

Abstract
Autophagy is a process by which cytoplasmic material is sequestered in a double-membrane vesicle destined for degradation. Nutrient deprivation stimulates the pathway and the number of autophagosomes in the cell increases in response to such stimulus. In the current report we have demonstrated that actin is necessary for starvation-mediated autophagy. When the actin cytoskeleton is depolymerized, the increase in autophagic vacuoles in response to the starvation stimulus was abolished without affecting maturation of remaining autophagosomes. In addition, actin filaments colocalized with ATG14, BECN1/Beclin1 and PtdIns3P-rich structures, and some of them have a typical omegasome shape stained with the double FYVE domain or ZFYVE1/DFCP1. In contrast, no major colocalization between actin and ULK1, ULK2, ATG5 or MAP1LC3/LC3 was observed. Taken together, our data indicate that actin has a role at very early stages of autophagosome formation linked to the PtdIns3P generation step. In addition, we have found that two members of the Rho family of proteins, RHOA and RAC1 have a regulatory function on starvation-mediated autophagy, but with opposite roles. Indeed, RHOA has an activatory role whereas Rac has an inhibitory one. We have also found that inhibition of the RHOA effector ROCK impaired the starvation-mediated autophagic response. We propose that actin participates in the initial membrane remodeling stage when cells require an enhanced rate of autophagosome formation, and this actin function would be tightly regulated by different members of the Rho family.
Keywords: autophagosome formation, starvation, actin, RHOA, RAC1, ROCK
Introduction
Macroautophagy (in this report referred hereafter as autophagy) is one of the major cellular degradation processes, by which cytoplasmic material is degraded into a double-membrane compartment.1 This process begins with a double membrane which closes to form a vesicle (i.e., autophagosome), sequestering cytoplasmic material such as protein aggregates, ribosomes or, in some cases, entire organelles. Subsequently, the autophagosome acquires degradative components by fusion with late endosomes and lysosomes, becoming an autophagolysosome where the sequestered material is degraded.2 In the last decade, numerous studies carried on in yeast have led to the identification of several of the proteins that orchestrate a series of steps implied in the autophagic pathway. Most of these proteins have mammalian orthologs, which is evidence that autophagy is highly conserved through evolution.3 In mammals some of the main players are the Ulk1 kinase complex (consisting of ULK1, RB1CC1/FIP200, ATG13 and C12orf44/Atg101), the class III phosphatidylinositol 3-kinase (composed by PIK3C3/Vps34, PIK3R4/Vps15, BECN1/Beclin1 and ATG14), the ATG12-ATG5-ATG16L1 complex and the Atg8 mammalian-homolog MAP1LC3.4
Initially, autophagosome formation requires membrane remodeling to generate a vesicle. Afterwards, vesicles need to be transported in the cytoplasm and the maturation of these vesicles involves fusion with other compartments.4,5 The actin cytoskeleton could potentially participate in each of these steps. For example, actin polymerization and subsequent depolymerization occurs at the plasma membrane during the phagocytic process.6 It is also known that different motor proteins bind to the actin cytoskeleton to mediate vesicle transport.7 In addition, there is evidence showing actin participation in phagosome/lysosome fusion.8,9
The proteins of the Rho family are monomeric GTPases that regulate a wide diversity of cellular activities. Some of them are tightly related to the actin cytoskeleton remodeling (membrane protrusion, cell adhesion and motility) and others to the regulation of the cell cycle and gene transcription.10 These proteins cycle between two states, inactive and active conformations. In the inactive state the small GTPase is mainly bound to GDP and it is sequestered in the cytoplasm by another protein called guanosine nucleotide dissociation inhibitors (GDI).11 In the active form the GTPase binds GTP and is translocated to the membrane. This activation occurs through the interaction with a guanine exchange factor (GEF) that exchanges the nucleotide GDP for GTP, increasing the affinity for the effectors up to 100 folds.12 Due to the GTPase activity of the Rho proteins, GTP is hydrolyzed and turns into GDP, an activity that is enhanced by the interaction with a protein called GTPase activating protein (GAP).12
RHOA, RAC1 and CDC42 are the three better characterized members of the Rho family and each one governs different actin-based processes.13 RAC1 and CDC42 regulate the formation of lamellipodia and filopodia, respectively, and promote protrusive activities, whereas RHOA regulates stress fibers and contractile rings formation.13 Both the stress fibers and the contractile ring are formed by actomyosin bundles with antiparallel actin filaments cross-linked by myosin. RHOA regulates these structures through stimulation of actin polymerization and activation of myosin. The Rho effectors involved in this function are mainly two: ROCK and DIAPH1/hDia1.14 Whereas DIAPH1 catalyzes actin nucleation and polymerization, the Ser/Thr protein kinase ROCK increases myosin light chain phosphorylation stimulating cross-linking between actin and myosin, which enhance actomyosin contractility.14
Autophagy occurs at a basal level in the majority of the cells, but different stimulus, like starvation, markedly increases the level of autophagosome formation.15 It has been postulated that the main function of basal autophagy is to enforce intracellular quality control by eliminating toxic proteins aggregates or damaged organelles, a process that has been called quality control autophagy.16 On the other hand, starvation-mediated autophagy supplies essential macromolecules to cells subjected to different stress conditions, by a nonselective degradation of cytosolic contents and organelles.16
A large number of factors involved in autophagosome formation have been identified, but the process is not yet fully dissected at the molecular level. In this study we show that actin filaments and regulatory molecules are required for starvation-mediated autophagy. When actin polymerization was abolished, the typical increase in the autophagosome number due to a starvation stimulus was impaired, and this effect was a consequence of a defect in the formation of autophagosomes at a very early stage. In addition, we show that RHOA and RAC1, two small GTPases of the Rho family of proteins, are involved in starvation-mediated autophagy but with opposite roles: RHOA had an activatory role in the pathway and RAC1 an inhibitory one. Furthermore, we also show that the RHOA effector ROCK participates in the regulation of starvation-induced autophagy.
Results
Depolymerization of actin filaments affects autophagy activation induced by starvation or rapamycin treatment
In order to address whether the actin cytoskeleton participates in the autophagic pathway, we tested the effect of the actin depolymerizing agent Latrunculin B on MAP1LC3 processing. It is known that conversion of cytosolic MAP1LC3 (MAP1LC3-I) to membrane-bound phosphatidylethanolamine (PE)–conjugated MAP1LC3 (MAP1LC3-II) occurs during autophagy induction, and that the amount of MAP1LC3-II correlates with the number of autophagosomes.17 Likewise, the number of MAP1LC3-labeled dots represents autophagic vesicles in different stage of maturation. CHO cells stably expressing GFP-MAP1LC3 were incubated under full-nutrient or starvation conditions in the presence or absence of Latrunculin B and then, the number of autophagosomes (i.e., punctate MAP1LC3 structures) in each cell was analyzed. In full-nutrient conditions no differences in autophagosome number between cells incubated without (control condition, Fig. 1A, a) or with Latrunculin B (Fig. 1A, d) were observed. Starvation increased the autophagosome number in cells incubated in the absence of Latrunculin B (Fig. 1A, b); however, in cells treated with Latrunculin B, that increase was abolished (Fig. 1A, e; see also quantifications in Fig. 1B).

Figure 1. Actin filament disruption prevented the autophagic response upon starvation or rapamycin treatment. (A) CHO cells stable expressing EGFP-MAP1LC3 were incubated for 2 h at 37°C in full nutrient medium (Ctrl) (a, d, c and f) or starvation medium (Stv) (b and e); in the presence (d–f) or absence (a–c) of the actin depolymerizing agent Latrunculin B (LatB) (10 μM), and/or 100 μM of Rapamycin (Rap) (b and e). Subsequently, cells were fixed in 3% paraformaldehyde and processed for immunofluorescence. (B) The EGFP-MAP1LC3 dots were quantified from max intensity projection of a confocal z-stack and the mean+SEM of the number of dots per cell is shown. Data evaluated correspond to three independent experiments. Scale bars: 10 μm.
To confirm these results we also tested Cytochalasin B, another actin depolymerizing agent and similar to the results obtained with Latrunculin B, inhibition of MAP1LC3 puncta formation under starvation condition was observed (Fig. S1A and S1B).
Surprisingly, the stabilization of actin cytoskeleton by Jasplakinolide did not produce any inhibitory effect on autophagosome formation induced by the starvation stimulus (Fig. S2A and S2B) indicating that actin depolymerization but not its stabilization is critical for the process.
Another potent stimulus for autophagy is the treatment with rapamycin, which inhibits the kinase MTOR, releasing the repressing effect that this kinase has over the autophagic pathway.18 As expected, when cells were incubated in control media in the presence of rapamycin the GFP-MAP1LC3 dots incremented (Fig. 1A, c). Similar to the results obtained in starved cells, such increment was abolished when cells were incubated with Latrunculin B (Fig. 1A, f). The quantification of the Latrunculin B inhibitory effect on the accumulation of MAP1LC3 dots under starvation conditions or rapamycin treatment is shown in Figure 1B. These results suggest that de novo polymerization is necessary at certain steps during the autophagic process induced by starvation or rapamycin.
The actin cytoskeleton participates in the formation of autophagosomes upon starvation induction
The inhibitory effect of Latrunculin B or Cytochalasin B on autophagy activation could be at different stages of the process such as formation or maturation of the autophagosomes (i.e., acidification and fusion with lysosomes). To differentiate between these possibilities we analyzed both, the acidification of autophagosomes, identified as MAP1LC3-positive dots, and fusion of these vesicles with later compartments of the endocytic pathway. When the acidification state of autophagosomes with the lysosomotropic probe Lysotracker was analyzed, a similar proportion of acidic GFP-MAP1LC3 positive dots near to 80% (Fig. 2B) in both, control (Fig. 2A, a, b and inset) and Latrunculin B treated cells (Fig. 2A, c, d and inset) was observed.

Figure 2. Latrunculin B-treatment does not affect autophagosome maturation. (A) HeLa cells overexpressing GFP-MAP1LC3 (a and c) were incubated in starvation media in the presence (c and d) or absence of 10 μM Latrunculin B (LatB) (a and b). After 1.5 h incubation, LysoTracker (b and d) was added to the medium and the cells were incubated for additional 15 min. Then cells were washed twice with PBS, and starvation media with or without Latrunculin B was added. Confocal images of cells in vivo were acquired and the number of MAP1LC3-positive dots showing LysoTracker staining was quantified (B). (C) HeLa cells overexpressing GFP-MAP1LC3 (a and c) were incubated for 2 h at 37°C with dextran-Texas Red (b and d), washed, and the internalized probe was chased for additional 2 h. Afterwards, cells were subjected to starvation for 1.5 h in the presence (c and d) or absence of 10 μM Latrunculin B (LatB) (a and b). Confocal images of cells in vivo were acquired and the number of MAP1LC3-positive dots showing dextran-Texas Red staining was quantified (D). The graph shows the mean+SEM of the percentage of colocalization corresponding to three independent experiments. Scale bars: 10 μm.
Next, we analyzed the role of the actin cytoskeleton in the fusion of autophagosomes with later compartments of the endocytic pathway. To address this issue, HeLa cells were incubated for 2 h at 37°C with dextran-Texas Red. Subsequently, cells were washed and the probe was chased for additional 2 h in order to allow the arrival of dextran to later compartments (i.e., late endosomes/lysosomes). Afterwards, cells were subjected to starvation in the presence or absence of Latrunculin B and colocalization between GFP-MAP1LC3 and dextran-Texas Red was analyzed. As depicted in Figure 2C (a–d) and in the quantification shown in Figure 2D, no differences between cells treated or untreated with Latrunculin B were observed. This result suggests that actin depolymerization does not seem to affect the maturation step of autophagosomes.
As another approach, we used bafilomycin A1, an inhibitor of the proton ATPase that hampers the acidification of autophagosomes and causes an increase in autophagosome number due to a block of the autophagic flux.19 As expected, when bafilomycin A1 was added to starved cells, the MAP1LC3-positive dots increased four times compared with starved cells incubated without the drug (Fig. 3A). In contrast, in cells treated with bafilomycin A1 in the presence of Latrunculin B this increase was dramatically reduced (Fig. 3A). These results suggest that Latrunculin B blocks autophagosome formation rather than maturation.
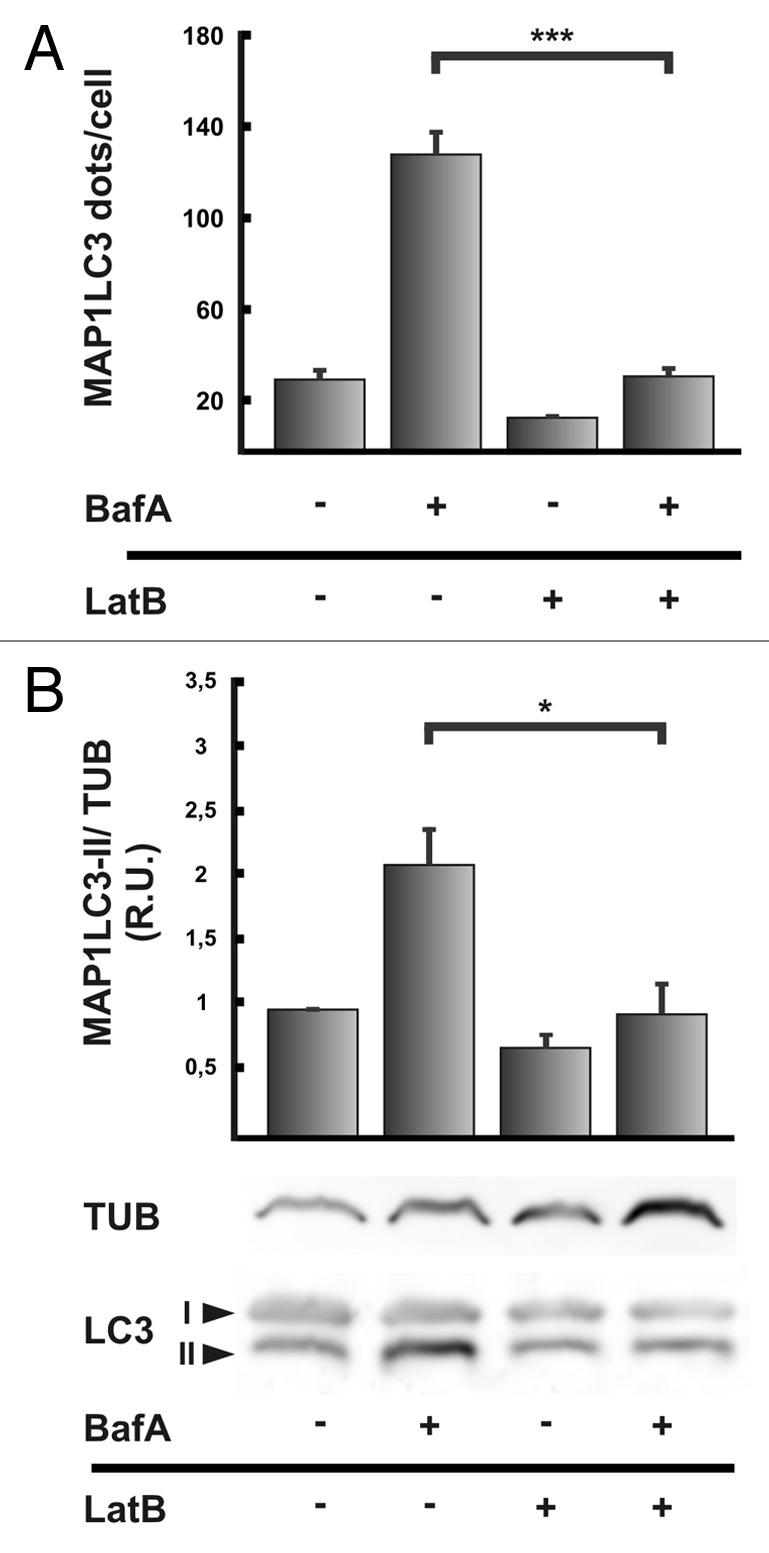
Figure 3. Actin filaments are required for autophagosome formation. (A) HeLa cells overexpressing RFP-MAP1LC3 were incubated 2 h at 37°C in starvation medium in the presence or absence of 10 μM Latrunculin B (LatB), with or without 100 nM bafilomycin A1 (BafA). Then, cells were fixed in 3% paraformaldehyde and processed for immunofluorescence. The EGFP-MAP1LC3 dots were quantified from max intensity projection of a confocal z-stack, and the mean+SEM of the number of dots per cell is shown. (B) HeLa cells were incubated in starvation medium in the presence or absence of 10 μM Latrunculin B (LatB), with or without 100 nM bafilomycin A1 (BafA) for 2 h at 37°C. Afterwards, cells were lysed in RIPA buffer and the samples were subjected to western blot analysis using a rabbit anti-MAP1LC3 and a mouse anti-TUB/Tubulin antibody and the corresponding HRP-labeled secondary antibodies, and subsequently developed with an enhanced chemiluminescence detection kit. The bands intensity was quantified with ImageJ software (gel analyzer plugin), and the MAP1LC3-II/TUB ratio was calculated. Western blot shown is representative of three independent experiments. Data evaluated in (A and B) corresponds to three independent experiments.
As indicated above, one of the hallmarks of the autophagic pathway is the processing of MAP1LC3-I to MAP1LC3-II.17 A diminution in MAP1LC3-II levels may suggest that autophagy has not been induced. However, since MAP1LC3-II is present in the autophagosome both in the outer and inner membrane, MAP1LC3-II is in part degraded after autophagosome-lysosome fusion. Thus, a very high degradation capacity leads to a quick disappearance of MAP1LC3-II, which might be misinterpreted as a defect in autophagosome synthesis.20 On the other hand, increased MAP1LC3-II levels may represent enhanced autophagosome formation or a block in MAP1LC3-II degradation.21 The inhibition of lysosomal degradation using a protease inhibitor (pepstatin or E64) or the vacuolar proton ATPase inhibitor bafilomycin A1 is useful to differentiate whether a reduced level of MAP1LC3-II is due to a inhibition of MAP1LC3 processing or to a high degradation rate.20 In Figure 3B we observed by western blot MAP1LC3-II accumulation when starved cells were treated with bafilomycin A1 (compare lines 1 and 2), but this accumulation was prevented by Latrunculin B addition (compare lines 2 and 4). A similar inhibitory effect in MAP1LC3-II accumulation was observed when cells were treated with Cytochalasin B (Fig. S1C). In agreement with the previous observations on the MAP1LC3 dots number, cell treatment with Jasplakinolide caused no alterations in the accumulation of MAP1LC3-II (Fig. S2C). These results indicate that actin depolymerization but not its stabilization is affecting the generation of MAP1LC3-II (i.e., autophagosome formation) rather than increasing autophagosome degradation rate.
Taken together, these results allow us to conclude that depolymerization of actin has an inhibitory effect on autophagosome formation, without affecting subsequent maturation steps.
Actin polymerization is involved in early steps of autophagosome formation
In recent years, cumulative evidence has indicated that the formation of an autophagosome involves the recruitment of several protein complexes to specialized areas of the endoplasmic reticulum. This leads to a membrane deformation generating a protrusion called omegasome that develops a phagophore or isolation membrane, which finally closes and forms a vesicle.22,23 Since proper functional actin filaments are required for autophagosome formation in response to starvation, we analyzed the ultrastructure of starved cells incubated in the presence or absence of Latrunculin B (Fig. S1A). Surprisingly, we found an accumulation of curved membranous structures (Fig. S3A, a and b) that may represent aberrant phagophores or isolation membranes (Fig. S3A, c and d). Our quantification data indicate that these structures were present in 60% of the cells treated with Latrunculin B (Fig. S3B). The idea that this curved membranous structures are autophagic-related structures is supported by the fact that wortmannin treatment decreased the percentage of cells depicting this kind of structures (please see quantification in Fig. S3B).
These results suggest that actin depolymerization is affecting very early steps of autophagosome formation. To address this hypothesis, we performed a colocalization analysis between actin fibers (using phallodin-rhodamine, a probe that marks only F-actin) and a set of proteins that participate in different steps of the autophagic pathway. Particularly, to test early steps of autophagosome formation, we used the proteins ULK1 and ULK2, members of ULK1 complex and the protein ATG14 and BECN1, which are components of the class III phosphatidylinositol 3-kinase complex, present at the omegasome and in the phagophore.24,25 This kinase is responsible for generating phosphatidyl inositol 3 phosphate (PtdIns3P), a lipid that is recognized by different proteins involved in membrane deformation such as ZFYVE1 and WIPI2.25 The presence of PtdIns3P can be also monitored using a specific probe (i.e., 2xFYVE) which is useful to identify areas of autophagosome formation.23 A subsequent step in the autophagosome biogenesis is the elongation of the phagophore that implies the recruitment of several proteins. The complex formed by ATG5, ATG12 and ATG16L is recruited to the phagophore membrane at this stage.24,25 Thus, to analyze this intermediate step in autophagosome formation we used the protein ATG5 that is an excellent phagophore marker since the ATG5-ATG12-ATG16L1 complex is released before the phagophore closure and it is not present in the autophagosome.25 Finally, we used the protein MAP1LC3 that is involved in the last steps of autophagosome formation and it remains partly associated to vesicles, even when the autophagosomes mature.25
As shown in Figure S4 actin fibers labeled with phallodin-rhodamine sporadically colocalized with EGFP-MAP1LC3 (Fig. S4, a–d), and even though ATG5-positive structures frequently appear near to the actin fibers, no colocalization was observed between these two kind of structures (Fig. 4, e–h). In contrast, we found a strong colocalization between ATG14 and actin fibers, in reticular-like structures in the cytoplasm and in some punctate structures close to the plasma membrane (Fig. 4, a–d). Also, a comparable colocalization with BECN1 was observed, in similar type of structures (Fig. 4, e–h).

Figure 4. The actin participation in the autophagic pathway is at the early steps of the pathway. HeLa cell overexpressing GFP-ATG14 (a–d), Flag-BECN1 (e–h), GFP-FYVE (i–p) or GFP-ZFYVE1 (q–t) were incubated in starvation medium for 2 h at 37°C. Subsequently, cells were fixed in 3% paraformaldehyde and processed for immunofluorescence and actin filaments were stained using Phalloidin-Rhodamine. To stain FLAG-BECN1, a mouse anti-flag antibody and a secondary anti-mouse Alexa Fluor 488 antibody were used. Scale bars: 10 μm. Arrowheads indicate colocalization sites.
We next used GFP-2xFYVE, a probe that labels PtdIns3P, the PtdIns-3 kinase product, and the colocalization with actin fibers was assessed. Similar to the results obtained with ATG14, we found in all cells analyzed FYVE punctated structures that colocalized with actin (Fig. 4, i–l). Surprisingly, we also found actin associated with donut-shaped structures and unclosed rings colocalizing with the FYVE signal (Fig. 4, m–p). These kinds of structures known as omegasomes have been previously described,23 and correspond to early steps in the formation process of some autophagosomes. To confirm that the structures that colocalized are autophagic structures we also used the protein ZFYVE1, a FYVE domain-containing protein that is present at the omegasomes.23 Similar to the FYVE probe, ZFYVE1 also colocalized with actin fibers (Fig. 4, q–t).
On the other hand no colocalization was found with the early autophagic markers ULK1 (Fig. S4, i–l) or ULK2 (Fig. S4, m–p). These results reinforce the idea that actin is involved in very early steps of autophagosome formation related to the PtdIns3P generation.
RHOA is able to activate the autophagic pathway
GTPases of the Rho family are important proteins that regulate actin dynamics.26 The most studied proteins of this family are RHOA, CDC42 and RAC1. We first examined the role of RHOA in autophagosome formation. HeLa cells coexpressing RFP-MAP1LC3 and the vector EGFP, EGFP-RHOA wt or the mutants -RHOA V14 (a constitutive active mutant) or -RHOA N19 (a constitutive inactive mutant) were incubated for 2 h at 37°C either in control or starvation medium. As expected HeLa cells overexpressing the vector EGFP showed a significantly increase in autophagosome number in response to the starvation stimuli (Fig. 5A, a–d and Fig. 5B). When either EGFP-RHOA WT (Fig. 5A, e–h) or EGFP-RHOA V14 (Fig. 5A, i–l) were overexpressed, the autophagosome number increased at the same level that in starved conditions even in the control cells incubated in full-nutrient medium (Fig. 5B). Interestingly, the overexpression of the dominant negative mutant EGFP-RHOA N19 (Fig. 5A, m–p) did not affect the autophagosome number in cells incubated in control medium, but it was able to abolish the increase produced by the starvation stimulus (Fig. 5B).

Figure 5. RHOA is able to modulate autophagy. (A) HeLa cells were cotransfected with pRFP-MAP1LC3 and pEGFP alone (a and c), pEGFP-RHOA WT (e and g), pEGFP-RHOA V14 (i and k), or pEGFP-RHOA N19 (m and o). Afterwards, cells were incubated in control full-nutrient or starvation medium for 2 h at 37°C. Then, cells were fixed and processed for immunofluorescence. (B) The RFP-MAP1LC3 dots were quantified from max intensity projection of a confocal z-stack and the mean+SEM of the number of dots per cell is shown. (C) HeLa cells were pre-incubated with the C3 toxin or the toxin vehicle alone for 4 h at 37°C. Cells were washed twice with PBS and incubated in starvation medium in the presence or absence of the C3 toxin for additional 2 h at 37°C. Afterwards, cells were lysed in RIPA buffer and the samples were subjected to western blot analysis using a rabbit anti-MAP1LC3 and a mouse anti-TUB antibodies and the corresponding HRP-labeled secondary antibodies the corresponding HRP-labeled secondary antibody, and subsequently developed with an enhanced chemiluminescence detection kit. The bands’ intensity was quantified with ImageJ software (gel analyzer plugin), and the MAP1LC3-II/TUB ratio was calculated. (D) HeLa cells were transfected with a siRNA against RHOA according to the manufacturer indication. Afterwards, cells were lysed in RIPA buffer and the samples were subjected to western blot analysis using a mouse anti-RHOA and a mouse anti-TUB antibodies and the corresponding HRP-labeled secondary antibody, and subsequently developed with an enhanced chemiluminescence detection kit. The bands’ intensity was quantified with ImageJ software (gel analyzer plugin), and the RHOA/TUB ratio was calculated. (E) HeLa cells were transfected with RFP-MAP1LC3 alone or cotransfected with a siRNA against RHOA. After 48 h transfection, cells were incubated in control full-nutrient or starvation medium for 2 h at 37°C. Subsequently, cells were fixed and processed for immunofluorescence. The RFP-MAP1LC3 dots were quantified from max intensity projection of a confocal z-stack and the mean+SEM of the number of dots per cell is depicted. (F) HeLa cells were transfected with a siRNA against RHOA. Untransfected cells or cells transfected with the siRNA were incubated in starvation medium in the presence or absence of 100 nM bafilomycin A1 (BafA) for 2 h at 37°C. Afterwards, cells were lysed in RIPA buffer and the samples were subjected to western blot analysis using a rabbit anti-MAP1LC3 or mouse anti-TUB and the corresponding HRP-labeled secondary antibody, and subsequently developed with an enhanced chemiluminescence detection kit. The bands’ intensity was quantified with ImageJ software (gel analyzer plugin), and the MAP1LC3-II/TUB ratio was calculated. The data evaluated and western blots shown are representative of three independent experiments. Scale bars: (A) 15 μm.
One of the major problems of analyzing transfected cells is that, in general, only a fraction of the cells are actually transfected and express the protein of interest. Thus, in order to analyze the role of RHOA in the processing of MAP1LC3 by western blot we used an alternative approach. Cells subjected to starvation were incubated with a cell permeable variant C3 toxin of Clostridium botulinum that inhibits RHOA, B and C.27,28 As shown in Figure 5C, treatment of starved cells with the C3 toxin produced a diminution in the MAP1LC3-II levels, supporting the requirement of active RHOA in the autophagic pathway.
To confirm the role of RHOA in the autophagic process we depleted the protein using a siRNA approach. Similar to the effects obtained with the overexpression of the dominant negative mutant RHOA N19 and with the C3 toxin, the RHOA knockdown (Fig. 5D) prevented MAP1LC3 dots accumulation due to the starvation stimulus (Fig. 5E, compare lines 2 and 4). The silencing of RHOA also abolished the accumulation of MAP1LC3 II in starved cells treated with BafA (Fig. 5F, compare lines 2 and 4).
Due to the similarities observed between the results obtained with the actin depolymerizing agent and with the inhibition or knockdown of RHOA we hypothesized that this protein is involved in the same step that the actin filaments. To test this, we analyzed the colocalization between RHOA and proteins involved in different steps of autophagosome formation. We have found that RHOA, similar to the actin fibers, colocalized with BECN1 (Fig. S5) but not with MAP1LC3, ULK1 or ULK2 (data not shown). Taken together, our results suggest that the actin regulator RHOA is involved in starvation-induced autophagy.
The activity of the kinase ROCK is required for starvation-mediated autophagy
One of the most studied effectors of RHOA is the kinase ROCK.14 Thus, we decided to study if this kinase has a role in starvation-activated autophagy. We assayed Y-27632, a compound that has been extensively used to inhibit ROCK activity.29 When HeLa cells were incubated in starvation medium, in the presence of the ROCK inhibitor we observed a decrease in the number of RFP-MAP1LC3-positive dots (Fig. 6A, compare a and d). Quantification of the number of dots indicates a significantly diminution of the RFP-MAP1LC3-positive dots, that reached a level comparable to the basal condition of autophagy (Fig. 6B). This result suggests that ROCK is likely a downstream RHOA effector whose activity is required for autophagy.

Figure 6. Inhibition of ROCK abolished the autophagy induction mediated by starvation, but not by the overexpression of the constitutive active mutant RHOA V14. (A) HeLa cells were transfected with pRFP-MAP1LC3 (a and d) or cotransfected with pRFP-MAP1LC3 (b and e) and pEGFP-RHOA V14 (c and f). Afterwards, cells were incubated in starvation medium in the absence (a–c) or presence (d–f) of the ROCK inhibitor Y27632 (10 μM) for 2 h at 37°C. Then, cells were fixed and processed for immunofluorescence. (B) The RFP-MAP1LC3 dots were quantified from max intensity projection of a confocal z-stack and the mean+SEM of the number of dots per cell is shown. The data evaluated correspond to three independent experiments. (C) HeLa cells were transfected with a siRNA against RHOA according to the manufacturer's instructions. Afterwards, cells were lysed in RIPA buffer and the samples were subjected to western blot analysis using a mouse anti-ROCK1 and a mouse anti-TUB and the corresponding HRP-labeled secondary antibody, and subsequently developed with an enhanced chemiluminescence detection kit. The bands’ intensity was quantified with ImageJ software (gel analyzer plugin), and the ROCK1/TUB ratio was calculated. (D) HeLa cells were transfected with RFP-MAP1LC3 alone or cotransfected with the siRNA against ROCK1. After 48 h transfection, cells were incubated in control full-nutrient or starvation medium for 2 h at 37°C. Then, cells were fixed and processed for immunofluorescence. The RFP-MAP1LC3 dots were quantified from max intensity projection of a confocal z-stack and the mean+SEM of the number of dots per cell is shown. (E) HeLa cells were transfected with the siRNA against ROCK1. Untransfected cells or cells transfected with the siRNA were incubated in starvation medium in the presence or absence of 100 nM bafilomycin A1 (BafA) for 2 h at 37°C. Afterwards, cells were lysed in RIPA buffer and the samples were subjected to western blot analysis using a rabbit anti-MAP1LC3 or mouse anti-TUB and the corresponding HRP-labeled secondary antibody, and subsequently developed with an enhanced chemiluminescence detection kit. The bands’ intensity was quantified with ImageJ software (gel analyzer plugin), and the MAP1LC3-II/TUB ratio was calculated. The data evaluated and western blots shown are representative of three independent experiments. Scale bars: (A) 20 μm.
The data obtained in the previous section indicate that under full-nutrient conditions the sole overexpression of the constitutive active mutant RHOA V14 increased the number of autophagosomes at a similar level than the starvation stimulus (Fig. 5A). Thus, we next analyzed the effect of ROCK inhibition in cells overexpressing the active mutant RHOAV14. Interestingly, treatment of transfected cells with Y-27632 had no effect in the number of RFP-MAP1LC3-positive dots (Fig. 6A, b and e; and Fig. 6B).
We also tested a siRNA against the kinase ROCK. siRNA-mediated knockdown of ROCK (Fig. 6C) caused an impairment of the typical increase in autophagosome numbers due to the starvation stimulus (please see Fig. 6D) as well as in MAP1LC3 II accumulation determined by western blot analysis (Fig. 6E).
Taken together, our results indicate that the kinase ROCK is participating in the activation of autophagy by starvation, but its inhibition is not able to inhibit the signal triggered by the overexpression of the active mutant RHOA V14, suggesting that other downstream effectors are also activated by this GTPase.
RAC1 has an inhibitory effect in the induction of autophagy by starvation
Other important proteins implied in actin dynamics regulations are CDC42 and RAC1.10 We tested if the overexpression of different variants of RAC1, wild type, the constitutive active mutant V12 or the constitutive inactive mutant N17 altered autophagosome formation. HeLa cells coexpressing RFP-MAP1LC3 and EGFP, EGFP-RAC1 wt or the mutants V12 or N17 were incubated for 2 h either in control full-nutrient or starvation medium. As shown in Figure 7A, the behavior of cells coexpressing RAC1 wt (Fig. 7A, e–h) both, in control or in the starved situation, showed no significant differences (Fig. 7B) compared with cells coexpressing the vector EGFP (Fig. 7A, a–d). In contrast, in cells coexpressing RFP-MAP1LC3 and the constitutive active mutant EGFP-RAC1 V12 the starvation stimulus was not able to increase de RFP-MAP1LC3 positive dots (in Fig. 7A, compare j with l) showing a statistically significant difference with cells expressing EGFP subjected to starvation (Fig. 7B).

Figure 7. RAC1 negatively modulates autophagy. (A) HeLa cells were cotransfected with pRFP-MAP1LC3 and pEGFP alone (a and c), pEGFP-RAC1A WT (e and g), pEGFP-RAC1 V12 (i and k), or pEGFP-RHOA N17 (m and o). Afterwards, cells were incubated for 2 h at 37°C in control full-nutrient or starvation medium. Cells were fixed and processed for immunofluorescence. (B) The RFP-MAP1LC3 dots were quantified from max intensity projection of a confocal z-stack and the mean+SEM of the number of dots per cell is shown. (C) HeLa cells were transfected with a siRNA against RAC1 according to the manufacturer's instructions. After 48 h transfection, cells were lysed in RIPA buffer and the samples were subjected to western blot analysis using a mouse anti-RAC1 and a mouse anti-TUB antibodies and the corresponding HRP-labeled secondary antibody, and subsequently developed with an enhanced chemiluminescence detection kit. The bands intensity was quantified with ImageJ software (gel analyzer plugin), and the RAC1/TUB ratio was calculated. (D) HeLa cells were transfected with RFP-MAP1LC3 alone or cotransfected with the siRNA against RAC1. After 48 h transfection, cells were incubated in control full-nutrient or starvation medium for 2 h at 37°C. Then, cells were fixed and processed for immunofluorescence. The RFP-MAP1LC3 dots were quantified from max intensity projection of a confocal z-stack and the mean+SEM of the number of dots per cell is depicted. The data evaluated correspond of three independent experiments. Scale bars: (A) 20 μm.
Interestingly, the sole overexpression of the constitutive inactive mutant RAC1 N17 in cells incubated in control media was enough to augment the number of MAP1LC3-positive dots (Fig. 7A, m and p). Quantification of the MAP1LC3-positive dots indicates that the levels in control and starvation cells expressing RAC1 N17 were similar to that observed in cells expressing EGFP alone in the starved situation (Fig. 7B).
As another approach to demonstrate the role of RAC1 in the autophagic pathway we used siRNA-mediated silencing of RAC1 (Fig. 7C). Similar to the results obtained with the overexpression of the negative mutant RAC1N17, the knockdown of RAC1, caused an increase in the number of autophagosomes in cells incubated in completed medium at a similar level to the starvation stimulus (Fig. 7D).
On the other hand, when we overexpressed different variants of CDC42 (wild type, and the constitutive active mutant V14 or the constitutive inactive mutant N17) no effect was observed in either full-nutrient or starvation conditions (data not shown).
Taken together, these results allow us to conclude that RAC1 in its active form is able to inhibit the starvation-induced autophagic response and that inhibition or depletion of this protein can moderately activate the autophagic pathway.
Discussion
The cytoskeleton has been associated with different roles in the autophagic pathway. Particularly, microtubules and their associated motors have been studied in mammalian cells. It has been shown that autophagosome movement in the cytoplasm is dependent on microtubules,30,31 through an association of the autophagosome with the motor complex dynein-dinactin.32 This type of movement is required to direct autophagosomes to the centrosome region, where the autophagosomes fuse with lysosomes.32 More recently other proteins that collaborate in the relationship between autophagosomes and microtubules have been described. One example is FYCO1, a protein that forms a complex with MAP1LC3 and RAB7, and it is necessary to mediate the microtubule-depending movement of autophagosomes.33
In the present report we have analyzed the role of actin in autophagy. Treatment of CHO and HeLa cells with the actin depolymerizing agent Latrunculin B or Cytochalasin B abolished the rapamycin- and starvation-dependent increase of MAP1LC3-positive dots and the MAP1LC3-II levels, without affecting basal autophagy. In addition, the same effects of Latrunculin B-treatment were observed in starved cells incubated with bafilomycin A1, indicating that the actin cytoskeleton is involved in the formation of autophagosomes rather than in the maturation steps. Indeed, we confirmed that maturation was not impaired using two assays, the acidification of the autophagosome and the fusion with late endocytic/lysosomal compartments. When we compared cells incubated with or without Latrunculin B, both showed a similar percentage of acidic EGFP-MAP1LC3 dots, positive for LysoTracker; and the colocalization between EGFP-MAP1LC3 and TexasRed-Dextran incorporated by endocitosis was not altered.
In mammalian cells, evidence about the participation of actin in autophagy has been reported, but its role has been poorly characterized. An accumulation of proteins was observed in the liver of rats treated with phalloidin, a toxin that binds to actin filaments and blocks their depolymerization.34 In addition, the typical increase of autolysosome density produced by leupeptin/E64 treatment was hindered by phallodin treatment. With these data the authors hypothesized that the stabilization of actin affects autophagosome formation. The apparent discrepancy with our results using Jasplakinolide, an actin stabilizing agent, could be due to that in contrast to Phalloidin, Jasplakinolide may also promote actin nucleation;35 reflecting the necessity of de novo actin polymerization. In another report it has been shown that the depolymerization of actin using Citochalasin B or D decreases the degradation of long-lived proteins and also prevents the accumulation of autophagic related structures.36 Our findings about the role of actin in autophagosome formation are consistent with these previous observations and expand those results by determining in which stage of the autophagic pathway actin is involved.
In yeast, it has been noted that actin is required for the Cvt pathway and also pexophagy.37 Atg9, a transmembrane protein required for autophagosome formation, cycles between the structure called PAS and the mitochondria.38 Interestingly, it has been shown that actin participates in the transport of Atg9. This process is mediated by the interaction with Atg1139 and requires the activity of Arp 2/3, an actin nucleator.40 Similar to yeast, in mammals ATG9 has a cyclic behavior, but it cycles between the Golgi apparatus and the endosomes. Recently MYO2/myosin II, a motor associated to the actin filaments, has been related to the transport of ATG941 which represents an indirect evidence that actin participates in some steps of autophagy. It has been postulated that ATG9 is involved in the regulation of autophagosome size at an early stage of the autophagosome formation process.42 These data are in agreement with our observations that actin filaments colocalized with ATG14 and BECN1, two members of PI3Kinase complex, which has an important function at the initial steps of autophagosome formation.25
In addition we also found actin fibers associated to omegasomes (evidenced by the colocalization between actin filaments and the 2xFYVE domain and ZFYVE1), a membranous structure enriched in PtdIns3P, that has been identified as an autophagosome precursor.23 In contrast, actin showed no colocalization with markers of later steps of autophagosome formation like ATG5 or MAP1LC3 confirming a very early requirement of actin in autophagosome biogenesis particularly linked to the PtdIns3P formation step, since no colocalization was found between actin and ULK1/2.
Several signaling molecules drive actin polymerization/depolymerization cycles depending on the particular process in which microfilaments are involved. In the process of autophagosome formation induced by starvation, we have established the participation of RHOA and one of its effectors, the kinase ROCK. It has been previously described that RHOA triggers a series of signals, with the participation of ROCK, leading to actin polymerization and MYO2 activation. Interestingly, even though our results indicate that ROCK is a component of the signaling pathway involved in autophagy, inactivation of this kinase was unable to prevent the effect of a permanently activated RHOA, suggesting that other downstream effectors are also activated by this GTPase.14
On the other hand, we have found that RAC1 but not CDC42 is also involved in the regulation of the autophagic pathway, but it has an inhibitory role. Consistently, in a paper recently published by Zhu and collaborators43 it has been demonstrated, using a siRNA approach, that RAC3 but not the RAC1 or RAC2 isoforms, has an inhibitory effect on autophagy. These differences in isoform requirement are likely due to the cell lines used. In the publication by Zhu et al. the siRNA against RAC1 was used in HCT116, a cell line that has a low level of expression of this protein compared with RAC2 and RAC3, whereas in the HeLa cells used in our work, RAC1 is the main expressed isoform. An interestingly observation comes from a paper showing that MAP1LC3 binds to SOS1, a GEF for RAC1, impairing RAC1 activation due to inhibition of the GEF activity of SOS1.44 Taken together, those observations in conjunction with our results suggest that signals involved in the autophagic pathway and the RAC signaling pathway are mutually regulated.
In the present manuscript we have demonstrated a role for the actin cytoskeleton in autophagosome formation. Early steps in autophagosome formation, like membrane deformation to generate the omegasome and subsequent elongation of the phagophore, have a defined kinetic. It is likely that when an enhanced activity of autophagosome formation is needed, such as by starvation or rapamycin stimulation, the whole autophagosome formation process needs to be activated. Thus, when cells require an increased rate of autophagosome formation, this kinetics could be enhanced taking advantage of the actin cytoskeleton that may actively participate in membrane remodeling. On the other hand, it is tempting to hypothesize that the opposite roles of RHOA and RAC1 in the starvation-mediated autophagy ensure a tight regulation of the process, depending on which of them are activated in a given situation. Further studies are necessary to unveil the participation of both actin modulatory proteins in keeping a precise regulation of the autophagic activity.
Materials and Methods
Materials
Dulbecco’s Modified Eagle Medium (D-MEM, 12100), (α-MEM, 11900-024) fetal bovine serum (FBS, 16000-044), Earle’s Balanced Salt Solution (EBSS, 14155063) and penicillin and streptomycin (15640055) were obtained from Gibco BRL/Life technologies.
The plasmid encoding GFP-MAP1LC3 was kindly provided by Drs. Noboru Mizushima (Tokio Medical and Dental University) and Tamotsu Yoshimori (Osaka University). The pRFP-MAP1LC3 construct was generated in our lab and it has been previously described.45 The plasmid pGFP-FYVE was provided by Dr. Sergio Grinstein (Hospital of Sick’s Kids). Plasmids encoding EGFP-RAC1, -CDC42 and -RHOA and theirs mutants were kindly provided by Dr. Philippe Chavrier (Centre National de la Recherche Scientifique/Institut Curie) and Mark R. Phillips (Laboratory of Molecular Rheumatology, NYU, School of Medicine). The plasmid EGFP-ZFYVE1 was provided by Nicholas Ktistakis (Babraham Institute). Plasmids encoding myc-ULK1 and ULK2 were kindly provided by Sharon Tooze (London Research Institute). pSG5-FLAG- epitope-tagged human BECN1 was provided by Dr. Beth Levine (Southwestern Medical Center). EGFP-ATG5 and EGFP-ATG14 was provided by Tamotsu Yoshimori (Osaka University). siRNA against RHOA (1129127), ROCK1 (1130663) and RAC1 (1126011) was purchased to Bioneer (Alameda).
The polyclonal rabbit antibody against MAP1LC3 (L7543), and the inhibitors Latrunculin B (L5288), bafilomycin A1 (B1793), wortmannin (w1628), Jasplakinolide (j4580), and the mouse anti-myc (M4439), mouse anti-flag (F1804) and mouse anti-TUB (T9026) antibodies were purchased from Sigma-Aldrich. The polyclonal secondary antibody goat anti-Rabbit conjugated with HRP (111-035-003) and goat anti-Mouse conjugated Alexa Fluor 488 (715-545-150) was purchased from Jackson ImmunoResearch Laboratories, Inc.
Cell culture and transfection
HeLa cells were grown in DMEM and CHO cells in α-MEM (Gibco Laboratories, Invitrogen), both supplemented with 10% heat inactivated fetal bovine serum, 2.2 g/l sodium bicarbonate, 2 mM glutamine and 0.1% penicillin/streptomycin at 37°C under 5% CO2.
Autophagy was induced by amino acid and serum deprivation. Briefly, cells were washed three times with PBS and incubated in EBSS at 37°C for different periods of time in the presence or the absence of different drugs, as indicated in the figure legends.
For transient expression, Lipofectamine 2000 (Invitrogen, 11668019) was used according to the manufacturer’s instructions.
Fluorescence staining
HeLa or CHO cells were fixed with 3% paraformaldehyde solution in PBS for 10 min at 37°C, washed with PBS, and blocked with 50 mM NH4Cl in PBS. Subsequently, cells were permeabilized with 0.05% saponin (Sigma-Aldrich, S4521) in PBS containing 0.5% BSA (Sigma-Aldrich, A2153) and mounted in MOWIOL (Sigma-Aldrich, 10853).
Confocal microscopy
For immunofluorescence, cells were grown on coverslips overnight to 50–80% confluence. After incubation under different experimental conditions, cells were fixed with 3% paraformaldehyde in PBS for 15 min at room temperature, washed with PBS, blocked with 50 mM NH4Cl in PBS and subsequently permeabilized with PBS containing 0.05% saponin and 0.2% bovine serum albumin for 15 min. The coverslips were mounted with Mowiol. Fixed cells were imaged using an Olympus Fluoview 1000 confocal microscope using objective 60× PlanApo oil, numerical aperture (NA) 1.42.
Image processing
All the images were processed using ImageJ software (Wayne Rasband, National Institutes of Health). Briefly, the images were deconvolved with Parallelal Spectral Deconvolution plugin (Piotr Wendykier) using a theorical PSF generated by Diffraction PSF 3D plugin (Robert Dougherty). Background was eliminated using the Subtract Background plugin. For MAP1LC3 dots quantification, the sum of images from a z-stack was obtained using the Z Project plug-in. After the image binarization using a defined threshold, the dots number was quantified using the Particle Analyzer plugin. For the colocalization analysis between GFP-MAP1LC3 and LysoTracker or dextran-TexasRed a single plane image from each channel was acquired, processed as described before and then, the number of particles that have signals in both channels was quantified.
Western blot
The cells were lysed for 30 min at 4°C with RIPA buffer supplemented with the following inhibitors: 10 μg/mL aprotinin, 10 μg/mL leupeptin, 5 μg/mL pepstatin A, 1 mM sodium orthovanadate and 1 mM sodium fluoride. The lysates were treated with Laemmli’s buffer and separated by electrophoresis in polyacrylamide. After electrophoresis, the proteins were transferred to nitrocellulose in a wet system at 200 mAmp for 1 h. The protein bands were immunodetected with the adequate primary antibodies incubated for 1 h at room temperature. The primary antibodies were revealed with horseradish peroxidase (HRP)-conjugated secondary antibodies, visualized by enhanced chemiluminescence (ECL) (General Electric, RPN2232) and analyzed with Fujifilm LAS-4000 equipment. Densitometric analysis of bands was performed using the ImageJ software.
Transmission electron microscopy
Cells were fixed with 2% glutaraldehyde in PBS. After 1 h, the samples were scraped and centrifuged for 15 min at 500 x g, and the pellets were processed for transmission electron microscopy using conventional techniques.
Statistical analysis
Results are presented as the mean ± SEM from at least two independent experiments. The comparisons were performed using ANOVA in conjunction with Tuckey and Dunnett tests. Significant differences: *p < 0.01; **p < 0.005; ***p < 0.001.
Supplementary Material
Acknowledgment
We are grateful to Drs. Nickolas Ktistakis, Tamotsu Yoshimori, Sharon Tooze, Noburu Mitsushima, Sergio Grinstein, Phillipe Chavrier, Mark R. Phillips and Beth Levine for the material provided. We also thank Alejandra Medero for technical assistance with tissue culture, Marcelo Furlan for technical assistance with electron microscopy sample preparation and Graciela Gutierrez for technical assistance. This work was partly supported by grants from Agencia Nacional de Promoción Científica y Tecnológica (PICT 2005 38420 and PICT 2008 0192) and from SeCTyP (Universidad Nacional de Cuyo) to M.I.C.
Glossary
Abbreviations:
- ULK1
Unc-51-like kinase 1
- FIP200
focal adhesion kinase family interacting protein of 200 kD
- ATG
autophagy-related gene
- VPS
vacuolar protein sorting
- GTP
guanosine triphosphate
- GDP
guanosine diphosphate
- GDI
guanosine nucleotide dissociation inhibitors
- GEF
guanine exchange factor
- GAP
GTPase activating protein
- RHOA
Ras homolog gene family, member A
- RAC1
RAS-related C3 botulinum toxin substrate
- CDC42
cell division control protein 42 homolog
- ROCK
RHO-associated protein kinase
- EGFP
enhanced green fluorescent protein
- MAP1LC3
microtubule-associated protein 1 light chain 3
- PtdIns3P
phosphatidyl inositol 3 phosphate
- Baf
bafilomycin A1
- LatB
Latrunculin B
- Rap
rapamycin, FYCO, FYVE and coiled-coil domain-containing protein, DFCP1, double FYVE containing protein1
- WIPI
WD repeat domain, phosphoinositide interacting
- MTOR
mechanistic target of rapamycin
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials can be found at: www.landesbioscience.com/journals/autophagy/article/21459
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/21459
References
- 1.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- 3.Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tooze SA, Jefferies HB, Kalie E, Longatti A, McAlpine FE, McKnight NC, et al. Trafficking and signaling in mammalian autophagy. IUBMB Life. 2010;62:503–8. doi: 10.1002/iub.334. [DOI] [PubMed] [Google Scholar]
- 5.Noda T, Fujita N, Yoshimori T. The late stages of autophagy: how does the end begin? Cell Death Differ. 2009;16:984–90. doi: 10.1038/cdd.2009.54. [DOI] [PubMed] [Google Scholar]
- 6.May RC, Machesky LM. Phagocytosis and the actin cytoskeleton. J Cell Sci. 2001;114:1061–77. doi: 10.1242/jcs.114.6.1061. [DOI] [PubMed] [Google Scholar]
- 7.DePina AS, Wöllert T, Langford GM. Membrane associated nonmuscle myosin II functions as a motor for actin-based vesicle transport in clam oocyte extracts. Cell Motil Cytoskeleton. 2007;64:739–55. doi: 10.1002/cm.20219. [DOI] [PubMed] [Google Scholar]
- 8.Gopaldass N, Patel D, Kratzke R, Dieckmann R, Hausherr S, Hagedorn M, et al. Dynamin A, Myosin IB and Abp1 couple phagosome maturation to F-actin binding. Traffic. 2012;13:120–30. doi: 10.1111/j.1600-0854.2011.01296.x. [DOI] [PubMed] [Google Scholar]
- 9.Marion S, Hoffmann E, Holzer D, Le Clainche C, Martin M, Sachse M, et al. Ezrin promotes actin assembly at the phagosome membrane and regulates phago-lysosomal fusion. Traffic. 2011;12:421–37. doi: 10.1111/j.1600-0854.2011.01158.x. [DOI] [PubMed] [Google Scholar]
- 10.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–69. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 11.DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005;15:356–63. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Spiering D, Hodgson L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh Migr. 2011;5:170–80. doi: 10.4161/cam.5.2.14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–5. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- 14.Narumiya S, Tanji M, Ishizaki T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009;28:65–76. doi: 10.1007/s10555-008-9170-7. [DOI] [PubMed] [Google Scholar]
- 15.Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol. 2011;7:9–17. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- 16.Lee JY, Yao TP. Quality control autophagy: A joint effort of ubiquitin, protein deacetylase and actin cytoskeleton. Autophagy. 2010;6:555–7. doi: 10.4161/auto.6.4.11812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 20.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 21.Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, et al. In search of an “autophagomometer”. Autophagy. 2009;5:585–9. doi: 10.4161/auto.5.5.8823. [DOI] [PubMed] [Google Scholar]
- 22.Walker S, Chandra P, Manifava M, Axe E, Ktistakis NT. Making autophagosomes: localized synthesis of phosphatidylinositol 3-phosphate holds the clue. Autophagy. 2008;4:1093–6. doi: 10.4161/auto.7141. [DOI] [PubMed] [Google Scholar]
- 23.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–76. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–56. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- 26.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–35. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 27.Aktories K, Rösener S, Blaschke U, Chhatwal GS. Botulinum ADP-ribosyltransferase C3. Purification of the enzyme and characterization of the ADP-ribosylation reaction in platelet membranes. Eur J Biochem. 1988;172:445–50. doi: 10.1111/j.1432-1033.1988.tb13908.x. [DOI] [PubMed] [Google Scholar]
- 28.Rubin EJ, Gill DM, Boquet P, Popoff MR. Functional modification of a 21-kilodalton G protein when ADP-ribosylated by exoenzyme C3 of Clostridium botulinum. Mol Cell Biol. 1988;8:418–26. doi: 10.1128/mcb.8.1.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narumiya S, Ishizaki T, Uehata M. Use and properties of ROCK-specific inhibitor Y-27632. Methods Enzymol. 2000;325:273–84. doi: 10.1016/S0076-6879(00)25449-9. [DOI] [PubMed] [Google Scholar]
- 30.Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem. 2006;281:36303–16. doi: 10.1074/jbc.M607031200. [DOI] [PubMed] [Google Scholar]
- 31.Köchl R, Hu XW, Chan EY, Tooze SA. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–45. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- 32.Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33:109–22. doi: 10.1247/csf.08005. [DOI] [PubMed] [Google Scholar]
- 33.Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253–69. doi: 10.1083/jcb.200907015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueno T, Watanabe S, Hirose M, Namihisa T, Kominami E. Phalloidin-induced accumulation of myosin in rat hepatocytes is caused by suppression of autolysosome formation. Eur J Biochem. 1990;190:63–9. doi: 10.1111/j.1432-1033.1990.tb15546.x. [DOI] [PubMed] [Google Scholar]
- 35.Bubb MR, Spector I, Beyer BB, Fosen KM. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J Biol Chem. 2000;275:5163–70. doi: 10.1074/jbc.275.7.5163. [DOI] [PubMed] [Google Scholar]
- 36.Aplin A, Jasionowski T, Tuttle DL, Lenk SE, Dunn WA., Jr. Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J Cell Physiol. 1992;152:458–66. doi: 10.1002/jcp.1041520304. [DOI] [PubMed] [Google Scholar]
- 37.Reggiori F, Monastyrska I, Shintani T, Klionsky DJ. The actin cytoskeleton is required for selective types of autophagy, but not nonspecific autophagy, in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:5843–56. doi: 10.1091/mbc.E05-07-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reggiori F, Shintani T, Nair U, Klionsky DJ. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy. 2005;1:101–9. doi: 10.4161/auto.1.2.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He C, Song H, Yorimitsu T, Monastyrska I, Yen WL, Legakis JE, et al. Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. J Cell Biol. 2006;175:925–35. doi: 10.1083/jcb.200606084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monastyrska I, He C, Geng J, Hoppe AD, Li Z, Klionsky DJ. Arp2 links autophagic machinery with the actin cytoskeleton. Mol Biol Cell. 2008;19:1962–75. doi: 10.1091/mbc.E07-09-0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang HW, Wang YB, Wang SL, Wu MH, Lin SY, Chen GC. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J. 2011;30:636–51. doi: 10.1038/emboj.2010.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Webber JL, Tooze SA. New insights into the function of Atg9. FEBS Lett. 2010;584:1319–26. doi: 10.1016/j.febslet.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 43.Zhu WL, Hossain MS, Guo DY, Liu S, Tong H, Khakpoor A, et al. A role for Rac3 GTPase in the regulation of autophagy. J Biol Chem. 2011;286:35291–8. doi: 10.1074/jbc.M111.280990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Furuta S, Miura K, Copeland T, Shang WH, Oshima A, Kamata T. Light Chain 3 associates with a Sos1 guanine nucleotide exchange factor: its significance in the Sos1-mediated Rac1 signaling leading to membrane ruffling. Oncogene. 2002;21:7060–6. doi: 10.1038/sj.onc.1205790. [DOI] [PubMed] [Google Scholar]
- 45.Zoppino FC, Militello RD, Slavin I, Alvarez C, Colombo MI. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic. 2010;11:1246–61. doi: 10.1111/j.1600-0854.2010.01086.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.