mechanism of regulation, but conclusive data supporting this hypothesis have yet to be shown.23
Knock-In Mouse Models: Stealing the Spotlight
The development of Mdm2 and MdmX knock-out models is invaluable in demonstrating the essential and nonredundant roles these 2 proteins play in p53 regulation, and while useful in identifying their general importance, the development of a number of Mdm2 and MdmX knock-in mouse models has allowed for a more focused look at the mechanisms through which Mdm2 and MdmX are functioning to control p53. The RING finger domain of Mdm2 confers its ability to function as an E3 ubiquitin ligase for p53.24 The introduction of a single point mutation in the RING finger domain, cysteine to alanine, at the 462–amino acid residue of Mdm2 (Mdm2C462A), disrupts Mdm2 E3 ubiquitin ligase activity.25 Homozygous mutation of Mdm2 in this manner results in embryonic lethality that, similarly to deletion of Mdm2, can be rescued with concomitant deletion of p53.25 Despite leaving the p53-interacting N-terminus of Mdm2 intact and functional, the Mdm2C462A mutation leads to stabilization of p53 and p21, suggesting that Mdm2:p53 interaction in the absence of Mdm2 E3 ubiquitin ligase activity is not sufficient for proper regulation of p53 activity.25 While in vitro studies have shown that Mdm2 inhibits p53 by a dual mechanism involving both binding to p53 to inhibit its transcriptional activity and ubiquitinating p53 to target it for degradation, the lethality of the Mdm2 C462A/C462A knock-in mouse implicates the RING finger domain, presumably its E3 ubiquitin ligase activity, to be the critical mechanism through which Mdm2 is functioning. This finding highlights the importance of in vivo models in determining the true effect of regulatory mechanisms; while in vitro studies open the door to potential functions of different proteins and complexes, it is not until interactions are observed at physiological levels that the foundation for a true understanding of a protein’s function can be laid; that is, just because a protein is capable of a specific function in vitro, this function will not necessarily be carried out in vivo at endogenous levels.
In addition to disrupting Mdm2 E3 ubiquitin ligase activity, it was later found that the Mdm2C462A mutation also disrupts Mdm2:MdmX interaction. Because of the fact that the Mdm2C462A mutation disrupts both E3 ubiquitin ligase activity as well as Mdm2:MdmX binding, from this model, it cannot be determined whether one or both of these RING finger domain functions is essential. The recent development of 2 MdmX mutant mouse models serves to address the significance and the potential function of Mdm2:MdmX interaction. Introduction of a cysteine-to-alanine point mutation at the 462–amino acid residue of MdmX (MdmXC462A) results in disruption of Mdm2:MdmX binding.26 Similarly, a second MdmX mutant lacking the RING domain, MdmX∆RING, also results in disruption of Mdm2:MdmX interaction.27 Because both MdmXC462A and MdmX∆RING maintain the ability to interact with p53 via the N-terminus, and as Mdm2 is left untouched and should be able to maintain its activity towards p53, these models allow for a closer look at whether Mdm2 and MdmX work together or independently to regulate p53 and if it is necessary for the 2 proteins to interact with one another in order to function. Surprisingly, both MdmX C462A/C462A and MdmX ∆RING/∆RING homozygosity result in lethality by embryonic day 9.5.26,27 These embryos demonstrate increased levels of a number of p53 downstream targets, including Mdm2, p21, Bax, and Puma, at both the transcriptional and protein levels, suggesting that disruption of the Mdm2:MdmX heterodimer allows for activation of p53.26,27 Simultaneous deletion of p53 rescues the lethality observed in MdmXC462A and MdmX∆RING homozygotes, again suggesting that the lethality is due to overexpression and overactivity of p53.26,27 Furthermore, the lethality of MdmX∆RING homozygotes is also rescued when the mutation is placed in the transcriptionally inactive p53 515A/515A background as well as the transcriptionally hypomorphic p53 Neo/Neo background, further supporting that the primary function of MdmX, at least during embryonic development, is to restrain p53.27 Of note, while the lethality observed in MdmX ∆RING/∆RING mice can be rescued by being placed in a p53 Neo/Neo background, which expresses approximately 15% of wild-type p53 levels, Mdm2 knock-out mice are not similarly rescued, suggesting a difference in the mechanism and/or severity of p53 misregulation between Mdm2 –/– and MdmX ∆RING/∆RING mice.28 While the lethality of these MdmX RING finger mutants is indicative of a critical role for the Mdm2:MdmX heterodimer in p53 regulation, the ability of reduced p53 expression to rescue MdmX ∆RING/∆RING, but not Mdm2 –/–, lethality suggests that an additional level of regulation is likely at play. Moreover, embryonic lethality indicates only that heterodimerization plays a role in p53 regulation during embryogenesis but not necessarily during later stages of development. Induction of the MdmX∆RING mutation in adult conditional Mdm4 flxRING/∆RING CreER mice with tamoxifen resulted in viable animals, with only a slight upregulation of the p53 target, p21, and no change in Mdm2 or PUMA mRNA levels.27 The viability of adult mice with the MdmX∆RING mutation suggests that heterodimerization is not essential for p53 regulation in adult animals and that differing mechanisms likely exist to regulate p53 in embryogenesis and later in life.
Mdm2:MdmX Heterodimerization: A Rising Star
The combined pool of data from the knock-out and knock-in mouse models points to the Mdm2:MdmX heterodimer as a critical bastion of p53 regulation. While this idea is not new, through the characterization of these mouse models, it has been made clear that in vivo Mdm2:MdmX interaction plays a critical role in proper control of p53 stability and activity. The development of each of these mouse models has allowed for a more complete picture to emerge, and combined analysis of these models may allow for a deeper understanding of p53 regulation in vivo and how the Mdm2:MdmX heterodimer may be functioning at the crux of this pathway (Table 1). One tempting hypothesis is that heterodimerization affects the physical structure of the complex and with that affects its affinity toward p53. In vitro, Mdm2 and MdmX form a tight 1:1 complex via their respective C-terminal RING finger domains, and further, heterodimers form preferentially to Mdm2:Mdm2 or MdmX:MdmX homodimers.29,30 In light of the homology that exists between Mdm2 and MdmX, it is unclear why heterodimers and homodimers cannot function interchangeably, but some evidence suggests that structural differences may exist in the RING finger of the Mdm2:MdmX heterodimer compared to the Mdm2 monomers of a homodimer.31 It is possible that the slight structural differences that exist in Mdm2:MdmX heterodimers affect the binding affinity of these proteins toward p53; however, problems with solubility and stability make it difficult to address this possibility directly through crystal structuring.31 In mouse embryonic fibroblasts (MEFs), however, no difference is observed between the binding affinity of wild-type MdmX and MdmXC462A, which lacks heterodimerization, with p53.26 While it does not appear as if loss of heterodimerization affects p53:MdmX binding in MEFs, it is still possible that it exerts an effect on p53:Mdm2 binding.
Table 1.
Effects of p53, Mdm2, and MdmX Deletion and Mutation on Mouse Development in a Wild-Type (WT) and p53-Null Background
| Genotype | Model | WT background | p53 –/– |
|---|---|---|---|
| p53 –/– |
|
Developmentally normal; early susceptibility to tumor formation | N/A |
| Mdm2 –/– |

|
Lethal between E4.5 and E7.5; increased apoptosis | Viable |
| MdmX –/– |
|
Lethal between E7.5 and E8.5; decreased cellular proliferation | Viable |
| Mdm2 C462A |
|
Lethal prior to E7.5; decreased cellular proliferation | Viable |
| MdmX C462A |
|
Lethal at ~E9.5; decreased cellular proliferation; increased apoptosis | Viable |
| MdmX ∆RING |
|
Lethal at ~E9.5 | Viable |
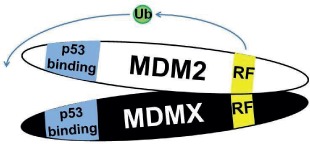
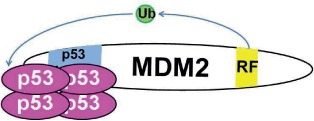
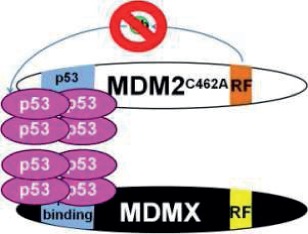
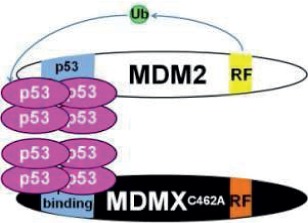
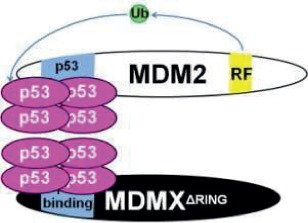
Note: RF (yellow) indicates the RING finger domain, p53 binding (blue) indicates the p53 binding domain, and Ub (green) represents ubiquitin ligase activity. In models where Mdm2:MdmX interaction is disrupted, Mdm2 and MdmX are shown to interact with separate tetramers of p53, as the current data leave it unclear whether noninteracting Mdm2 and MdmX can share interaction with one p53 tetramer. N/A = Not applicable.
A second functional role for heterodimerization may be regulation of the subcellular localization of MdmX. MdmX lacks a nuclear localization signal and nuclear export signal, but interaction with Mdm2 and p53 can allow for its translocation into the nucleus.32,33 The MdmX∆RING mutation, which disrupts Mdm2:MdmX heterodimerization, results in increased cytoplasmic MdmX, while the distributions of Mdm2 and p53 remain similar to that observed in the wild-type MEFs.27 Enrichment of cytoplasmic MdmX is also observed in Mdm2C462A MEFs, suggesting that dissociation of Mdm2 and MdmX may increase the level of cytoplasmic MdmX (Figure 1). As MdmX is thought to regulate p53 primarily through its ability to directly bind p53 and inhibit its function as a transcription factor, changes in the distribution of MdmX may affect its interaction with p53. Neither MdmX∆RING nor Mdm2C462A appear to affect p53 localization in MEF cell culture, discouraging the possibility that these changes in MdmX localization result in sequestration of p53 into the cytoplasm. One possible interpretation is that disruption of Mdm2:MdmX interaction may enhance cytoplasmic MdmX, potentially decreasing MdmX:p53 interaction in the nucleus. Another possibility is that changes in the ratio of cytoplasmic-to-nuclear MdmX affect the frequency of Mdm2:MdmX interaction, decreasing the E3 ubiquitin ligase potential of Mdm2.
Figure 1.
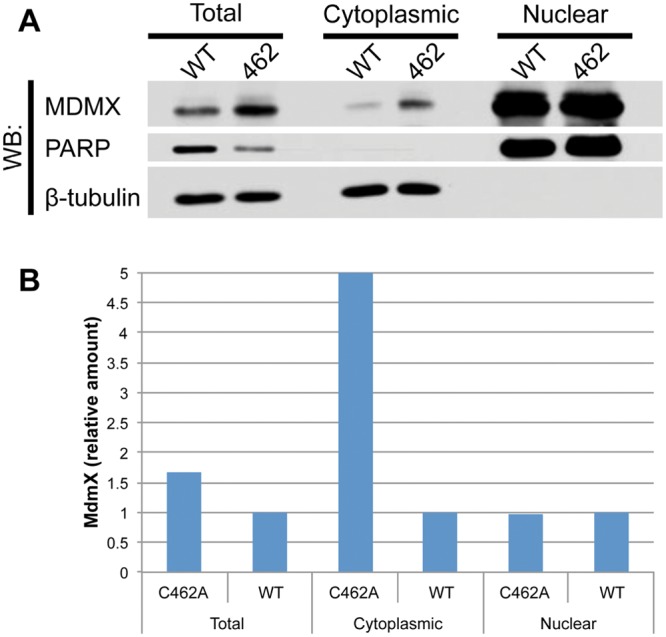
(A) Subcellular localization of Mdm2+/+, p53ER/– and Mdm2C462A/C46A, and p53ER/– MEFs. All cells were treated with 4-OHT for 24 hours. PARP and β-tubulin were used as nuclear and cytoplasmic markers, respectively. (B) Quantitation of MdmX as measured by ImageJ. Total and cytoplasmic values were normalized to β-tubulin, and nuclear signal was normalized to PARP. All values are expressed as relative to the wild-type value for the respective subcellular fraction.
Another role of the Mdm2:MdmX heterodimer that has been explored, and is perhaps the best supported, is the idea that the dimer of Mdm2 and MdmX, rather than Mdm2 alone, functions as the E3 ubiquitin ligase for p53.26 Ubiquitination of Mdm2 is reduced in the presence of MdmXC462A compared to wild-type MdmX, which Huang et al. extrapolate as indication that disruption of Mdm2:MdmX binding is capable of hindering the E3 ligase activity of Mdm2 toward p53 as well.26 A similar phenomenon has been described in vitro; 3 mutations—Mdm2T488D, Mdm2Y489A, and Mdm2Y489D—each disrupt Mdm2 E3 ubiquitin ligase activity toward p53, but upon co-expression with MdmX, they regain the ability to degrade p53, adding further support to the idea that the importance of Mdm2:MdmX interaction lays in the ability of MdmX to influence Mdm2 E3 ubiquitin ligase activity.18,34 In addition, other RING finger proteins, such as the BRCA1-BARD1 heterodimer, demonstrate enhancement of E3 ubiquitin ligase activity upon heterodimer formation.35 On the flip side, the presence of the MdmX∆RING mutation, which also disrupts Mdm2:MdmX binding, does not affect the half-lives of Mdm2 and p53 or p53 ubiquitination status.27 Additionally, while Mdm2 is capable of autoubiquitination in vitro, the ability of Mdm2C462A to be degraded normally, despite losing the ability to degrade p53, suggests that Mdm2 and p53 are not degraded in the same manner and that an outside E3 may be responsible for ubiquitination of Mdm2 in vivo.25,36,37 In light of this, the fact that MdmXC462A disrupts the ubiquitination of Mdm2 is not necessarily a reflection of changes in the ability of p53 to be ubiquitinated and subsequently degraded, as ubiquitination of endogenous Mdm2 and p53 may not occur through the same mechanism.
The interconnected nature of Mdm2, MdmX, and p53 degradation and activation can make it difficult to parse out causes and effects. Rather than directly influencing how Mdm2 and MdmX interact with p53, heterodimerization may be key due to the effects of interaction on the stability of Mdm2 and MdmX, which then ultimately affects p53 stability and activity. The relative levels of Mdm2, MdmX, and p53 have been shown to be important in whether interaction between these proteins results in degradation, stabilization, or promotion of transcription of one another.31 Despite its general characterization as an inhibitor of p53 activity, upon DNA damage, MdmX is also capable of enhancing p53 stability and activity.38 MdmX interaction with p53 specifically stimulates activity toward the Mdm2 promoter, and thus, downregulation of MdmX can result in downregulation of Mdm2.39 While MdmX is associated with enhanced Mdm2 transcription, simultaneously, Mdm2 is responsible for the degradation of MdmX.40-42 The Mdm2:MdmX interaction plays a central role in MdmX degradation, as MdmX rather than Mdm2 is ubiquitinated in the heterodimer, and proper Mdm2:MdmX heterodimerization is necessary for degradation of MdmX following DNA damage.31,43 In vitro studies also show that MdmX can serve as a competing substrate for Mdm2 E3 ligase activity, and thus, when Mdm2:MdmX binding is disrupted and MdmX no longer competes with Mdm2 for Mdm2 E3 ligase activity, Mdm2 E3 ligase activity toward itself is increased, resulting in overall lower levels of Mdm2.31 This competition model directly contradicts evidence based on the MdmXC462A mouse model, in which disruption of heterodimerization results in decreased ubiquitination of Mdm2 and increased levels of p53 and Mdm2, and further disagrees with data from the Mdm2C462A mouse model, in which Mdm2 does not autoubiquitinate, together serving as additional examples of the disagreement that exists between in vitro and in vivo modeling of the Mdm2: MdmX:p53 regulatory pathway.25,26 Due to the interconnected and multiple levels of interaction of Mdm2 and MdmX regulation, the relative level of these proteins becomes increasingly important, further emphasizing the necessity for in vivo studies.
Conclusions and Future Directions
Despite the advances that in vivo models have allowed, much remains unknown about the specifics of Mdm2:MdmX:p53 interaction and regulation, in part due to incongruences that exist among the current models. Additionally, many of the current models offer negative data; that is, deletion or mutation results in a nonviable animal. While this information is very useful in broadening our knowledge, embryonic lethality limits the kind of studies that can be performed and, with that, what can be further extrapolated. To address the issue of embryonic lethality, many of the Mdm2 and MdmX models are placed into an inducible, transcriptionally dead, or hypomorphic p53 background (i.e., p53ER/–, p53515A, p53Neo), and while this does allow for further investigation, it also introduces additional complications into an already intricate regulatory network. For example, while p53 is generally characterized to be primarily nuclear, p53515A is equally represented in the cytoplasmic and nuclear fractions.27,44 The level of cytoplasmic p53 appears to be increased in the presence of the MdmX∆RING mutant, but because the subcellular distribution of p53 is already distorted, it is difficult to conclude whether the changes in localization are due to the MdmX∆RING mutant increasing cytoplasmic retention or nuclear export of p53 or whether it is an effect of the p53515A mutation itself independent of MdmX mutation status.
Even with disagreements among the various models, as well as contradictions between in vivo and in vitro studies, the combined analyses of the models at hand have further clarified the p53 regulatory network. One idea that is supported consistently by each of these models is that Mdm2:MdmX heterodimerization is a critical component of proper p53 regulation in vivo. A few key questions remain: 1) What is the functional importance of Mdm2:MdmX heterodimerization in p53 regulation, and why are Mdm2:Mdm2 or MdmX:MdmX homodimers unable to function in this same manner? 2) How is Mdm2 degraded in vivo? 3) What is the relationship between p53 stabilization and p53 activity? In vitro studies suggest that loss of Mdm2 E3 ligase activity may stabilize p53; however, loss of Mdm2:p53 binding is necessary to activate p53, returning us to the idea that Mdm2 functions via a dual mechanism to regulate p53.43 In contrast, the Mdm2C462A mouse model suggests that p53 binding alone is not sufficient for inhibiting p53 activity in vivo, but as noted earlier, the Mdm2 C462A mutation disrupts Mdm2:MdmX binding in addition to Mdm2 E3 ligase activity, and the significance of this loss of heterodimerization on p53 regulation may be greater than initially believed.25 The idea of a dual mechanism for Mdm2 regulation of p53 may feel full circle, but rather than Mdm2:p53 binding alone or Mdm2 E3 ligase function playing the critical role in this dual mechanism, it now appears as if Mdm2 interaction via its RING finger domain with MdmX may be at the heart of its ability to properly and efficiently regulate p53 stability and activity. Through further experimentation with the models previously developed, as well as the development of additional mouse models, specifically, a model evaluating the effect of disrupting Mdm2 E3 ligase activity alone without affecting Mdm2:MdmX binding, these questions can be addressed, taking us one step closer to understanding how p53, a protein critical in tumor suppression, metabolism, and reproduction, is regulated at physiological levels. The use of mouse models allows for a closer look at how the p53 regulatory network is functioning at the level of the organism and under conditions of endogenous protein expression, allowing for a greater understanding of the misregulation of this pathway in cancer and other diseases, thereby opening the door for better potential therapeutic targets.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institutes of Health to Y.Z.: CA100302, CA127770, and CA155235.
References
- 1. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323-31 [DOI] [PubMed] [Google Scholar]
- 2. Hainaut P, Soussi T, Shomer B, et al. Database of p53 gene somatic mutations in human tumors and cell lines: updated compilation and future prospects. Nucleic Acids Res. 1997;25:151-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Danovi D, Meulmeester E, Pasini D, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004;24:5835-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26:3453-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ramos YF, Stad R, Attema J, Peltenburg LT, van der Eb AJ, Jochemsen AG. Aberrant expression of HDMX proteins in tumor cells correlates with wild-type p53. Cancer Res. 2001;61:1839-42 [PubMed] [Google Scholar]
- 6. Deisenroth C, Zhang Y. The ribosomal protein-Mdm2-p53 pathway and energy metabolism: bridging the gap between feast and famine. Genes Cancer. 2011;2:392-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol. 2011;12:259-65 [DOI] [PubMed] [Google Scholar]
- 8. Lane D, Levine A. p53 research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol. 2010;2:a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The Mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237-45 [DOI] [PubMed] [Google Scholar]
- 10. Wu X, Bayle JH, Olson D, Levine AJ. The p53-Mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126-32 [DOI] [PubMed] [Google Scholar]
- 11. Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to Mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev. 1994;8:1235-46 [DOI] [PubMed] [Google Scholar]
- 12. Yang Y, Li CC, Weissman AM. Regulating the p53 system through ubiquitination. Oncogene. 2004;23:2096-106 [DOI] [PubMed] [Google Scholar]
- 13. Leng RP, Lin Y, Ma W, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779-91 [DOI] [PubMed] [Google Scholar]
- 14. Dornan D, Wertz I, Shimizu H, et al. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429:86-92 [DOI] [PubMed] [Google Scholar]
- 15. Rajendra R, Malegaonkar D, Pungaliya P, et al. Topors functions as an E3 ubiquitin ligase with specific E2 enzymes and ubiquitinates p53. J Biol Chem. 2004;279:36440-4 [DOI] [PubMed] [Google Scholar]
- 16. Shvarts A, Steegenga WT, Riteco N, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996;15:5349-57 [PMC free article] [PubMed] [Google Scholar]
- 17. Jackson MW, Berberich SJ. MdmX protects p53 from Mdm2-mediated degradation. Mol Cell Biol. 2000;20:1001-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Poyurovsky MV, Priest C, Kentsis A, et al. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 2007;26:90-101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206-8 [DOI] [PubMed] [Google Scholar]
- 20. Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in Mdm2-deficient mice by deletion of p53. Nature. 1995;378:203-6 [DOI] [PubMed] [Google Scholar]
- 21. Parant J, Chavez-Reyes A, Little NA, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92-5 [DOI] [PubMed] [Google Scholar]
- 22. Terzian T, Wang Y, Van Pelt CS, Box NF, Travis EL, Lozano G. Haploinsufficiency of Mdm2 and Mdm4 in tumorigenesis and development. Mol Cell Biol. 2007;27:5479-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006;13:927-34 [DOI] [PubMed] [Google Scholar]
- 24. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25-7 [DOI] [PubMed] [Google Scholar]
- 25. Itahana K, Mao H, Jin A, et al. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell. 2007;12:355-66 [DOI] [PubMed] [Google Scholar]
- 26. Huang L, Yan Z, Liao X, et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc Natl Acad Sci U S A. 2011;108:12001-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pant V, Xiong S, Iwakuma T, Quintas-Cardama A, Lozano G. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc Natl Acad Sci U S A. 2011;108:11995-2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Y, Suh YA, Fuller MY, et al. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest. 2011;121:893-904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 1999;447:5-9 [DOI] [PubMed] [Google Scholar]
- 30. Kostic M, Matt T, Martinez-Yamout MA, Dyson HJ, Wright PE. Solution structure of the Hdm2 C2H2C4 RING, a domain critical for ubiquitination of p53. J Mol Biol. 2006;363:433-50 [DOI] [PubMed] [Google Scholar]
- 31. Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008;15:841-8 [DOI] [PubMed] [Google Scholar]
- 32. Marine JC, Jochemsen AG. Mdmx as an essential regulator of p53 activity. Biochem Biophys Res Commun. 2005;331:750-60 [DOI] [PubMed] [Google Scholar]
- 33. Li C, Chen L, Chen J. DNA damage induces MDMX nuclear translocation by p53-dependent and -independent mechanisms. Mol Cell Biol. 2002;22:7562-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uldrijan S, Pannekoek WJ, Vousden KH. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J. 2007;26:102-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hashizume R, Fukuda M, Maeda I, et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001;276:14537-40 [DOI] [PubMed] [Google Scholar]
- 36. Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275:8945-51 [DOI] [PubMed] [Google Scholar]
- 37. Honda R, Yasuda H. Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene. 2000;19:1473-6 [DOI] [PubMed] [Google Scholar]
- 38. Di Conza G, Mancini F, Buttarelli M, Pontecorvi A, Trimarchi F, Moretti F. MDM4 enhances p53 stability by promoting an active conformation of the protein upon DNA damage. Cell Cycle. 2012;11:749-60 [DOI] [PubMed] [Google Scholar]
- 39. Biderman L, Poyurovsky MV, Assia Y, Manley JL, Prives C. MdmX is required for p53 interaction with and full induction of the Mdm2 promoter after cellular stress. Mol Cell Biol. 2012;32:1214-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. de Graaf P, Little NA, Ramos YF, Meulmeester E, Letteboer SJ, Jochemsen AG. Hdmx protein stability is regulated by the ubiquitin ligase activity of Mdm2. J Biol Chem. 2003;278: 38315-24 [DOI] [PubMed] [Google Scholar]
- 41. Kawai H, Wiederschain D, Kitao H, Stuart J, Tsai KK, Yuan ZM. DNA damage-induced MDMX degradation is mediated by MDM2. J Biol Chem. 2003;278:45946-53 [DOI] [PubMed] [Google Scholar]
- 42. Pan Y, Chen J. MDM2 promotes ubiquitination and degradation of MDMX. Mol Cell Biol. 2003;23:5113-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wade M, Li YC, Matani AS, et al. Functional analysis and consequences of Mdm2 E3 ligase inhibition in human tumor cells. Oncogene. Epub 2012. January 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O’Brate A, Giannakakou P. The importance of p53 location: nuclear or cytoplasmic zip code? Drug Resist Updat. 2003;6:313-22 [DOI] [PubMed] [Google Scholar]