

Abstract
Lysosome-associated membrane protein type 2A (LAMP2A) is a key protein in the chaperone-mediated autophagy (CMA) pathway. LAMP2A helps in lysosomal uptake of modified and oxidatively damaged proteins directly into the lumen of lysosomes for degradation and protein turnover. Elevated expression of LAMP2A was observed in breast tumor tissues of all patients under investigation, suggesting a survival mechanism via CMA and LAMP2A. Reduced expression of the CMA substrates, GAPDH and PKM, was observed in most of the breast tumor tissues when compared with the normal adjacent tissues. Reactive oxygen species (ROS) mediated oxidative stress damages regulatory cellular components such as DNA, proteins and/or lipids. Protein carbonyl content (PCC) is widely used as a measure of total protein oxidation in cells. Ectopic expression of LAMP2A reduces PCC and thereby promotes cell survival during oxidative stress. Furthermore, inhibition of LAMP2A stimulates accumulation of GAPDH, AKT1 phosphorylation, generation of ROS, and induction of cellular apoptosis in breast cancer cells. Doxorubicin, which is a chemotherapeutic drug, often becomes ineffective against tumor cells with time due to chemotherapeutic resistance. Breast cancer cells deficient of LAMP2A demonstrate increased sensitivity to the drug. Thus, inhibiting CMA activity in breast tumor cells can be exploited as a potential therapeutic application in the treatment of breast cancer.
Keywords: PCC, TUNEL, autophagy, CMA, tumor, tissues, doxorubicin
Introduction
Lysosome-associated membrane protein type 2A (LAMP2A) is a key protein in the chaperone-mediated autophagy pathway (CMA). It is a selective autophagy that is responsible for the lysosomal degradation of approximately 30% of the modified and oxidatively damaged soluble cytosolic proteins that contain the recognizable peptide sequence motif (KFERQ). Molecular chaperones, such as HSPA8/HSC70, recognize the oxidatively modified proteins in the cytoplasm, and translocate them to the lysosomal membrane. The damaged proteins then bind to LAMP2A for lysosomal membrane uptake into the lysosomal lumen and are subsequently degraded by lysosomal proteases.1-3 It is well established that modulation of CMA underlies the pathogenesis of certain human diseases such as neurodegenerative disorders, Danon disease,4-7 Mucolipidosis type IV (MCOLN1) lysosomal storage disorder.8 Therefore, there is a growing interest to investigate CMA activity under different physiological and pathological conditions, such as cancer. Alternative splicing of LAMP2 produces three splice variants, LAMP2A, LAMP2B and LAMP2C that are expressed in different tissues.9 LAMP2A and LAMP2B differ in sequence at the extreme C-terminus.10 LAMP2A isoform is highly expressed in placenta, lung and liver but has low expression in the brain and skeletal muscle, and was not reported to be expressed in breast tissues. This manuscript demonstrates the expression of LAMP1, LAMP2A and LAMP2B in both breast tumors and normal adjacent breast tissues. Lysosome-associated membrane protein (LAMP1) is the membrane protein of the same class, which is translocated to endosomes from trans Golgi before being transported to lysosomes upon activation. Both LAMP1 and LAMP2 are highly glycosylated, and are thought to have overlapping functions in tumor cell metastasis.12-15 Levels of LAMP2A are controlled by the dynamic distribution of LAMP2A between the lumen of the lysosome and its membrane.16
Autophagy acts as a double-edged sword. It is a stress response mechanism that protects cancer cells from low nutrient supply or oxidative insults, but in contrast the autophagy process is also involved in the removal of tumor cells by triggering a nonapoptotic/apoptotic cell death program, signifying a negative role in tumor growth.17 Defective or inhibition of autophagy (loss of its prosurvival role) causes DNA damage, and subsequently triggers genomic instability in mammary tumors in vivo.18 CMA is known to be activated due to nutrient deprivation and oxidative stress, and it is essential for the cellular response to stress.19 Recently it was shown that Eps8, which is one of four family members of a family of Actin barbed end capping proteins, was involved in CMA activity in cancer cells,20 but a link between CMA and cancer has yet to be established. While this manuscript was under review, a report was published suggesting the role of CMA in tumor growth.21 Exploring the yet-unknown relationship between altered autophagy and other breast cancer-promoting functions such as upregulation of LAMP2A, may provide valuable insight into the pathogenesis of breast cancer. This manuscript is focused on the role of LAMP2A in regulating CMA in breast tumors and cancer cells during oxidative stress.
Results
Effect of LAMP2A expression on CMA substrates in breast cancer cells
CMA process can be monitored indirectly by investigating the abundance of its targeted proteins such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and M2 isoform of pyruvate kinase (PKM), which are typically not regulated by transcriptional mechanisms and contain a recognizable KFERQ motif.19,22 LAMP2A expression was either upregulated or downregulated in both T47D (Fig. 1) and MCF-7 (Fig. S1) breast adenocarcinoma cells, and lysates from these cells were fractionated on a 4–20% tris glycine gradient SDS PAGE and subjected to immunoblotting. Typical expression of transfected LAMP2A tagged with GFP and control GFP in MCF-7 cells is shown in Figure S1A. Transfection efficiency was reduced with the LAMP2A plasmid compared with the background, and the efficiencies varied from 25–40% in both MCF-7 and T47D cell lines. The expression of the CMA substrates was reduced in the cells where LAMP2A was overexpressed, suggesting an increased CMA activity in treated T47D cells. There was no change in the expression levels of nonCMA substrate such as ACTB in the cultured cells (Fig. 1A). Surprisingly, there was no significant difference in GAPDH/PKM abundance observed between control siRNA-treated cells and LAMP2A knocked down cells (Fig. 1B). Densitometric analyses from three independent experiments for both upregulated (Fig. 1A) and downregulated (Fig. 1B) LAMP2A are shown with standard error of measurements (SEM) on the right of each panel. A similar observation was found with GAPDH in MCF-7 cells (Fig. S1B and S1C).

Figure 1. Effect of LAMP2A expression on known CMA substrates. Proliferating T47D cells were either transfected with LAMP2A or empty pcDNA3 vector for 48 h (A) and with LAMP2A-siRNA or control siRNA for 72 h (B). The cells were then harvested, and protein lysates were fractionated on a 4–20% Tris Glycine SDS PAGE and immunoblotted with indicated antibodies. Densitometric analyses with respect to ACTB from three independent experiments are shown on the right of each panel. Error bars are SEM and asterisk represents a statistically significant difference (*p < 0.05, ANOVA; Tukey test). LAMP2A downregulated T47D cells were either treated with proteasome inhibitor, MG132 (C) or incubated with lysosomal inhibitor, bafilomycin A1 (BAFA1) (D) as described under methods and followed by SDS PAGE and immunoblotting with indicated antibodies. DMF and DMSO were the solvents for the inhibitors and thus serve as controls.
To understand the contribution of other proteolytic pathways, such as ubiquitin proteasome system (UPS) and macroautophagy, T47D cells with downregulated LAMP2A were incubated with either MG132 (ubiquitin proteasome inhibitor)23 (Fig. 1C) or bafilomycin A1 (BAFA1; lysosomal inhibitor)24 (Fig. 1D) to limit the targeted signaling pathways. No significant differential expression of GAPDH or PKM was observed in the LAMP2A downregulated cells compared with controls in either of the blocked pathways. Although it was reported that CMA activity is specific to the cell type, and upregulation of the CMA occurs during the catalytic inhibition of proteasome.25 Similar results were obtained when MCF-7 cells with downregulated LAMP2A were incubated with the above inhibitors (Fig. S1D and S1E) as well as when the same cells were incubated with another macroautophagy inhibitor, 3 methyl adenine (3MA)26 (Fig. S1F).
Turnover of GAPDH in breast cancer cells
A direct approach was applied to measure the association of purified GAPDH with active intact lysosomes isolated in the presence of protease inhibitor from LAMP2A overexpressed and control transfected cells. Lysosomes isolated from LAMP2A overexpressed MCF-7 and T47D cells demonstrated increased lysosomal association of GAPDH, suggesting higher CMA activity when compared with the control cells (Fig. S2). Next, changes in the stability of GAPDH due to the expression levels of LAMP2A in both the breast cancer cell lines were measured via pulse-chase experiment using 35[S] Methionine. The steady-state levels of exogenously expressed LAMP2A revealed elevated degradation of GAPDH after 48 h in MCF-7 cells, whereas it was clearly detected in control pcDNA3 transfected cells at the same time points (Fig. 2A). On the other hand, initial analysis revealed reduced degradation of GAPDH in LAMP2A knockdown T47D cells between 24 and 48 h compared with control knockdown T47D cells (Fig. 2C). There is no significant change in the degradation levels of GAPDH in the control siRNA-treated cells during the same time period. Analysis of GAPDH degradation during LAMP2A overexpression revealed the half-life of GAPDH to be approximately 36.28 ± 2.73 h compared with 69.57 ± 5.24 h in the case of background pcDNA3 transfection (Fig. 2B). Alternatively, analysis of GAPDH degradation during LAMP2A knockdown revealed an increased half-life of 107.9 ± 4.01 compared with control siRNA-treated cells (70.92 ± 2.99) (Fig. 2D). Thus, it indicates a direct involvement of LAMP2A in modulating CMA activity in breast cancer cells. Figure S3 demonstrates the same pulse-chase experiment for GAPDH as with other sets of cells. Analysis of these data revealed that the half life of GAPDH in LAMP2A overexpressed T47D cells, and control transfected cells were 31.87 ± 3.17 and 62.48 ± 4.43, respectively. Alternatively, the half life of GAPDH in LAMP2A siRNA-treated, and control siRNA-treated MCF-7 cells were 111.93 ± 1.73 and 64.85 ± 2.81 respectively. The half lives were calculated from the equations of the trend lines drawn through the data points (shown on the figure).

Figure 2. Effect of LAMP2A expression on the half-life of GAPDH. Proliferating MCF-7 cells were transfected with LAMP2A or pcDNA3 for 24 h (A), and similarly proliferating T47D cells were transfected with LAMP2A siRNA or control siRNA (C). Representative autoradiograms of 35[S]-Methionine labeled GAPDH at the indicated time points from MCF7 (A) and T47D (C) cells are shown following immunoprecipitation with anti-GAPDH antibody in a pulse-chase experiment (see methods). Lanes are labeled according to treatments and the duration of hours after removal of 35[S]-Methionine (chase period). ‘0’ represents initial labeling. Quantifications of the labeled bands from three independent experiments were performed using Image J software and are shown in (B) (MCF-7) and (D) (T47D) with error bars (SD). Linear trend lines were drawn through the time points, and the equations of the lines are shown on the graph.
Expression of LAMP2A is elevated in breast tumors suggesting elevated CMA activity
To understand the physiological significance of LAMP2A in breast cancer; tissues from breast cancer patients, as well as adjacent normal tissues from the same patients were investigated. This was an attempt to document the physiological significance of LAMP2A expression between normal and invasive ductal breast carcinoma tissues in patients that may indirectly reflect the CMA activity in patients. Commercially available OncoPair INSTA-BlotTM that contains breast tumor and adjacent normal tissue lysates from seven different breast cancer patients (P1-P7) was used for this purpose. Figure 3A demonstrates the amido black staining of the blot to determine the total protein loading in each well. This is preferable because many of the common housekeeping genes used for loading controls differ in expression in tumor tissues.27 The description of the loaded samples and lane markings are given in Figure 3B. The blot was probed with specific antibodies as indicated (Fig. 3C). Although a total of 14 μg of tissue lysates were loaded in each well, the expression of the housekeeping genes, ACTB and TUBB, were not consistent. This difference in housekeeping gene expression between tumor tissues and adjacent normal tissues has been well documented.28 Three independent tissue blots were immunoblotted with the indicated antibodies, and densitometric analysis with respect to ACTB expression are given in Figure 3 D–I. Apparently, it appeared that ACTB loading is higher in invasive tissues compared with the normal tissues, but this is not accurate as amido black staining of the same blot indicate relatively equal loading. Thus, densitometric analyses of all the bands with respect to ACTB were needed to equate the differential expression of ACTB between the samples. Analysis of the same immunoblotting data with respect to TUBB results in a similar experimental outcome as ACTB and is shown in Figure S4A–F. Both ACTB and TUBB serve two purposes; first they do not contain the required KFERQ motif to be a CMA substrate, and second they can be used as loading controls for quantification.

Figure 3. Breast tumors express higher levels of LAMP2A. The breast cancer OncoPair INSTA-BlotTM that contains 14 lanes of paired breast tumor, and matched normal adjacent tissue lysates from seven different patient donors was used. (A) Amido black staining of the blot. (B) Table portraying the type, grades, stages of breast tumors, sex and age of the breast cancer patients. (C) Patient tissue blot was immunoblotted with indicated antibodies. (D–I) Protein expression bands (as indicated) were quantified by densitometry, and the levels were normalized to ACTB (arbitrary units). The values are means ± SEMs of at least three independent experiments, and asterisk represents a statistically significant difference between samples (*p < 0.05).
Analysis of the patient tissue immunoblots revealed that both the expression of LAMP2A and HSPA8 (key proteins of CMA pathway) in breast cancer tissue samples were higher than the corresponding normal breast adjacent tissues from the same patient, indirectly suggesting higher CMA activity in tumor tissues (Fig. 3D and E). As LAMP2A is the only focus of this manuscript, close members of LAMP2A such as LAMP2B and LAMP1 were also investigated. LAMP2B and LAMP1 were differentially expressed in the tissue pairs but distinctly different from the LAMP2A expression (Fig. 3F and G). The specificity of the LAMP2A antibody was determined by immunoblotting and is shown in Figure S5. Next, the same tissue blot was further immunoblotted with antibodies against GAPDH and PKM, which are the known substrates of the CMA process. Four patients (P2, P4, P5 and P7) showed a significant reduction in GAPDH expression, in their invasive tumors, compared with normal tissue (Fig. 3H). Similarly, PKM expressions were downregulated in P1, P2, P3, P5 and P7 patient tumors compared with normal adjacent tissues (Fig. 3I). Further studies are needed to understand why some breast tumors do not show CMA activity with high LAMP2A expression.
LAMP2A protects cells from oxidative damage and reduces oxidative modification of cellular proteins in breast cancer cells
ROS are formed during normal metabolism and in higher levels during hazardous environmental exposure as well as under pathological conditions such as cancer. Tumors are always under the influence of oxidative stress rendered by ROS, which might be one of the causes of cancer progression. Expression of LAMP2A in breast tumor tissues prompted an investigation about its role in regulating oxidative stress. T47D and MCF-7 breast adenocarcinoma cells were transiently transfected either with LAMP2A to overexpress LAMP2A or by LAMP2A siRNA to knock down the expression of LAMP2A. Cells were then subjected to various concentrations of H2O2 to induce oxidative stress as described previously.29,30 Cell survivability was measured as described under ‘Methods.’ Overexpression of LAMP2A, in both MCF-7 and T47D cells, induced cell survivability approximately by 15–20%, when compared with background vector and vehicle (Fig. 4A and C). Similarly, underexpression of LAMP2A in the same cells abolishes the protection offered by LAMP2A to around 20–25% when compared with control siRNA and vehicle (Fig. 4B and D). Cell survivability assay with overexpressed LAMP1 and LAMP2B showed little protection against oxidative stress in T47D cells (Fig. 4E).

Figure 4. LAMP2A overexpression stimulates breast cancer cell survival during oxidative stress. Proliferating MCF-7 (A and B) and T47D (C and D) cells were either transfected with LAMP2A, or empty pcDNA3 vector for 48 h (A and C), as well as transfected with LAMP2A siRNA or control siRNA for 72 h (B and D). (E) demonstrates T47D cells overexpressed with LAMP1 and LAMP2B. Treatment with H2O2 was performed in the final 24 h of incubation and cell survival assay was performed as described under methods. Cell viability values are means + SEM of three independent experiments and are normalized to the control (vehicle only) treatment. Significant induction in survival due to LAMP2A and reduction due to LAMP2A siRNA are indicated by an asterisk (*p < 0.05, ANOVA; Tukey test).
ROS also takes part in oxidative damage to cellular proteins, forming protein carbonyls. Protein carbonyl content (PCC) is arguably a well accepted marker of oxidative modification of cellular proteins. Breast cancer cells transfected with either LAMP2A or LAMP2A siRNA were used to investigate the effect of LAMP2A on modulating PCC of the cells in the presence of 150 nM H2O2. PCC was reduced by 50–60% in T47D cells that harbor ectopic expression of LAMP2A when compared with the control pcDNA3 expressing cells (Fig. 5A). Similarly, a 30–40% increase in PCC was observed in LAMP2A knocked down cells (Fig. 5B). Densitometric analyses from three independent experiments, separately with overexpression and underexpression of LAMP2A are given in Figure 5C and D along with the SEM. Western blot showing the expression of LAMP2A is given in Figure 5E and F. MCF-7 cells also show similar results and they are shown in Figure S6.
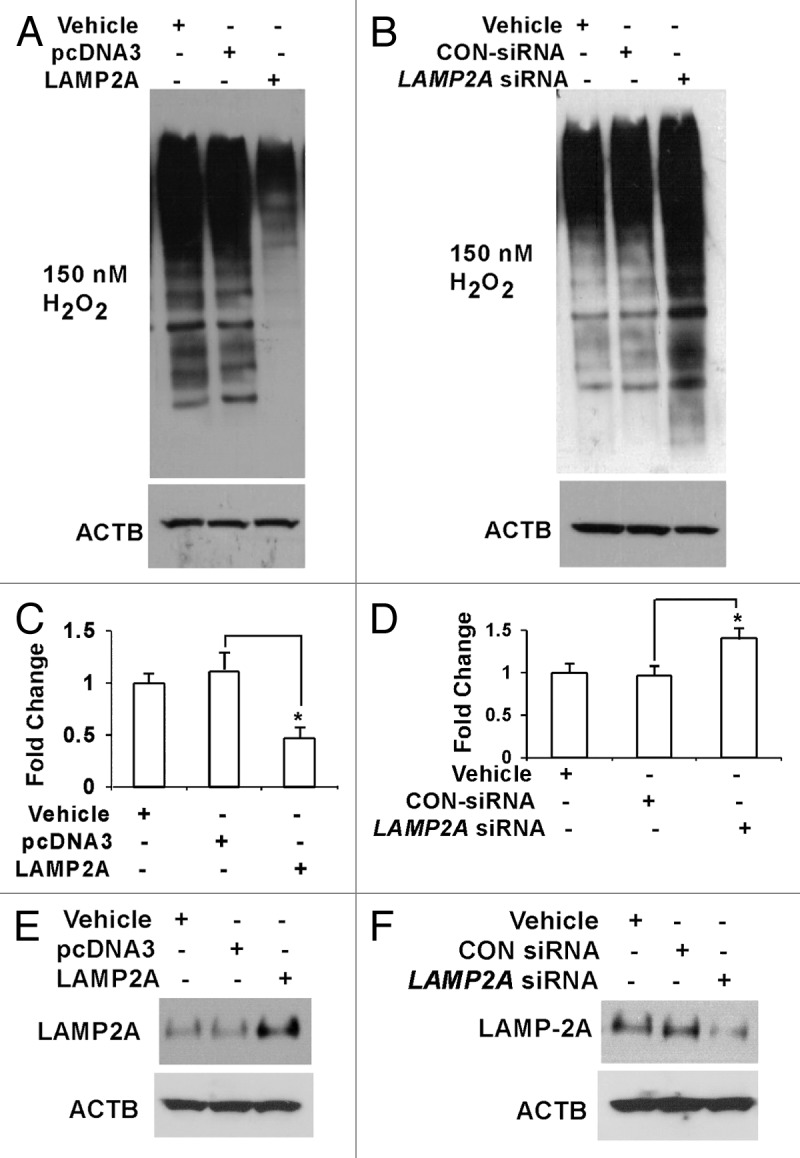
Figure 5. Ectopic expression of LAMP2A inhibits protein carbonyl content (PCC) formation. Proliferating T47D cells were transiently transfected with LAMP2A or empty pcDNA3 vector for 48 h (A and C) as well as transfected with LAMP2A siRNA or control siRNA for 72 h (B and D). Cytosolic extracts were prepared; and 50 μg aliquots were tested for their PCC. (C and D) represent the quantitative estimation of PCC, expressed as means ± SEMs of three independent experiments. Significant changes in the PCC contents are indicated by an asterisk (*p < 0.05 ANOVA; Tukey test). Western blots showing the protein levels of LAMP2A and ACTB in different transfection conditions are provided in (E and F).
LAMP2A modulates PtdIns3K/AKT1 signaling pathway
PtdIns3K/AKT1 and NFKB1 signaling pathways have been previously implicated in various types of tumors including breast, prostate, ovarian and thyroid cancer,31,32 as well as in proteolysis.33-35 Initial studies with MCF-7 cells with either upregulated or downregulated LAMP2A protein expression did not show any differential expression of the NFKB1 signaling pathway proteins (NFKB1A and RELA-NFKB1) in the total cellular extracts between the experimental and control cells (Fig. S7A and S7B). Next, the LAMP2A upregulated cells were subjected to subcellular fractionation, and both the cytoplasmic and the nuclear fraction were fractionated in SDS PAGE followed by immunoblotting with the RELA-NFKB1 antibody. As observed in the above experiments, LAMP2A overexpression in MCF-7 cells did not show any differential expression of RELA-NFKB1 in both cytoplasm and nuclear fraction (Fig. S7C). Ectopic expression of LAMP2A in T47D cells, however, demonstrated reduced expression of RELA-NFkB1 in the cytoplasmic fraction of the LAMP2A overexpressed cells and simultaneously there was an increase in the expression of the same in the nuclear fraction (Fig. S7D). The samples were then subjected to a more sensitive and direct approach to investigate the activation of NFKB1 via pNFKB1-MetLuc luciferase assay kit (Clonetech, 631742). LAMP2A overexpressed T47D cells significantly induced, whereas LAMP2A-siRNA treated cells significantly decreased the NFKB1 luciferase activity suggesting that LAMP2A could modulate NFKB1 signaling pathway in T47D cells (Fig. S7E and S7F). It is possible that due to existing high activity of NFKB1 signaling in MCF-7 cells, LAMP2A failed to produce any significant differences in the activity.
Next, the potential contribution of AKT1 pathway was investigated with the above experimental cells. T47D cells overexpressed with LAMP2A, showed reduced expression of the phosphorylated form of AKT1 [pAKT1 (Ser473)] (Fig. 6A), whereas an increased expression of pAKT1 (Ser473) was observed in LAMP2A knocked down cells (Fig. 6B). Addition of 0.5 μM of wortmannin, which inhibits AKT1 activation, abolished the induction of pAKT1 in LAMP2A knocked down cells (Fig. 6B). In all cases, there was no change in total AKT1 expression (Fig. 6A and B, bottom). An essential function of AKT1 is the regulation of glycogen synthesis through phosphorylation and inactivation of GSK3Α and GSK3B. This property of AKT1 has been utilized, and immobilized phospho-AKT1 (Ser473) monoclonal antibody was used to immunoprecipitate AKT1 from all the sample extracts. Then, an in vitro kinase assay was performed using GSK3 fusion protein as a substrate and measured the phosphorylation of GSK3 by western blot using p-GSK3Α/Β (Ser21/9) antibody. AKT1 activation was inhibited in LAMP2A overexpressed cells as demonstrated by downregulation of GSK3 expression (Fig. 6C). Alternatively, the expression of GSK3 was higher in the cells when LAMP2A was downregulated, indicating activation of AKT1 signaling pathway (Fig. 6D). Similar results were obtained when cells were treated with H2O2, to induce oxidative stress, followed by AKT1 kinase assay (Figs. 6E and F). Densitometric analyses from three independent pGSK3 expressions are shown in Figure 6E and F (bottom). Similar results were obtained with MCF-7 cells, which are shown in Figure S8. CMA is known to be activated during starvation. To understand whether starvation leads to the AKT1 activation or not, both MCF-7 and T47D cells were starved as described under ‘Methods’ followed by immunoblotting with the indicated antibodies (Fig. 6G and H). There was no significant change in the expression of the phosphorylated form of AKT1 [pAKT1 (Ser473)] in the starved cells, when compared with control. Additionally, there was no change in the total AKT1 expression suggesting that starvation might not lead to the activation of AKT1 signaling.

Figure 6. Knockdown of LAMP2A triggers AKT1 phosphorylation at Ser473. Proliferating T47D cells were either transfected with LAMP2A, or empty pcDNA3 vector for 48 h (A, C and E) and with LAMP2A siRNA or control siRNA for 72 h (B, D and F). (A and B) showing western blots analysis of total AKT1 and phospho AKT1 (Ser 473). (C and D) demonstrate AKT1 kinase activity using AKT1 substrate GSK3 fusion protein as a measure of AKT1 activation. (E and F) demonstrate the AKT1 substrate assay under oxidative stress condition rendered by 150 nM H2O2. Densitometric analyses from (E and F) are given under each panel. Data are mean ± SEMs, and significant changes are indicated by an asterisk (*p < 0.05 ANOVA; Tukey test). “#” represents the sample lanes that were treated with 1 μM Wortmannin. (G and H) Starvation does not promote AKT1 phosphorylation. Both MCF-7 and T47D cells were starved as described under methods and the cells were harvested after 48 h followed by SDS PAGE, and immunoblotted with indicated antibodies.
Inhibition of LAMP2A triggers ROS generation and promotes apoptosis in breast cancer cells
Proliferating T47D cells were transfected with the indicated vector(s), exposed to H2O2 (150 nM) for 24 h, and assayed for carboxy-DCF fluorescence, which is a ROS indicator, by flow cytometry. We previously reported that transfection with empty pcDNA3 vector increased the carboxy-DCF fluorescence, indicating that transfection itself causes oxidative stress.29 A reduction in generation of ROS due to LAMP2A overexpression in T47D cells was observed (Fig. 7A), whereas an increase in ROS production was seen in cells where LAMP2A had been knocked down (Fig. 7B). Analyses of ROS using the same dye in live T47D cells via confocal microscopy demonstrate similar effects. Uptake of carboxy-DCF appeared to be low in LAMP2A overexpressed cells compared with pcDNA3 transfected cells (Fig. S9A; Carboxy-DCF; green), whereas the dye uptake was increased in LAMP2A downregulated cells compared with control siRNA-treated cells (Fig. S9B; Carboxy-DCF; green). Mitochondria is a source of ROS generation, so all the treated cells were simultaneously stained with MitoTracker dye [CMXRos (Invitrogen)] to visualize mitochondria (Fig. S9A and S9B; mitochondria; red) and the merged pictographs showing the extent of overlapping between green and red fluorescence are shown in Figure S9A and S9B (merged). Quantitative measurements of green fluorescence (Carboxy-DCF) in our experimental samples were analyzed by Metamorph imaging software and represented as bar diagrams (Fig. 7C and D).

Figure 7. LAMP2A modulates ROS generation in breast cancer cells. Proliferating T47D cells were either transfected with LAMP2A or empty pcDNA3 vector for 48 h (A and C) and with LAMP2A siRNA or control siRNA for 72 h (B and D) in the presence of 150 nm H2O2 for the last 24 h. (A and B) Cells were assayed for carboxy-DCF fluorescence by flow cytometry, and the total geometric means of the carboxy-DCF tagged cells were plotted in each case. (C and D) Cellular uptakes of carboxy-DCF by the indicated cells were imaged live using confocal microscopy (Fig. S9). The green fluorescence due to carboxy DCF among different samples was analyzed using Metamorph imaging software and represented as bar diagram. Error bars are SEM and asterisk represents a statistically significant difference (*p < 0.05, ANOVA; Tukey test).
To check the apoptotic status, flow cytometry-based TUNEL apoptosis assay was performed with the above samples that were treated with or without H2O2. The results illustrate that apoptosis was inhibited in LAMP2A overexpressed cells when compared with control transfections (Fig. 8A), suggesting that LAMP2A was able to rescue the cells from the deleterious influence of oxidative stress and thereby stimulate cell viability and promote tumorigenesis. On the other hand, when LAMP2A was downregulated, this inhibition was lost, and elevated levels of apoptosis were observed in the cells compared with the controls (Fig. 8B). Further investigations with some key proteins in the apoptosis pathway, such as CASP3 (activator protein in apoptosis), BCL2 (anti-apoptotic protein) and BAX (pro-apoptotic protein) were performed with the LAMP2A manipulated cells. No changes in the expression of total CASP3 were observed in both LAMP2A over and underexpressed cells (Fig. 8C and D). Activation of CASP3 requires proteolytic processing of its inactive zymogen into activated p17 and p12 fragments. Immunoblotting the same membrane with antibody specific to cleaved 17/19 fragment of CASP3 revealed that LAMP2A overexpressed cells had reduced cleaved CASP3 fragments (Fig. 8C), whereas LAMP2A downregulated cells have elevated levels of the same (Fig. 8D). Moreover, downregulation of BAX and simultaneous upregulation of BCL2 was observed when LAMP2A was overexpressed in T47D cells (Fig. 8E). The opposite phenomenon was observed when LAMP2A was knocked down by LAMP2A siRNA in the same T47D cells (Fig. 8F). Densitometric analysis from three or more experiments is given in Figure 8G and H along with SEM demonstrating modulation of apoptotic proteins by LAMP2A.
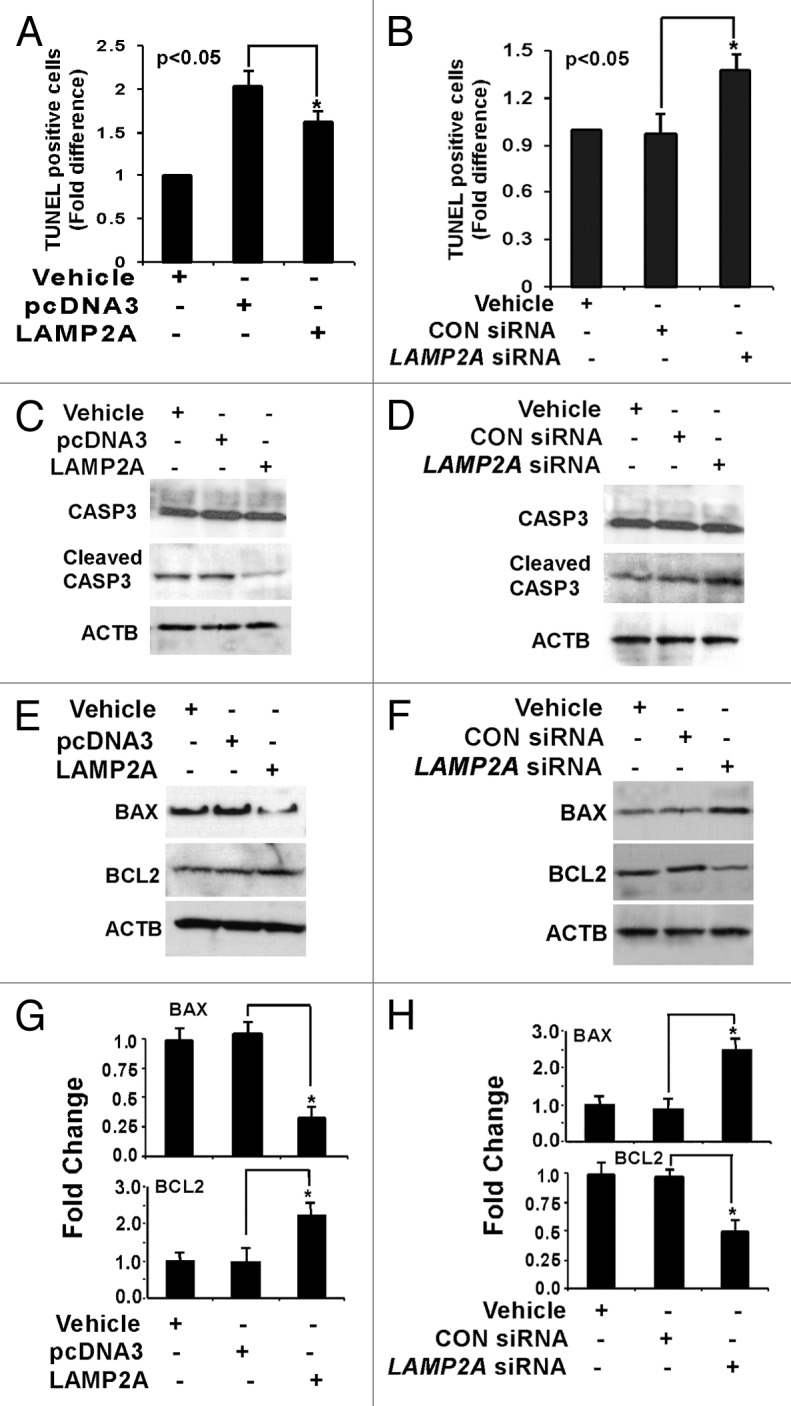
Figure 8. Downregulation of LAMP2A triggers apoptosis in breast cancer cells. Proliferating T47D cells were either transfected with LAMP2A or empty pcDNA3 vector for 48 h or transfected with LAMP2A siRNA or control siRNA for 72 h followed by incubation with 150 nm H2O2 for the last 24 h. The cells were then assayed using Apo-Direct apoptosis kit. Percentage of FITC positive cells from both LAMP2A overexpressed (A) and underexpressed (B) samples were represented as a bar diagram. (C and D) demonstrate immunoblotting with CASP3 and cleaved CASP3 fragments in the indicated samples. Protein expression levels of BAX and BCL2 in LAMP2A overexpressed cells, as well as LAMP2A knock down cells, are shown in (E and F). The densitometric analyses of the indicated proteins with respect to ACTB from three independent experiments are shown as bar diagram in (G and H). Values are means ± SEMs from three independent experiments. Significant changes are indicated by an asterisk (*p < 0.05 ANOVA; Tukey test).
Inhibition of LAMP2A sensitizes tumor cells against doxorubicin
Doxorubicin is a cancer chemotherapeutic drug, which is also known as Adriamycin. It is widely used to treat early stage, node positive, HER2 positive and metastatic breast cancer.36 The drug intercalates the DNA of the cancer cell and prevents replication and protein synthesis. As shown previously, inhibition of LAMP2A induces ROS and apoptosis; thus the combined effect of LAMP2A inhibition and doxorubicin treatment could be beneficial to treat breast cancer patients. This hypothesis was tested with both MCF-7 and T47D cells where LAMP2A was downregulated using LAMP2A siRNA. Cells were treated with or without doxorubicin and cell survivability assay was performed. Cells downregulated for LAMP2A expression induced 15–20% of cell death in both MCF-7 (Fig. 9A) and T47D (Fig. 9C) in the absence of doxorubicin treatments. The same LAMP2A downregulated cells when treated with 2.5 μM of doxorubicin for 24 h revealed enhanced cell killing in both MCF-7 (Fig. 9B) and T47D cells (Fig. 9D). Parental cells without doxorubicin treatment were considered as 100% survival. These data suggest that treatment with DNA damaging drugs in combination with inhibition of CMA may increase treatment efficacy in breast cancer patients.
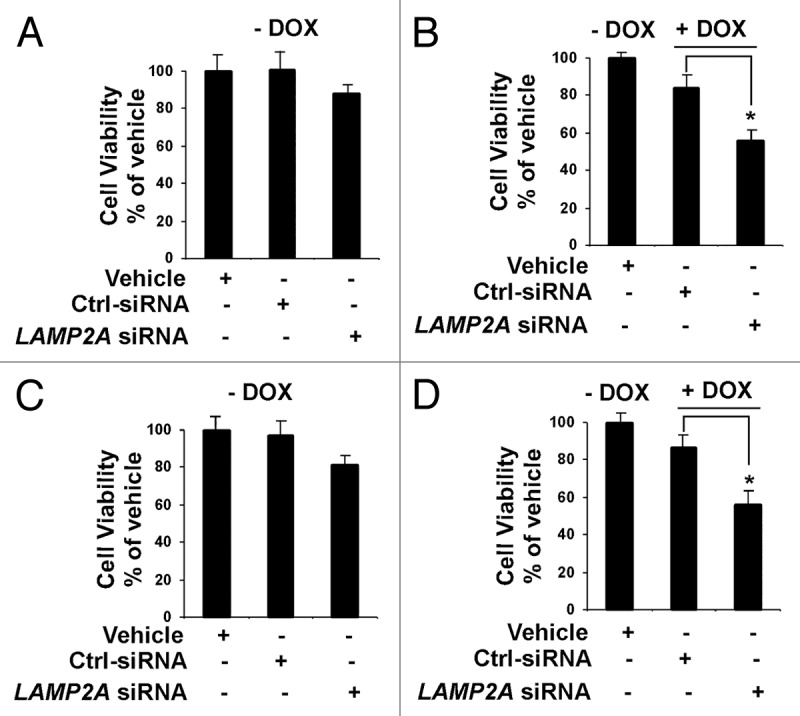
Figure 9. Inhibition of LAMP2A sensitizes breast tumor cells to DNA damaging drug. Proliferating MCF-7 cells (A and B) and T47D cells (C and D) were transfected with LAMP2A siRNA or control siRNA for 72 h and treated/untreated with 2.5 μM doxorubicin for the last 24 h of transfections. The cells were then subjected to cell survivability assay as described under methods. – DOX indicates no doxorubicin treatments where as + DOX indicates cells treated with doxorubicin. The DOX treatments were compared with no DOX treatment in (B) for MCF-7 cells and (D) for T47D cells. Error bars representing SEM and significant changes is indicated by an asterisk (*p < 0.05 ANOVA; Tukey test).
Discussion
During starvation, growth factor deprivation, and under metabolic/oxidative stress, the process of autophagy induces survival mechanisms37,38 that protect tumor cells against cellular death.39 The latest developments in the study of autophagy suggest that it might play a crucial role in apoptosis, but the cell death-promoting conditions of autophagy vs. cell survivability are still unknown.40 Studies with macroautophagy-deficient mouse mammary epithelial cells exhibit increased DNA damage in response to oxidative stress,18 raising the possibility that breast tumor cells with autophagy defects may be particularly sensitive to DNA damaging agents. Moreover, it was shown previously that when macroautophagy was blocked in cells, CMA activity was induced, which leads to the survival of the cells.41 When both macroautophagy and CMA are blocked in breast cancer cells, tumor cells can be sensitized to DNA damaging agents like cisplatin, carboplatin or doxorubicin. This strategy has been used with BRCA1 mutant cells that have a defective DNA repair system. These cells exhibit exquisite sensitivity to certain DNA damaging agents, as well as to agents interfering with DNA damage repair, such as poly (ADP-ribose) polymerase (PARP) inhibitors.42 Thus, CMA-deficient breast tumor cells might also exhibit high sensitivity to agents causing DNA damage or responsive to drugs that inhibit DNA repair system. My work has demonstrated that doxorubicin effectively sensitize LAMP2A downregulated cells than the drug alone treatment to the parental cells. Thus, inhibiting CMA activity in breast tumor cells can be exploited as a potential therapeutic application in the treatment of breast cancer.
One of the hallmarks of cancer cells is to adopt survival mechanisms by avoiding apoptosis, and continuing cell proliferation. This manuscript demonstrates that inhibition of CMA activity, promotes the generation of ROS, and induces apoptosis in breast cancer cells. Thus, treatment of these kinds of breast tumors with a CMA inhibitor or a LAMP2A inhibitor might sensitize tumor cells to a variety of anticancer agents. A similar approach has already been established in a Myc-induced lymphoma model in which inhibition of autophagy with either chloroquine or ATG5 short hairpin RNA (shRNA) enhanced the ability of an alkylating drug to stimulate cancer cell death.43 Thus, concurrent inhibition of autophagy and reactivation of apoptosis might provide a more competent way to treat breast cancer patients.
Pathologic importance of LAMP2A is well established in aging and neurodegenerative diseases, but a link between cancer and CMA is not yet thoroughly established. The data obtained from the patient tissues demonstrate the physiological importance of this work. All the breast cancer tissues that were under investigation showed increased expression of LAMP2A and HSPA8 and some of them show reduced expression of the CMA substrates (GAPDH and PKM). LAMP2A and HSPA8 are the key components of CMA, so apparently this study indicates that breast cancer tissues have high CMA activity that leads to cancer cell survival. To understand the molecular mechanism of survival, a similar situation was created by transfecting LAMP2A in both MCF-7 and T47D breast cancer cells. Expression of GAPDH was reduced in LAMP2A overexpressed cells, but there was no significant accumulation of GAPDH in LAMP2A knockdown cells.
The development of breast carcinogenesis from in situ malignancy to metastatic stage requires changes in signaling pathways that foster enhanced tumor cell survival and cell motility. A remarkable increase in AKT1 kinase activity has been found in nearly 30–40% of the breast cancer patients.44,45 Studies with 252 human breast carcinoma cases showed that 33.3% of the tissues exhibited upregulated pAKT1 expression.46 Higher AKT1 activity has been reported to inhibit apoptosis, but results in this manuscript with the MCF-7 and T47D cells demonstrated that LAMP2A inhibition in breast cancer cells triggers AKT1 activation that promotes ROS generation and induces apoptosis. The most likely explanation is that the tumor tissues that are under investigation fall beyond the 30–40% range, where the AKT1 activation could be neutral or reduced. More research is needed to understand the mechanisms.
Furthermore, it was also shown that oxidative stress induced by H2O2 in cardiac myocytes and vascular smooth muscle cells induces the AKT1 signaling pathway.47,48 Recent studies suggest that under normal conditions, AKT1 increases intracellular ROS levels by stimulating oxidative metabolism in the mitochondria and by repressing FOXO phosphorylation. Studies also reveal that when AKT1 signaling was downregulated, ROS levels were reduced due to both decreased mitochondrial activity and increased FOXO-mediated expression of antioxidant enzymes.49 Conversely, AKT1 hyperactivation, which occurs in several cancer cell types, raises metabolic activities in the mitochondria and inhibits FOXO transcriptional activity, resulting in highly increased ROS levels.50 In conclusion, experiments with breast tumor tissues indirectly indicated an increased activity of CMA pathway that probably contributes to cell survival. Identification of a LAMP2A inhibitor and other strategies that might lead to reduced CMA activity remain to be investigated, which could have remarkable prognostic and therapeutic implications for breast cancer patients.
Materials and Methods
Patient tissue blot
OncoPair INSTA-BlotTM was purchased from Imgenex (Imgenex, IMB-130a), which was a ready-to-use PVDF western blot membrane containing denatured protein lysates from paired breast tumor and matched normal adjacent tissue lysates from 7 patient donors. Approximately 14 μg/lane of protein was loaded in each well. The blot was stained with Amido Black to visualize sample loadings. The blot was then immunoblotted with indicated antibodies. At least three immunoblotting experiments were performed to statistically analyze the experimental findings.
Cell lines
MCF-7 and T47D human mammary adenocarcinoma cell lines were obtained from the American Type Culture Collection (ATCC, HTB-22, HTB-133) and cultured as described previously.30 MCF-7 cells were isolated from a 69-y-old Caucasian woman suffering from invasive breast ductal carcinoma. This epithelial lineage cell line has wild-type TP53, posseses both estrogen and progesterone receptors and is only tumorigenic in mice if supplemented with estrogen. Similarly, T47D cell line is also isolated from 54-y-old woman with an infiltrating ductal breast carcinoma. This epithelial origin cell line also has receptors to 17 β-estradiol, other steroids and calcitonin, and expresses mutant TP53 protein. Both the cell lines were cultured in complete DMEM medium containing 10% FBS, 1% Penn/Strep, 1% nonessential amino acids. Starvation experiments with these cell lines were performed in complete DMEM medium containing 0.5% FBS for 24 h before being harvested for downstream processes.
Expression vectors and reagents
LAMP2A expression plasmid was purchased from ORIGENE, (SC118738) that harbors a full length open reading frame (ORF) and driven by the cytomegalovirus (CMV) promoter. Dimethylsulfoxide (DMSO), H2O2, β-mercaptoethanol and all the other chemicals were obtained from the Sigma Chemical Co., unless otherwise stated.
Transient transfections and treatments
Transient transfections were done as described previously.30,51-53 For overexpression, cells were transfected with LAMP2A plasmid (tagged with or without GFP) for 48 h before being harvested. In contrast, for underexpression of LAMP2A using LAMP2A siRNA, cells were harvested after 72 h post-transfection and utilized in downstream experiments. The following siRNAs were used in this study: control-siRNA [ON-TARGET plus nontargeting siRNA (Dharmacon, D-001810-01)] and LAMP2A siRNA custom made from Dharmacon (5′→ 3′CTGCAATCTGATTGATTATT). Indicated concentrations of H2O2 were used to induce oxidative stress in the cells in medium with 0.5% FBS for the final 24 h at 37°C. Treatments with the autophagy inhibitors, 3 methyl adenine (final concentration 10 mM) and bafilomycin A1 (final concentration 10 nM) were performed for the final 24 h post-transfection in each case. Both the inhibitors were from Sigma Aldrich (M9281 and B1793, respectively). Proteasome inhibitor, MG132 (final concentration 10 μM), was added to the cells in the final 3 h of post-transfection (Sigma, C2211). Doxorubicin treatment (final concentration 2.5 μM) was performed for the final 24 h of transfection (EMD Biosciences, 324380). In each case, the cells were harvested following the treatments and subjected to immunoblotting with indicated antibodies.
Western blotting
Whole cell lysates from transfected/nontransfected cells were prepared after the indicated treatments as described previously.29,30 The primary antibodies were as follows: LAMP2A (Abcam, ab18528, 1 μg/ml); LAMP2B, (Abcam, ab18529 1 μg/ml); LAMP1 (Cell Signaling, C54H11, 1:1000); HSPA8 (Santa Cruz Biotechnology, sc1059, 1:200); CASP3 (Imgenex, IMG-144A, 5 μg/ml); BAX (Santa Cruz Biotechnology, sc526, 1:200); BCL2 (Santa Cruz Biotechnology, sc-7382, 1:200); GAPDH (Imgenex, IMG5143A, 1 μg/ml); PKM (Cell Signaling, 3186, 1:1000); AKT1 (Cell Signaling, 9272, 1:1000); phospho-AKT1 (ser473) (Cell Signaling, 4051S, 1:1000); TUBB (Imgenex, IMG-5810A, 1 μg/ml) and ACTB (Imgenex, IMG-5142A, 1 μg/ml).
Pulse-chase analysis of GAPDH
MCF-7 and T47D cells were first transfected with either LAMP2A or LAMP2A-siRNA as stated above. Twenty-four hours post-transfection, cells were washed twice with prewarmed 1x PBS and were then starved by incubating with methionine-free DMEM medium containing 10% FBS for 1 h at 5% CO2/37°C. The cells were then pulsed with 35[S]-Methionine (50 μCi) (Perkin Elmer, EasyTag™ L-[35S]-Methionine Waltham, MA) for 4 h at 5% CO2/37°C. At the end of the incubation period, the medium was removed, and the cells were washed 3 times with prewarmed 1x PBS supplemented with 5 mM cold Methionine. The radioactivity incorporated into the cellular proteins was chased for 0–48 h following the addition of DMEM containing 10% FBS and nonradioactive methionine (5 mM). At each indicated time interval, cellular lysates were prepared and subjected to immunoprecipitation using the GAPDH antibody (Santa Cruz Biotechnology, sc-25778). 35[S]-Methionine labeled GAPDH protein from different samples was then fractionated in a 4–20% SDS-PAGE. The gel was then dried for 60 min at 60°C, and exposed for autoradiography. The radioactivity of the individual bands was then quantified using Image-J, Version 1.44 (NIH) and plotted as shown.
Protein carbonyl content measurements
Protein carbonyl content (PCC) was measured using assay kit (Cell Biolabs, STA-308) as per manufacturer instructions. Briefly, cytosolic cell lysates were prepared via sonication, and were electrophoresced with reducing SDS sample buffer followed by transfer to a PVDF membrane. The samples were then derivatized on the membrane, followed by incubation with 1x DNPH (2, 4 dinitrophenyl hydrazine) and finally washed with 2N HCl and 100% methanol. This was followed by immunoblotting with primary antibody; rabbit anti-DNP Antibody (1:1000) and subsequently by secondary antibody, Goat Anti-Rabbit IgG, HRP-conjugate, (1:1000) and visualized using the enhanced chemiluminescence detection system (Santa Cruz Biotechnology, sc-2048).
Cell viability assay
Cell survivability following oxidative stress and doxorubicin treatments were performed using CellTiter-Glo luminescent cell viability assay from Promega (G7570). Briefly, cells were first plated in a 24-well tissue culture dish. Next the cells were either overexpressed by transfection with LAMP2A expression vector or underexpressed by transfection with LAMP2A siRNA. Forty-eight hours post-transfection, one volume of CellTiter-Glo reagent was added to one volume of cell culture medium containing treated cells. The contents were mixed for 2 min on an orbital shaker to induce cellular lysis followed by incubation at room temperature for 10 min to stabilize the signal. The supernatants from different samples were transferred to 96-well opaque-walled multiwell plates, and the luminescence was recorded immediately.
Determination of AKT1 kinase activity
AKT1 kinase activity was determined using a nonradioactive IP-AKT1 Kinase Assay Kit (Cell Signaling, 9840) according to the manufacturer’s protocol. The assay system uses two different antibodies, one to selectively precipitate AKT1, and the second one is to detect AKT1-induced phosphorylation of GSK3. Using the bead immobilized AKT1 antibody, AKT1 kinase was selectively immunoprecipitated from whole-cell lysates (300 μg/sample) prepared from untreated cells and cells treated with either H2O2 and/or wortmannin (Cell Signaling, 9951). Immunocomplexes of AKT1 kinase were incubated with 1 μg of GSK3 fusion protein in the presence of 100 μM ATP and 1x kinase buffer provided in the kit for 30 min at room temperature. An SDS-loading buffer was added to terminate the kinase reaction. Samples were boiled at 90°C for 5 min, and then resolved via SDS–PAGE. GSK3 phosphorylation was detected with an anti-phospho-GSK3Α/Β (Ser21/9) antibody (Cell Signaling, 9331, 1:1000).
Determination of intracellular ROS levels
Intracellular reactive oxygen species (ROS) levels were monitored by a fluorescent dye 2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA) (Molecular Probes, D-399) as described before.29 Briefly, fixed or live cells were incubated with H2DCFDA (10 µM) for 60 min at 37°C in phenol free DMEM, and immediately analyzed via flow cytometry (FACS) or Olympus Fluoview-FV300 Laser Scanning Confocal System. The geometric means were obtained from FACS via FCS Express Version 3 software for analysis of FACS data (DeNovo Software) and were represented as a bar graph. Image-iT- LIVE Green Reactive Oxygen Species Detection Kit (Invitrogen, I36007) was used to measure ROS in live cells. MitoTracker Red CMXRos (Invitrogen, M7512) (50 nM for the last 30 min) was also used to stain mitochondria followed by incubation with Hoechst 33342 (Invitrogen, H3570) to stain the nuclei. ROS fluorescence (green) was quantified using Metamorph image analysis software (Molecular Devices) and expressed as means ± SEM (arbitrary units) of three independent experiments.
TUNEL assay for apoptosis
APO-DIRECT from Phoenix Flow Systems (Part No-AD-1001) was used to assess apoptosis in the transfected/nontransfected cells as per manufacturer’s instructions. The apoptosis parameter was measured with the green fluorescence channel (fluorescein isothiocyanate deoxyuridine triphosphate (FITC-dUTP); 520 nm), and the DNA content was measured with red fluorescence channel (propidium iodide/RNase; 623 nm). Cells were fixed using 1% (w/v) paraformaldehyde in PBS. The cells were then permeabilized by cold 70% ethanol (v/v) for 12 h in the freezer and then incubated with 50 μl of DNA labeling solution for 1 h at 37°C. The cells were then resuspended in 0.5 ml of the propidium iodide/RNase A solution and analyzed immediately by flow cytometry. Percentage of FITC positive cells from different samples were plotted as a bar diagram, and three independent experiments were performed to obtain variability and statistical implications.
Statistical methods
Statistical comparisons were made using one-way analysis of variance (ANOVA) via Tukey Test using Sigmastat statistical software (Ver 3 for Windows). Data are presented as arithmetic means with error bars representing standard error of the mean (SEM) calculated from at least three independent experiments. A value of p < 0.05 or less was considered significant for all experiments.
Supplementary Material
Acknowledgments
T.S. is grateful to the American Cancer Society (IRG #97-152-16-2) and the Fisher Center for Familial Cancer Center, Georgetown University, for financial support. T.S. would like to thank Prof. Eliot Rosen for laboratory space and Prof. Peter Shields, Prof. Stephen Byers, Dr. Michael Johnson and Prof. Robert Clark of Lombardi Cancer Center, Georgetown University and Dr. Melvin L. DePamphilis of NICHD, NIH for their continuing support. T.S. is grateful to Drs. Rabin Roy and Sujata Choudhury of Oncology, Lombardi Cancer Center, Georgetown University for helping out in activating the radioactive permit for pulse-chase experiment in their laboratory. Lastly T.S. would like to extend sincere thanks to Dr. Sushil Rane, principal investigator from NIDDK, NIH, who provided all the initial materials to execute the initial ideas. T.S. also acknowledges the support from microscopy and imaging shared resources, flow cytometry and cell sorting shared resources and tissue culture shared resource at Lombardi Comprehensive Cancer Center. T.S. thanks Dr. Asha Acharya, Dr. Vikas Rishi and Mrs. Jordan Woodrick for carefully editing this manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/21654
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/21654
References
- 1.Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2435–44. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Massey A, Kiffin R, Cuervo AM. Pathophysiology of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2420–34. doi: 10.1016/j.biocel.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Zhou D, Li P, Lin Y, Lott JM, Hislop AD, Canaday DH, et al. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. 2005;22:571–81. doi: 10.1016/j.immuni.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Di Blasi C, Jarre L, Blasevich F, Dassi P, Mora M. Danon disease: a novel LAMP2 mutation affecting the pre-mRNA splicing and causing aberrant transcripts and partial protein expression. Neuromuscul Disord. 2008;18:962–6. doi: 10.1016/j.nmd.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Massey AC, Zhang C, Cuervo AM. Chaperone-mediated autophagy in aging and disease. Curr Top Dev Biol. 2006;73:205–35. doi: 10.1016/S0070-2153(05)73007-6. [DOI] [PubMed] [Google Scholar]
- 6.Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci. 2006;29:528–35. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Saftig P, Tanaka Y, Lüllmann-Rauch R, von Figura K. Disease model: LAMP-2 enlightens Danon disease. Trends Mol Med. 2001;7:37–9. doi: 10.1016/S1471-4914(00)01868-2. [DOI] [PubMed] [Google Scholar]
- 8.Venugopal B, Mesires NT, Kennedy JC, Curcio-Morelli C, Laplante JM, Dice JF, et al. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J Cell Physiol. 2009;219:344–53. doi: 10.1002/jcp.21676. [DOI] [PubMed] [Google Scholar]
- 9.Eskelinen EL, Cuervo AM, Taylor MR, Nishino I, Blum JS, Dice JF, et al. Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP-2. Traffic. 2005;6:1058–61. doi: 10.1111/j.1600-0854.2005.00337.x. [DOI] [PubMed] [Google Scholar]
- 10.Gough NR, Fambrough DM. Different steady state subcellular distributions of the three splice variants of lysosome-associated membrane protein LAMP-2 are determined largely by the COOH-terminal amino acid residue. J Cell Biol. 1997;137:1161–9. doi: 10.1083/jcb.137.5.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cook NR, Row PE, Davidson HW. Lysosome associated membrane protein 1 (Lamp1) traffics directly from the TGN to early endosomes. Traffic. 2004;5:685–99. doi: 10.1111/j.1600-0854.2004.00212.x. [DOI] [PubMed] [Google Scholar]
- 12.Furuta K, Ikeda M, Nakayama Y, Nakamura K, Tanaka M, Hamasaki N, et al. Expression of lysosome-associated membrane proteins in human colorectal neoplasms and inflammatory diseases. Am J Pathol. 2001;159:449–55. doi: 10.1016/S0002-9440(10)61716-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. 2006;27:495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Eskelinen EL, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N, et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell. 2004;15:3132–45. doi: 10.1091/mbc.E04-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saftig P, Beertsen W, Eskelinen EL. LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy. 2008;4:510–2. doi: 10.4161/auto.5724. [DOI] [PubMed] [Google Scholar]
- 16.Cuervo AM, Dice JF. Regulation of lamp2a levels in the lysosomal membrane. Traffic. 2000;1:570–83. doi: 10.1034/j.1600-0854.2000.010707.x. [DOI] [PubMed] [Google Scholar]
- 17.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–16. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–35. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829–40. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Welsch T, Younsi A, Disanza A, Rodriguez JA, Cuervo AM, Scita G, et al. Eps8 is recruited to lysosomes and subjected to chaperone-mediated autophagy in cancer cells. Exp Cell Res. 2010;316:1914–24. doi: 10.1016/j.yexcr.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, et al. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. 2011;3:ra117. doi: 10.1126/scitranslmed.3003182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sooparb S, Price SR, Shaoguang J, Franch HA. Suppression of chaperone-mediated autophagy in the renal cortex during acute diabetes mellitus. Kidney Int. 2004;65:2135–44. doi: 10.1111/j.1523-1755.2004.00639.x. [DOI] [PubMed] [Google Scholar]
- 23.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/S0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 24.Shacka JJ, Klocke BJ, Roth KA. Autophagy, bafilomycin and cell death: the “a-B-cs” of plecomacrolide-induced neuroprotection. Autophagy. 2006;2:228–30. doi: 10.4161/auto.2703. [DOI] [PubMed] [Google Scholar]
- 25.Koga H, Martinez-Vicente M, Macian F, Verkhusha VV, Cuervo AM. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun. 2011;2:386. doi: 10.1038/ncomms1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stroikin Y, Dalen H, Lööf S, Terman A. Inhibition of autophagy with 3-methyladenine results in impaired turnover of lysosomes and accumulation of lipofuscin-like material. Eur J Cell Biol. 2004;83:583–90. doi: 10.1078/0171-9335-00433. [DOI] [PubMed] [Google Scholar]
- 27.Aldridge GM, Podrebarac DM, Greenough WT, Weiler IJ. The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J Neurosci Methods. 2008;172:250–4. doi: 10.1016/j.jneumeth.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubie C, Kempf K, Hans J, Su T, Tilton B, Georg T, et al. Housekeeping gene variability in normal and cancerous colorectal, pancreatic, esophageal, gastric and hepatic tissues. Mol Cell Probes. 2005;19:101–9. doi: 10.1016/j.mcp.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 29.Saha T, Rih JK, Rosen EM. BRCA1 down-regulates cellular levels of reactive oxygen species. FEBS Lett. 2009;583:1535–43. doi: 10.1016/j.febslet.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saha T, Rih JK, Roy R, Ballal R, Rosen EM. Transcriptional regulation of the base excision repair pathway by BRCA1. J Biol Chem. 2010;285:19092–105. doi: 10.1074/jbc.M110.104430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 33.Franch HA, Wang X, Sooparb S, Brown NS, Du J. Phosphatidylinositol 3-kinase activity is required for epidermal growth factor to suppress proteolysis. J Am Soc Nephrol. 2002;13:903–9. doi: 10.1681/ASN.V134903. [DOI] [PubMed] [Google Scholar]
- 34.Shen W, Brown NS, Finn PF, Dice JF, Franch HA. Akt and Mammalian target of rapamycin regulate separate systems of proteolysis in renal tubular cells. J Am Soc Nephrol. 2006;17:2414–23. doi: 10.1681/ASN.2005111157. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka K, Kawakami T, Tateishi K, Yashiroda H, Chiba T. Control of IkappaBalpha proteolysis by the ubiquitin-proteasome pathway. Biochimie. 2001;83:351–6. doi: 10.1016/S0300-9084(01)01237-8. [DOI] [PubMed] [Google Scholar]
- 36.Havelka AM, Berndtsson M, Olofsson MH, Shoshan MC, Linder S. Mechanisms of action of DNA-damaging anticancer drugs in treatment of carcinomas: is acute apoptosis an “off-target” effect? Mini Rev Med Chem. 2007;7:1035–9. doi: 10.2174/138955707782110196. [DOI] [PubMed] [Google Scholar]
- 37.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 38.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 39.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–10. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 41.Kaushik S, Massey AC, Mizushima N, Cuervo AM. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell. 2008;19:2179–92. doi: 10.1091/mbc.E07-11-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 43.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu W, Bagaitkar J, Watabe K. Roles of AKT signal in breast cancer. Front Biosci. 2007;12:4011–9. doi: 10.2741/2367. [DOI] [PubMed] [Google Scholar]
- 45.Shcherbakov AM, Gershtein ES, Anurova OA, Kushlinskii NE. Activated proteinkinase B in breast cancer. Bull Exp Biol Med. 2005;139:608–10. doi: 10.1007/s10517-005-0357-4. [DOI] [PubMed] [Google Scholar]
- 46.Tokunaga E, Kimura Y, Mashino K, Oki E, Kataoka A, Ohno S, et al. Activation of PI3K/Akt signaling and hormone resistance in breast cancer. Breast Cancer. 2006;13:137–44. doi: 10.2325/jbcs.13.137. [DOI] [PubMed] [Google Scholar]
- 47.Pham FH, Sugden PH, Clerk A. Regulation of protein kinase B and 4E-BP1 by oxidative stress in cardiac myocytes. Circ Res. 2000;86:1252–8. doi: 10.1161/01.RES.86.12.1252. [DOI] [PubMed] [Google Scholar]
- 48.Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, et al. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274:22699–704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 49.van den Berg MC, Burgering BM. Integrating Opposing Signals Toward Forkhead Box O. Antioxid Redox Signal. 2011;14:607–21. doi: 10.1089/ars.2010.3415. [DOI] [PubMed] [Google Scholar]
- 50.Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–70. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan S, Meng Q, Saha T, Sarkar FH, Rosen EM. Low concentrations of diindolylmethane, a metabolite of indole-3-carbinol, protect against oxidative stress in a BRCA1-dependent manner. Cancer Res. 2009;69:6083–91. doi: 10.1158/0008-5472.CAN-08-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saha T, Ghosh S, Vassilev A, DePamphilis ML. Ubiquitylation, phosphorylation and Orc2 modulate the subcellular location of Orc1 and prevent it from inducing apoptosis. J Cell Sci. 2006;119:1371–82. doi: 10.1242/jcs.02851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saha T, Smulson M, Rosen EM. BRCA1 regulation of base excision repair pathway. Cell Cycle. 2010;9:2471–2. doi: 10.4161/cc.9.13.12084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.