

Abstract
Ethanol is a neuroteratogen and neurodegeneration is the most devastating consequence of developmental exposure to ethanol. The mechanisms underlying ethanol-induced neurodegeneration are complex. Ethanol exposure produces reactive oxygen species (ROS) which cause oxidative stress in the brain. We hypothesized that ethanol would activate autophagy to alleviate oxidative stress and neurotoxicity. Our results indicated that ethanol increased the level of the autophagic marker Map1lc3-II (LC3-II) and upregulated LC3 puncta in SH-SY5Y neuroblastoma cells. It also enhanced the levels of LC3-II and BECN1 in the developing brain; meanwhile, ethanol reduced SQSTM1 (p62) levels. Bafilomycin A1, an inhibitor of autophagosome and lysosome fusion, increased p62 levels in the presence of ethanol. Bafilomycin A1 and rapamycin potentiated ethanol-increased LC3 lipidation, whereas wortmannin and a BECN1-specific shRNA inhibited ethanol-promoted LC3 lipidation. Ethanol increased mitophagy, which was also modulated by BECN1 shRNA and rapamycin. The evidence suggested that ethanol promoted autophagic flux. Activation of autophagy by rapamycin reduced ethanol-induced ROS generation and ameliorated ethanol-induced neuronal death in vitro and in the developing brain, whereas inhibition of autophagy by wortmannin and BECN1-specific shRNA potentiated ethanol-induced ROS production and exacerbated ethanol neurotoxicity. Furthermore, ethanol inhibited the MTOR pathway and downregulation of MTOR offered neuroprotection. Taken together, the results suggest that autophagy activation is a neuroprotective response to alleviate ethanol toxicity. Ethanol modulation of autophagic activity may be mediated by the MTOR pathway.
Keywords: alcohol, cerebellum, cerebral cortex, fetal alcohol spectrum disorders, mitophagy, neurodegeneration
Introduction
Fetal alcohol spectrum disorders (FASD) are caused by maternal alcohol consumption during pregnancy and are characterized by a spectrum of structural anomalies and neurocognitive/behavioral disabilities. Ethanol is a neuroteratogen and ethanol-induced neuronal loss in the developing central nervous system (CNS) accounts for many symptoms shown in FASD children.1 The underlying mechanisms, however, remain unclear. Oxidative stress, which is caused by excessive production of reactive oxygen species (ROS), has been proposed as a potential mechanism for ethanol-induced neuronal damage. ROS are generated during ethanol catabolism or from damaged mitochondria in response to ethanol exposure. At low levels, ROS are important regulators of cellular functions, but excessive ROS may cause oxidative stress, resulting in neurodegeneration.
Autophagy is the cellular catabolic process in which intracellular proteins/organelles are sequestered and then transported to and degraded in lysosomes to recycle cellular components under stress conditions or to eliminate damaged proteins/organelles. Constitutive clearance of cytosolic proteins by low-level basal autophagy is an important cytoprotective function. By selectively eliminating damaged mitochondria, the most important source of free radicals in the cell, autophagy may protect cells from oxidative stress.2 Autophagy is a dynamic process consisting of several sequential stages (initiation, elongation, maturation and degradation) controlled by a group of autophagy-related (Atg) genes as well as their respective Atg proteins. More than 30 Atg genes have been identified in yeast and most of them have homologs in mammalian cells. Downregulation of some Atg genes (such as VPS30/ATG6/BECN1 or ATG8/MAP1LC3) inhibits autophagy activity.3,4 Mechanistic target of rapamycin (MTOR), a serine/threonine protein kinase, is a convergence point of cell signaling pathways stimulated by cellular stress and is a well-known regulator of autophagy. An abundance of nutrients usually activates MTOR and suppresses autophagy, whereas nutrient deprivation and stressors inhibit MTOR and activate autophagy.
Excessive ROS generation from dysfunctional or damaged mitochondria may trigger autophagy, which acts to clean up damaged mitochondria.5 Therefore, under certain circumstances, autophagy is a cell self-defense mechanism used to alleviate cellular stress/damage. We hypothesized that autophagy would act as a protective response to ethanol-induced neuronal damage. With both in vitro and in vivo approaches, we demonstrated that ethanol triggered autophagy in neuronal cells. Furthermore, enhancement of autophagic flux alleviated ethanol-induced neuronal death by mitigating oxidative stress; contrarily, inhibition of autophagy exacerbated ethanol neurotoxicity. Our results indicated that ethanol modulation of autophagic activity may be mediated by the MTOR pathway.
Results
Ethanol increases the expression of markers for autophagy
First, we determined whether ethanol altered the expression of markers for autophagy in cultured neuronal cells. The conversion of MAP1LC3 (LC3) from the soluble form (LC3-I) to the autophagosome-associated form (LC3-II) implies an increase of autophagosomes within the cells.6 Sequestosome 1 (SQSTM1, also known as p62) is a protein associated with autophagosomes and is degraded in lysosomes after autophagosomes fuse with lysosomes.7 It appeared that serum contents in the medium affected the levels of LC3-II and SQSTM1, the two important markers of autophagy; the level of LC3-II was upregulated while SQSTM1 was downregulated when SH-SY5Y neuroblastoma cells were grown in a serum-free medium (Fig. 1A). Therefore, we examined the effect of ethanol on cells grown in medium containing 10% serum. Ethanol increased the level of LC3 lipidation in SH-SY5Y cells in a concentration-dependent manner while it decreased SQSTM1 levels (Fig. 1A). We examined the effect of ethanol on mRNA levels of LC3, SQSTM1 and BECN1. BECN1, also known as Vps30/Atg6 or Beclin 1, is a protein required for the initiation of autophagosome formation and is frequently used as a marker for autophagy. Ethanol increased the level of BECN1 mRNA, but had little effect on mRNA levels of LC3 and SQSTM1 (Fig. 1B). We investigated the effect of ethanol on the level of SQSTM1 protein. At the condition of either 2% or 0.1% SDS, ethanol significantly reduced the level of SQSTM1 (Fig. 1C). This result indicated the effect of ethanol was not caused by an increase in protein aggregation. We further examined the effect of ethanol on the levels of autophagic markers in the developing brain. As shown in Figure 1D and E, an injection of ethanol increased the levels of LC3-II and BECN1, but decreased SQSTM1s level in the cerebellum and cerebral cortex of 7-d-old mice.

Figure 1A–C. Effect of ethanol on the expression of autophagic markers. (A) SH-SY5Y cells were maintained in media containing 10% serum or 0% serum. The cells were exposed to ethanol (EtOH: 0, 0.4% or 0.8%) for 8 h. In some experimental groups, cells were treated with bafilomycin A1 (Baf: 10 nM). The levels of LC3-II and SQSTM1 were examined with immunoblotting (top panel). The experiment was replicated three times. The levels of LC3-II and SQSTM1 were quantified by densitometry and normalized to actin levels (bottom panels). *p < 0.05, **p < 0.01. (B) SH-SY5Y cells were exposed to ethanol (EtOH, 0.8%) for 8 h and the mRNA levels for LC3, SQSTM1 and BECN1 were analyzed by qRT-PCR as described under Materials and Methods. The experiment was replicated three times. *p < 0.05. (C) SH-SY5Y cells were exposed to ethanol (EtOH, 0.8%) for 8 h, and the proteins were extracted by RIPA buffer containing 2% or 0.1% SDS. The level of SQSTM1 was determined by immunoblotting (top panel) and quantified (bottom panel). *p < 0.05.

Figure 1D and E. Effect of ethanol on the expression of autophagic markers. (D and E) Mice of postnatal day 7 (PD7) were injected subcutaneously with ethanol (2.5 g/kg or saline) at 0 and 2 h as described under Materials and Methods. At 8 h after the first injection, the cerebellum (D, top panel) and cerebral cortex (E, top panel) were removed, protein was extracted and the levels of LC3, BECN1 and SQSTM1 were analyzed by immunoblotting. The results represent samples obtained from four control and four ethanol-exposed mouse pups. Relative levels of LC3-II, BECN1 and SQSTM1 were determined by densitometry and normalized to actin levels (bottom panels). *p < 0.05, **p < 0.01.
Ethanol increases autophagic flux
The dynamic process of autophagy includes initiation, elongation, maturation and degradation, which is also called autophagic flux. An increase of LC3 lipidation may result from increased formation of autophagosomes or decreased degradation of autophagosomes. Ethanol decreased SQSTM1 levels in SH-SY5Y cells (Fig. 1A). Bafilomycin A1, an inhibitor of autophagosome and lysosome fusion, increased SQSTM1 levels, and more importantly, ethanol plus bafilomycin A1 further upregulated SQSTM1 levels (Fig. 1A), suggesting that ethanol increased autophagic flux rather than blocked the fusion of autophagosomes with lysosomes. Bafilomycin A1 also potentiated ethanol-induced LC3 lipidation (Figs. 1A and 2A). To further confirm that ethanol enhanced autophagic flux, we treated SH-SY5Y cells with ethanol in the presence or absence of wortmannin (an inhibitor of autophagosome initiation) or rapamycin (an autophagy activator) and then examined LC3 lipidation. As shown in Figure 2A, wortmannin attenuated ethanol-induced LC3-II upregulation, whereas rapamycin further increased LC3 lipidation. Downregulation of BECN1 by a BECN1-specific shRNA abrogated ethanol-induced LC3 lipidation (Fig. 2A). Furthermore, rapamycin potentiated ethanol-induced downregulation of SQSTM1 levels (Fig. 2B). The formation of LC3 puncta is often observed in cells during autophagic activation.8,9 To visualize LC3 puncta, we transfected SH-SY5Y cells with a GFP-LC3 plasmid and examined its intracellular distribution. As shown in Figure 3, ethanol increased the number of GFP-LC3 puncta, and treatment of rapamycin or bafilomycin A1 further increased the levels of LC3 puncta. In contrast, wortmannin attenuated an ethanol-induced increase in GFP-LC3 puncta. Together, these results supported the finding that ethanol-induced LC3 lipidation was mediated by increasing autophagic flux rather than by inhibiting autophagosome degradation.

Figure 2. Effect of ethanol on autophagic flux in SH-SY5Y cells. SH-SY5Y cells were treated with ethanol (0 or 0.8%) in the presence/absence of wortmannin (Wort: 10 µM), bafilomycin A1 (Baf: 10 nM), rapamycin (Rap: 10 nM) or BECN1 shRNA (BECN1). The protein samples were collected 8 h after the treatment. The levels of LC3 (A) and SQSTM1 (B) were examined with immunoblotting (top panel). The experiment was replicated three times. Relative levels of LC3-II or SQSTM1 were determined by densitometry and normalized to actin levels (bottom panel). *p < 0.05, **p < 0.01. (C) Cells were transfected with BECN1 shRNA for 24 h and downregulation of BECN1 by shRNA was confirmed by immunoblotting (top panel). Relative BECN1 level was determined by densitometry and normalized to actin levels (bottom panel). **p < 0.01.

Figure 3. Effect of ethanol on the formation of GFP-LC3 puncta in SH-SY5Y cells. (A) SH-SY5Y cells were transfected with a GFP-LC3 plasmid and exposed to ethanol (EtOH, 0 or 0.8%) in the presence/absence of bafilomycin A1 (Baf: 10 nM) or wortmannin (Wort: 10 µM) for 6 h. In some experimental groups, cells were cotransfected with BECN1 shRNA (BEC). The formation of GFP-LC3 puncta was examined under a fluorescence microscope 6 h after ethanol exposure. (B) GFP-LC3 puncta/cell was quantified as described under Materials and Methods. The data represent the mean and SEM of three replications. *p < 0.05.
Oxidative stress is involved in ethanol-mediated autophagic activation
Autophagy is activated under oxidative stress situations;10 oxidative stress has been proposed as a potential mechanism of ethanol neurotoxicity.11 To explore the role of oxidative stress in ethanol-induced autophagy, we pretreated SH-SY5Y cells with catalase or N-acetyl-cysteine 30 min prior to ethanol exposure. Catalase is a major antioxidant enzyme in the cell and NAC is a general antioxidant. We have previously used catalase and NAC to alleviate cellular oxidative stress.12,13 As shown in Figure 4, pretreatment of catalase or NAC inhibited ethanol-induced LC3 lipidation. Since autophagy may be a self-defense response to eliminate excessive ROS, we sought to determine whether manipulation of autophagy will alter ethanol-induced production of ROS. In this study, we used rapamycin to activate autophagy and BECN1 shRNA and bafilomycin A1 to inhibit autophagy. As shown in Figure 5, rapamycin mitigated ethanol-induced ROS production, whereas BECN1 shRNA and bafilomycin A1 increased ethanol-induced ROS generation. This result supported the hypothesis that autophagy was activated to alleviate ethanol-induced oxidative stress.
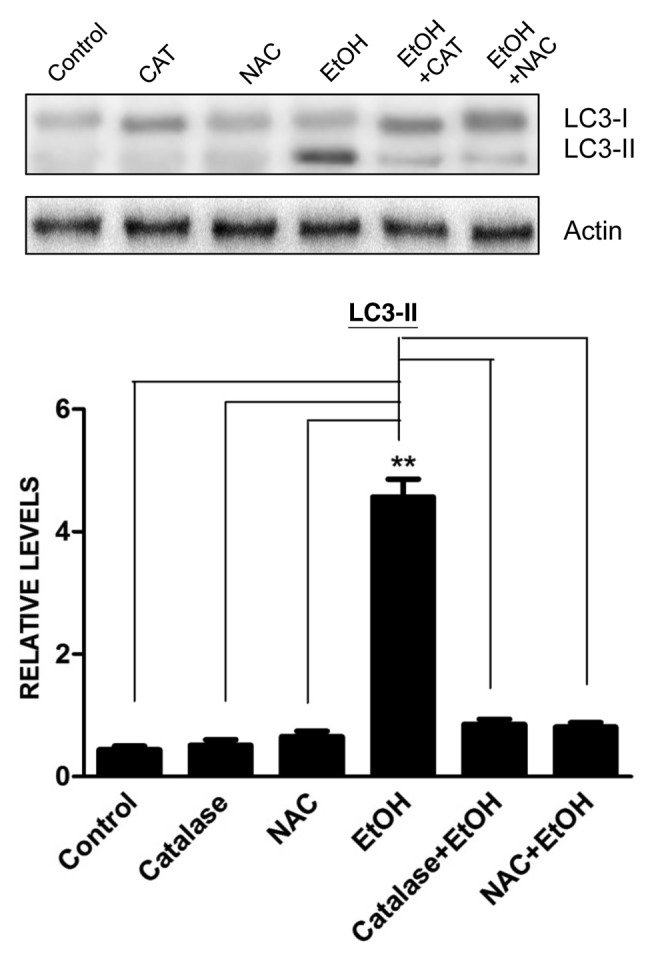
Figure 4. Effect of antioxidants on ethanol-induced LC3-II upregulation. SH-SY5Y cells were treated with ethanol (EtOH: 0.4%) with/without catalase (10,000 U/ml) or N-acetyl-cysteine (NAC: 10 mM). Cell lysates were collected 6 h after the treatment. The level of LC3 was determined by immunoblotting (top panel). The experiment was replicated three times. Relative LC3-II levels were determined by densitometry and normalized to actin levels (bottom panel). **p < 0.01.
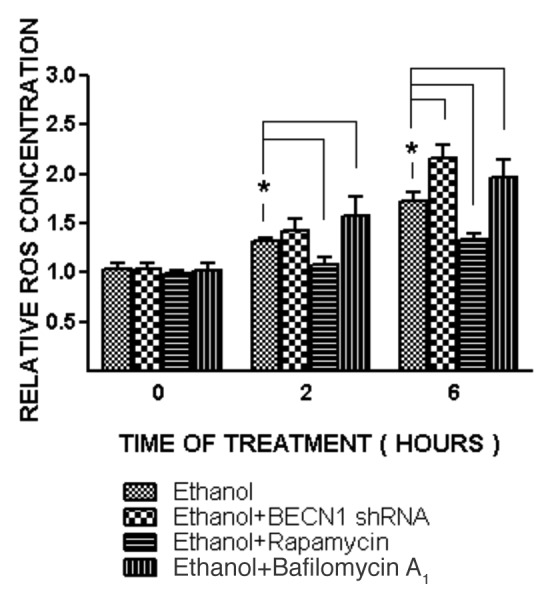
Figure 5. Effect of ethanol on ROS generation in SH-SY5Y cells. SH-SY5Y cells were treated with ethanol (0 or 0.8%) with/without rapamycin (10 nM) or bafilomycin A1 (10 nM) for 2 or 6 h. In some experimental groups, cells were treated with BECN1 shRNA to downregulate the expression of BECN1. ROS generation was measured as described under the Materials and Methods and relative amounts of ROS are presented. The data represent the mean and SEM of three replications. *p < 0.05.
Activation of autophagic flux ameliorates ethanol-induced cell death
Ethanol exposure causes neuronal death mainly in the form of apoptosis,14,15 and autophagy has been shown to modulate apoptosis.16 To determine the role of autophagy in ethanol-induced cell death, we manipulated autophagy in SH-SY5Y cells using rapamycin or wortmannin. As shown in Figure 6A, ethanol alone significantly reduced cell viability. Rapamycin offered protection against ethanol-induced cell death, whereas wortmannin exacerbated ethanol neurotoxicity, suggesting that autophagy was protective. To confirm this finding, we inhibited autophagy by treating cells with a BECN1 shRNA. Downregulation of BECN1 significantly enhanced ethanol-induced cell death (Fig. 6B). The protective effect of rapamycin was also observed in the developing brain. Injection of rapamycin attenuated ethanol-induced activation of CASP3 in the cerebral cortex of 7-d-old mice (Fig. 7). A similar effect was observed in the cerebellum (data not shown). These results suggested that activation of autophagy was protective against ethanol-induced neuronal death.

Figure 6. Effect of ethanol, rapamycin, wortmannin, catalase or BECN1 shRNA on the viability of SH-SY5Y cells. SH-SY5Y cells were treated with ethanol (0 or 0.8%) with/without rapamycin (10 nM), catalase (10,000 U/ml) or wortmannin (10 µM) for 48 h (A). In some experimental groups, cells were treated with a BECN1 shRNA to downregulate the expression of BECN1 (B). Cell viability was determined by MTT assay as described under Materials and Methods. The data represent the mean and SEM of three replications. *p < 0.05. SH-SY5Y cells were treated with 0.8% ethanol (EtOH) with/without 10 nM rapamycin (EtOH + Rap) and BECN1 shRNA (EtOH + BECN1). Mitochondria and lysosomes were detected by specific dyes as described under the Materials and Methods (MTG for mitochondria and LTR for lysosomes). The percentage of mitochondria that were colocalized with lysosomes under each treatment condition was calculated. The mean ± SEM of 120 cells for each treatment condition is presented (C). *p < 0.05.

Figure 7. Effect of ethanol and rapamycin on caspase-3 activation in the developing brain. PD7 mice were injected with ethanol (0 or 5 g/kg) and/or rapamycin (10 mg/kg or 20 mg/kg, Rapa: 10 or 20). The expression of active CASP3/caspase-3 in the cerebral cortex was examined 8 h after the injection by immunohistochemistry as described under Materials and Methods. Scale bar: 100 μm. The experiment was replicated three times.
We next determined the role of oxidative stress in ethanol-induced cell death. As shown in Figure 6A, catalase ameliorated ethanol-induced cell death, while treatment of BECN1 shRNA exacerbated ethanol-induced cell death. Catalase also eliminated BECN1 shRNA-meditated potentiation of ethanol neurotoxicity (Fig. 6B). Similarly, NAC protected cells against ethanol neurotoxicity (data not shown). These results suggested that ethanol-induced cell death as well as BECN1 shRNA potentiation of ethanol toxicity was at least partially mediated by enhanced ROS generation.
Damaged mitochondria are the major sources of ROS generation. Since these damaged mitochondria can be removed by autophagy by targeted degradation in lysosomes (mitophagy), we evaluated the effect of ethanol and rapamycin/beclin shRNA on the mitophagy in SH-SY5Y cells. The colocalization of mitochondria and lysosomes indicates mitochondrial turnover and is successfully used for the detection of mitophagy.17 In this study, the mitochondria were stained with MitoTracker Green FM (MTG), a mitochondrial tracker which stains both polarized and depolarized mitochondria. Lysosomes were stained with LysoTracker Red. As shown in Figure 6C, ethanol increased the mitochondrial turnover. BECN1 shRNA significantly decreased ethanol-induced mitochondria turnover, whereas rapamycin enhanced ethanol-induced mitochondrial turnover. The results suggest that mitophagy was activated to ameliorate ethanol-induced ROS generation.
MTOR signaling pathway is responsible for ethanol-induced autophagic response
The MTOR pathway is an important signaling pathway that regulates autophagic activity; generally, inhibition of this pathway results in autophagic activation.18 We determined the effect of ethanol on the MTOR pathway. As shown in Figure 8, ethanol inhibited the phosphorylation of ribosomal protein S6 kinase (RPS6K), a substrate of MTOR, in both SH-SY5Y cells and in the developing brain of PD7 mice. Ethanol caused a similar dephosphorylation of eukaryotic translation factor 4E binding protein 1 (EIF4EBP1), another downstream substrate of MTOR (data not shown). These results indicated that ethanol inhibited the MTOR pathway. As expected, rapamycin inhibited RPS6K phosphorylation; ethanol plus rapamycin caused a greater dephosphorylation of RPS6K (Fig. 8C). If autophagy is a protective response, downregulation of MTOR should mitigate ethanol neurotoxicity. As shown in Figure 8D, downregulation of MTOR by an MTOR-specific shRNA protected SH-SY5Y cells against ethanol-induced death.

Figure 8. Effect of ethanol on the MTOR pathway. (A) SH-SY5Y cells were treated with ethanol (0 or 0.4%) for the indicated times. The phosphorylation of RPS6K, a substrate of MTOR, was determined by immunoblotting (top panel). Relative level of RPS6K was quantified by densitometry and normalized to total RPS6K (bottom panel). **p < 0.01. (B) PD7 mice were injected with ethanol (0 or 5 g/kg) for 8 h as described above. The phosphorylation of RPS6K in the cerebral cortex was determined by immunoblotting (top panel) and was quantified by densitometry (bottom panel). **p < 0.01. (C) SH-SY5Y cells were treated with ethanol (0 or 0.4%) with/without rapamycin (Rap: 10 nM) for 6 h. The phosphorylation of RPS6K was determined by immunoblotting (top panel) and was quantified by densitometry (bottom panel). **p < 0.01. (D) The level of MTOR was knocked down by an MTOR shRNA as described under Materials and Methods. Cells were exposed to ethanol (0 or 0.8%) for 48 h and cell viability was determined by MTT assay. The data represent the mean and SEM of three replications. *p < 0.05. (E) Downregulation of MTOR by shRNA was confirmed by immunoblotting. **p < 0.01.
Discussion
Autophagy has recently received increasing attention because it is potentially involved in the pathogenesis of many diseases, including neurodegenerative diseases. Autophagy is implicated in ethanol-induced damage to the liver and heart.19-21 We demonstrate for the first time that ethanol enhances autophagic flux in neuronal cells and increased autophagic flux may be a cellular self-protective response to ethanol neurotoxicity. Our results indicate that mitophagy is activated to ameliorate ethanol-induced ROS generation. The study provides novel insight into the cellular mechanisms of ethanol neurotoxicity and potential future therapeutic strategies.
Ethanol stimulates autophagic flux
Autophagy is a dynamic process including the initiation, formation, maturation and degradation of autophagosomes. This dynamic flow is defined as autophagic flux.6 LC3 lipidation or visualization of autophagosomes using electronic microscopy has been commonly employed to measure autophagy activity. However, an increase in LC3-II may result from either an enhancement of autophagosomal formation or inhibition of autophagosomal degradation, or may be caused by autophagy-independent mechanisms.22 Therefore, both generation and degradation of autophagosomes must be taken into consideration when LC3-II is used to evaluate autophagy activity. In this study, we show that ethanol increased the level of LC3-II in vitro and in vivo. SQSTM1 is regulated by autophagy-dependent degradation. Ethanol decreases the level of SQSTM1, and bafilomycin A1, an inhibitor of autophagosome and lysosome fusion, enhances the SQSTM1 level. More importantly, the levels of SQSTM1 in cells treated with bafilomycin A1 and ethanol together are much higher than that treated by bafilomycin A1 alone. This suggests that ethanol increases the formation/maturation of autophagosomes rather than blocks the fusion of the autophagosomes and lysosomes. Furthermore, rapamycin, an initiator of autophagy, further increases ethanol-induced upregulation of LC3 lipidation and the formation of LC3 puncta. In contrast, wortmannin, an inhibitor of autophagic initiation/maturation, and BECN1-specific shRNA attenuated ethanol-promoted LC3 lipidation and LC3 puncta. These results provide additional evidence that ethanol promotes autophagic initiation and maturation. It is noted that serum deprivation increases the level of LC3-II, supporting the notion that nutrient deficiency may also induce autophagy. To avoid confounding conditions, we evaluated the effect of ethanol on cells cultured in media containing 10% serum. In all experiments, 0.4% and 0.8% ethanol were used and both were effective. Generally, ethanol at 0.8% produced a greater effect, therefore we present these data to better illustrate ethanol’s effect.
Autophagy is a protective response to ethanol-induced oxidative stress
Autophagy is a tightly regulated pathway involving the lysosomal degradation of cytoplasmic organelles or cytosolic components. This pathway can be stimulated by multiple forms of cellular stress, including nutrient or growth factor deprivation, hypoxia, oxidative stress, endoplasmic reticulum stress, DNA damage, protein aggregates, damaged organelles or intracellular pathogens.23,24 Ethanol causes oxidative stress in neurons in vitro and in vivo.25 We have previously demonstrated that ethanol induces ROS generation in SH-SY5Y cells.26 Antioxidants block ethanol-induced LC3 lipidation, suggesting that autophagy is activated in response to ethanol-induced oxidative stress.
The role of autophagy in the regulation of cell death has been controversial. Since the formation of autophagosomes is frequently associated with cellular stress and cell death, some believe that autophagy may promote cell death. The term “autophagic cell death” or “type II cell death” was coined to differentiate it from apoptotic (type I cell death) or necrotic (type III cell death).27 Activation of the autophagic pathway beyond a certain threshold may promote cell death directly by causing the collapse of cellular functions as a result of cellular atrophy. Alternatively, autophagy can lead to the execution of apoptotic or necrotic cell death programs, presumably via common regulators such as proteins from the BCL2 family.27
On the other hand, many view autophagy as a cellular self-defense response.28 Particularly, in neuropathies (Huntington, Alzheimer and Parkinson diseases) and ischemic heart disease, autophagy is more widely accepted as beneficial due to its role in eliminating ‘toxic assets’ and promoting cell viability.29 In fact, increasing evidence shows that autophagy is neuroprotective against various insults to the CNS.30 It is therefore likely the activation of autophagy is a double-edged sword. On one hand, autophagy is essential for provoking a protective response and enhancing cell adaptation to metabolic stresses and immunological challenges. On the other hand, excessive autophagic activation can give rise to pathogenic conditions by imbalanced degradation or recycling and disruption of critical cellular constituents leading to cell degeneration and toxicity.31
Our results support that autophagy is a cellular self-defense response to alleviate ethanol-induced oxidative stress and subsequent cell death. This is evident by our finding that antioxidants can alleviate ethanol-stimulated autophagy and rescue cells from ethanol-induced cell death. More importantly, stimulation of autophagy activity by rapamycin decreases ethanol-induced ROS production and offers neuroprotection. Contrarily, inhibition of autophagy promotes ROS generation and deteriorates ethanol-induced cell death. Furthermore, our results indicate that mitophagy is activated in response to ethanol exposure, suggesting that neuroprotection of autophagy may be mediated by removing damaged mitochondria and excessive cellular ROS.
Ethanol and MTOR signaling
The best characterized regulator for autophagy is mammalian target of rapamycin complex 1 (MTORC1) that consists of a serine/threonine kinase called mammalian target of rapamycin (MTOR), a regulatory associated partner of MTOR (raptor) and MLST8 (MTOR associated protein, LST8 homolog). MTOR functions as a convergence point for many upstream stimuli and pathways (such as insulin, growth factors and mitogens) to regulate autophagy as well as other cellular activities.18 An abundance of nutrients activates MTOR leading to suppression of autophagy, while stress conditions such as starvation, inhibit MTOR and consequently autophagy is activated.18 Rapamycin is a well-known inhibitor of MTOR. In our study, rapamycin not only increases basal levels of autophagy but also potentiates ethanol-induced autophagy in SH-SY5Y cells, confirming that inhibition of MTOR results in enhanced autophagy. Ethanol inhibits the phosphorylation of RPS6K and EIF4EBP1, two main substrates of MTOR in vitro and in vivo, indicating that ethanol suppresses MTOR activity. Rapamycin and ethanol together produce a greater dephosphorylation of RPS6K and EIF4EBP1 than when treated by either rapamycin or ethanol alone; the result is consistent with the finding that rapamycin potentiates ethanol-induced autophagy. Therefore it is likely that ethanol activates autophagy by inhibiting the MTOR pathway. Inhibition of MTOR by rapamycin or knockdown of MTOR offers protection against ethanol-induced cell death, further confirming the involvement of the MTOR pathway in ethanol-induced autophagy and cell death. The mechanisms underlying ethanol inhibition of the MTOR pathway, however, are unclear. The relationship between ethanol-induced oxidative stress and MTOR signaling, in particular, is worth further investigation.
Materials and Methods
Materials
The following materials were used: ethanol (Sigma-Aldrich, E7023), bafilomycin A1 (Sigma-Aldrich, B1793), wortmannin (Sigma-Aldrich, W1628), rapamycin (Sigma-Aldrich, R0395), anti-SQSTM1 antibody (Sigma-Aldrich, P0067), anti-actin antibody (Sigma-Aldrich, A5441), anti-LC3 antibody (Medical and Biological Laboratories, PM036), anti-BECN1 antibody (Abcam, ab55878), anti-RPS6K antibody (Cell Signaling Technology, 9202), anti-phospho-RPS6K antibody (Cell Signaling Technology, 9205), MitoTracker Green FM (Invitrogen, M7514) and LysoTracker Red (Invitrogen, L7528).
Animals and treatment
C57BL/6 mice were obtained from Harlan Laboratories and maintained at the animal facility of the University of Kentucky Medical Center. All procedures were performed in accordance with the guidelines set by the NIH and the Animal Care and Use Committee of the University of Kentucky. An acute ethanol exposure paradigm, which had been shown to induce robust neurodegeneration in infant mice14,32 was employed. Briefly, 7-d-old mice (postnatal day 7 or PD7) were injected subcutaneously with saline or ethanol (2.5 g/kg, 20% solution in saline) twice at time 0 and 2 h. For some experiments, rapamycin (10 or 20 mg/kg body weight) was intraperitoneally injected to the mice. Animals received two injections of rapamycin; they were administered at 24 and 2 h prior to ethanol exposure. Rapamycin was dissolved in DMSO (Sigma, D4540) at 25 mg/ml and diluted in saline to achieve the final concentrations. Eight hours after the ethanol injection, the brains were removed and processed for immunoblotting or immunohistochemical analysis.
Cell culture
Human neuroblastoma SH-SY5Y cells obtained from ATCC were grown in Eagle’s MEM containing 10% fetal bovine serum (FBS, Sigma, F2442), 2 mM L-glutamine, 25 µg/ml gentamicin (Invitrogen, 15710-064), 100 U/ml penicillin and 100 µg/ml streptomycin (Invitrogen, 15140-122) at 37°C with 5% CO2. For the analysis of cell survival, 3 × 104 cells/well and 15 × 104 cells/well were plated into 96-well culture trays and 24-well culture trays, respectively. Cells were incubated at 37°C in a humidified environment containing 5% CO2 for 24 h. After this incubation period, cells were cultured in serum-free medium overnight and then exposed to ethanol and other agents.
Ethanol exposure protocol
A method utilizing sealed containers was used to maintain ethanol concentrations in the culture medium. With this method, ethanol concentrations in the culture medium can be accurately maintained.33 A pharmacologically relevant concentration of 0.4% was used in this study. In general, the concentration for in vitro studies is higher than that required to produce a similar effect in vivo.33 In some experiments, 0.8% ethanol was used to better demonstrate the effect of ethanol.
Determination of cell viability
Cell viability was determined by MTT assay as previously described.34 The MTT assay is based on the cleavage of yellow tetrazolium salt MTT [3-(4,5-dimethylthiazol-2yl)-2,5-di-phenyl tetrazolium bromide] to purple formazan crystals by metabolically active cells. Briefly, the cells cultured in 96-well microtiter plates were exposed to ethanol for 48 h. After ethanol exposure, 10 µl of MTT labeling reagent (Roche, 11465007001) was added to each well, and the plates were incubated at 37°C for 4 h. The cultures were then solubilized, and spectrophotometric absorbance of the samples was detected by a microtiter plate reader. The wavelength to measure absorbance of formazan products is 570 nm, with a reference wavelength of 750 nm.
Immunoblotting
Cells were washed with phosphate-buffered saline (PBS; pH 7.4) and lysed with RIPA buffer [150 mM NaCl, 50 mM Tris (pH 8.0), 1% Nonidet P-40 (NP-40, Pierce, 28324), 0.1% sodium dodecyl sulfate (SDS), 0.5% deoxycholic acid sodium (Fluka, 30970), 0.1 mg/ml phenylmethylsulfonyl fluoride (Sigma, P7626), 1 mM sodium orthovanadate (Aldrich, 450243), and 3% aprotinin (Sigma, A6279)] on ice for 10 min; solubilized cells were centrifuged and the supernatant was collected. For some experiments, a buffer containing 2% SDS was used to evaluate insoluble proteins. The immunoblotting procedure has been previously described.34 Briefly, after the protein concentrations were determined, aliquots of the protein samples (20–40 µg) were loaded into the lanes of an SDS-polyacrylamide gel. The protein samples were separated by electrophoresis, and the separated proteins were transferred to nitrocellulose membranes. The membranes were blocked with 5% BSA (Sigma, A7906) in 0.010 M PBS (pH 7.4) and 0.05% Tween-20 (TPBS) at room temperature for 1 h. Subsequently, the membranes were probed with primary antibodies directed against target proteins for 2 h at room temperature or overnight at 4°C. After three quick washes in TPBS, the membranes were incubated with a secondary antibody conjugated to horseradish peroxidase (Amersham Biosciences, NA934V, NA931V) diluted at 1:2,000 in TPBS for 1 h. The immune complexes were detected by the enhanced chemiluminescence method (PerkinElmer, NEL105001EA). The blots were stripped and reprobed with an anti-actin antibody.
Quantitative RT-PCR
The mRNA expression was analyzed by quantitative real-time reverse transcription-PCR. Total RNA was extracted by a TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. The first-strand cDNA was synthesized using a ProtoScript AMV First Strand cDNA Synthesis Kit (New England Biolabs, E6550S). PCR was performed on a Roche LightCycler 480 by using a LightCycler 480 SYBR Green I Master Kit (Roche, 04707516001). The results were analyzed using the software of the LightCycler system. The relative expression level of a given mRNA was assessed by normalizing it to the housekeeping gene ACTB and was then compared with controls. Primers used in this experiment included ACTB (5′-agcacagagcctcgccttt-3′, 5′-agggtgaggatgcctctctt-3′), MAP1LC3 (5′-agcagcatccaaccaaaatc-3′, 5′-ctgtgtccgttcaccaacag-3′), BECN1 (5′-agctggagctggatgatgag-3′, 5′-cgacccagcctgaagttatt-3′) and SQSTM1 (5′-cacctgtctgagggcttctc-3′, 5′-cacactctccccaacgttct-3′).
Immunohistochemistry
After treatments, the mice were deeply anesthetized with chloral hydrate (350 mg/kg), then perfused with saline followed by 4% paraformaldehyde in 0.1 M potassium phosphate buffer (pH 7.2; Sigma-Aldrich, P6148). The brains were removed and post-fixed in 4% paraformaldehyde (Sigma-Aldrich, P6148) for an additional 24 h, then transferred to 30% sucrose (Sigma-Aldrich, S7903). Brains were sectioned at 40 μm with a sliding microtome (Leica Microsystems).
The procedure for immunohistochemistry staining has been described elsewhere.35 Briefly, free-floating sections were incubated in 0.3% H2O2 (Sigma-Aldrich, H0904) in methanol (Sigma-Aldrich, M3641) for 30 min at room temperature and then treated with 0.1% Triton X-100 for 10 min in PBS. The sections were washed with PBS three times, then blocked with 1% BSA and 0.01% Triton X-100 (Sigma-Aldrich, T9284) for 1 h at room temperature. The sections were incubated with an anti-active caspase-3 antibody (dilution of 1:1,000) overnight at 4°C. Negative controls were performed by omitting the primary antibody. After rinsing in PBS, sections were incubated with a biotinylated goat anti-rabbit IgG (1:200; Vector Laboratories, BA-1000) for 1 h at room temperature. The sections were washed three times with PBS, then incubated in avidin-biotin-peroxidase complex (1:100; Vector Laboratories, SP-2001) for 1 h and developed in 0.05% 3,3′-diaminobenzidine (DAB; Sigma-Aldrich, D3939) containing 0.003% H2O2 in PBS. The images were captured with an Olympus microscope (BX61) equipped with a DP70 digital camera.
GFP-LC3 puncta counting
Cells were transfected with a GFP-LC3 plasmid and examined and recorded under a Zeiss LSM 510 confocal microscope (Carl Zeiss). The numbers of GFP-LC3 puncta in each cell were manually counted. For each group, 50 cells were randomly selected. The results were presented as the mean ± SEM of three independent experiments.
Plasmids and transfection
A GFP-LC3 plasmid was a generous gift from Dr. Gutterman (University of Texas MD Anderson Cancer Center, Houston, TX), which has been described elsewhere.8 A BECN1 shRNA plasmid (SA Biosciences, KH05670N) and MTOR shRNA plasmid (Addgene, 1855) have been reported previously.36,37 Briefly, transfection of a GFP-LC3 or BECN1 shRNA or negative control shRNA into SH-SY5Y cells was performed by using Lipofectamine 2000 (Invitrogen, 11668-019) according to the manufacturer’s protocol. Forty-eight h after the transfection, the cells were subjected to immunofluorescent or immunoblotting analysis. Stable transfection of MTOR shRNA into SH-SY5Y cells was performed through lentivirus infection. Briefly, MTOR shRNA plasmids together with the psPAX2 packaging plasmids and pMD2.G envelope plasmids (Addgene, 12259) were transfected into HEK-293T by using FUGENE (Roche, 1181443001). Virus-containing supernatants were collected at 48 and 72 h after transfection. SH-SY5Y cells were infected twice with the virus-containing media in the presence of 8 µg/ml polybrene (Sigma-Aldrich, 107689); infected cells were selected for puromycin (8 µg/ml; Sigma-Aldrich, P7255) resistance and analyzed on the eighth day after infection.
Detection of intracellular ROS
Intracellular ROS levels were measured using the fluorescent dye carboxy-H2DCFDA staining method as previously described.26 Once incorporated into cells, H2DCFDA is converted into a nonfluorescent polar derivative (H2DCF) by cellular esterases. H2DCF is rapidly oxidized to the highly fluorescent 2,7-dichlorofluorescein (DCF) by intracellular ROS, mainly H2O2. Therefore, the intensity of the DCF signal reflects the quantity of intracellular ROS. Cells were treated with ethanol and/or other reagents for the indicated hours in 96-well plates. After treatment, the media were removed, 100 µl pre-warmed serum-free medium containing 5 µM carboxy-H2DCFDA (Invitrogen, C400) was added to each well and incubated for 45 min at room temperature. Cells were then washed with PBS twice and the intracellular ROS levels (DCF signals) were measured using a microtiter plate reader at an emission wavelength of 525 nm.
Assessment of mitochondria and lysosomes colocalization
To evaluate autophagic degradation of mitochondria (mitophagy), we assessed the colocalization of mitochondria and lysosomes. This method has been successfully used to monitor mitophagy.17 Briefly, cells were cultured in 35-mm glass bottom dishes (MatTek, P35G-1.5-14-C) and exposed to ethanol (0 or 0.8%) for 6 h. After treatment, the medium was replaced with fresh medium and the cells were stained with 100 nM MitoTracker Green FM (Invitrogen, M7514) and 200 nM LysoTracker Red DND-99 (Invitrogen, L7528) for 30 min according to the manufacturer’s instructions. Cells were washed in PBS and the colocalization of mitochondria and lysosomes were examined under an inverted IX81 microscope with a 60× oil lens (Olympus). The colocalization of mitochondria and lysosomes was quantified using Slidebook 5.0 software (Olympus). The colocalization was determined based on the analysis of 120 cells for each treatment condition.
Statistical analysis
Differences among treatment groups were tested by analysis of variance (ANOVA). p < 0.05 was considered statistically significant. In cases in which significant differences were detected, specific post-hoc comparisons between treatment groups were examined by Student-Newman-Keuls tests.
Acknowledgments
This research is supported by grants from the National Institutes of Health (AA019693 and AA015407).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/21376
References
- 1.Riley EP, McGee CL. Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp Biol Med (Maywood) 2005;230:357–65. doi: 10.1177/15353702-0323006-03. [DOI] [PubMed] [Google Scholar]
- 2.Tang D, Kang R, Zeh HJ, 3rd, Lotze MT. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal. 2011;14:1315–35. doi: 10.1089/ars.2010.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang C, Han LO. Knockdown of Beclin 1 inhibits vitamin K3‑induced autophagy, but promotes apoptosis of human hepatoma SMMC-7721 cells. Mol Med Report. 2010;3:801–7. doi: 10.3892/mmr.2010.347. [DOI] [PubMed] [Google Scholar]
- 4.Larsen KB, Lamark T, Øvervatn A, Harneshaug I, Johansen T, Bjørkøy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy. 2010;6:784–93. doi: 10.4161/auto.6.6.12510. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Gibson SB. Is mitochondrial generation of reactive oxygen species a trigger for autophagy? Autophagy. 2008;4:246–8. doi: 10.4161/auto.5432. [DOI] [PubMed] [Google Scholar]
- 6.Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol. 2009;452:1–12. doi: 10.1016/S0076-6879(08)03601-X. [DOI] [PubMed] [Google Scholar]
- 7.Ichimura Y, Komatsu M. Selective degradation of p62 by autophagy. Semin Immunopathol. 2010;32:431–6. doi: 10.1007/s00281-010-0220-1. [DOI] [PubMed] [Google Scholar]
- 8.Xu ZX, Liang J, Haridas V, Gaikwad A, Connolly FP, Mills GB, et al. A plant triterpenoid, avicin D, induces autophagy by activation of AMP-activated protein kinase. Cell Death Differ. 2007;14:1948–57. doi: 10.1038/sj.cdd.4402207. [DOI] [PubMed] [Google Scholar]
- 9.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285:10850–61. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777–90. doi: 10.1089/ars.2008.2270. [DOI] [PubMed] [Google Scholar]
- 11.Henderson GI, Chen JJ, Schenker S. Ethanol, oxidative stress, reactive aldehydes, and the fetus. Front Biosci. 1999;4:D541–50. doi: 10.2741/Henderson. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z, Wang X, Cheng S, Sun L, Son YO, Yao H, et al. Reactive oxygen species mediate arsenic induced cell transformation and tumorigenesis through Wnt/β-catenin pathway in human colorectal adenocarcinoma DLD1 cells. Toxicol Appl Pharmacol. 2011;256:114–21. doi: 10.1016/j.taap.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 13.Pan J, Chang Q, Wang X, Son Y, Zhang Z, Chen G, et al. Reactive oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel compound-induced apoptosis in BEAS 2B cells. Chem Res Toxicol. 2010;23:568–77. doi: 10.1021/tx9003193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Chen G, Ma C, Bower KA, Xu M, Fan Z, et al. Overexpression of glycogen synthase kinase 3beta sensitizes neuronal cells to ethanol toxicity. J Neurosci Res. 2009;87:2793–802. doi: 10.1002/jnr.22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nowoslawski L, Klocke BJ, Roth KA. Molecular regulation of acute ethanol-induced neuron apoptosis. J Neuropathol Exp Neurol. 2005;64:490–7. doi: 10.1093/jnen/64.6.490. [DOI] [PubMed] [Google Scholar]
- 16.Giansanti V, Torriglia A, Scovassi AI. Conversation between apoptosis and autophagy: “Is it your turn or mine?”. Apoptosis. 2011;16:321–33. doi: 10.1007/s10495-011-0589-x. [DOI] [PubMed] [Google Scholar]
- 17.Krebiehl G, Ruckerbauer S, Burbulla LF, Kieper N, Maurer B, Waak J, et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS One. 2010;5:e9367. doi: 10.1371/journal.pone.0009367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osna NA, Thomes PG, Jr TM. Involvement of autophagy in alcoholic liver injury and hepatitis C pathogenesis. World J Gastroenterol. 2011;17:2507–14. doi: 10.3748/wjg.v17.i20.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge W, Ren J. mTOR-STAT3-notch signalling contributes to ALDH2-induced protection against cardiac contractile dysfunction and autophagy under alcoholism. J Cell Mol Med. 2012;16:616–26. doi: 10.1111/j.1582-4934.2011.01347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Ren J. ALDH2 in alcoholic heart diseases: molecular mechanism and clinical implications. Pharmacol Ther. 2011;132:86–95. doi: 10.1016/j.pharmthera.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen G, Luo J. Anthocyanins: Are They Beneficial in Treating Ethanol Neurotoxicity? Neurotox Res. 2010;17:91–101. doi: 10.1007/s12640-009-9083-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen G, Ma C, Bower KA, Shi X, Ke Z, Luo J. Ethanol promotes endoplasmic reticulum stress-induced neuronal death: involvement of oxidative stress. J Neurosci Res. 2008;86:937–46. doi: 10.1002/jnr.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: that is the autophagic question. Curr Mol Med. 2008;8:78–91. doi: 10.2174/156652408783769616. [DOI] [PubMed] [Google Scholar]
- 28.Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7:457–65. doi: 10.4161/auto.7.5.14226. [DOI] [PubMed] [Google Scholar]
- 29.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huett A, Goel G, Xavier RJ. A systems biology viewpoint on autophagy in health and disease. Curr Opin Gastroenterol. 2010;26:302–9. doi: 10.1097/MOG.0b013e32833ae2ed. [DOI] [PubMed] [Google Scholar]
- 31.Liang C. Negative regulation of autophagy. Cell Death Differ. 2010;17:1807–15. doi: 10.1038/cdd.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res. 2002;133:115–26. doi: 10.1016/S0165-3806(02)00279-1. [DOI] [PubMed] [Google Scholar]
- 33.Luo J, Lindström CL, Donahue A, Miller MW. Differential effects of ethanol on the expression of cyclo-oxygenase in cultured cortical astrocytes and neurons. J Neurochem. 2001;76:1354–63. doi: 10.1046/j.1471-4159.2001.00129.x. [DOI] [PubMed] [Google Scholar]
- 34.Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004;18:1162–4. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- 35.Ke ZJ, Calingasan NY, Karuppagounder SS, DeGiorgio LA, Volpe BT, Gibson GE. CD40L deletion delays neuronal death in a model of neurodegeneration due to mild impairment of oxidative metabolism. J Neuroimmunol. 2005;164:85–92. doi: 10.1016/j.jneuroim.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 37.Airoldi I, Di Carlo E, Cocco C, Taverniti G, D’Antuono T, Ognio E, et al. Endogenous IL-12 triggers an antiangiogenic program in melanoma cells. Proc Natl Acad Sci U S A. 2007;104:3996–4001. doi: 10.1073/pnas.0609028104. [DOI] [PMC free article] [PubMed] [Google Scholar]