

Abstract
Autophagosome formation is a complex cellular process, which requires major membrane rearrangements leading to the creation of a relatively large double-membrane vesicle that directs its contents to the lysosome for degradation. Although various membrane compartments have been identified as sources for autophagosomal membranes, the molecular mechanism underlying these membrane trafficking steps remains elusive. To address this question we performed a systematic analysis testing all known Tre-2/Bub2/Cdc16 (TBC) domain-containing proteins for their ability to inhibit autophagosome formation by disrupting a specific membrane trafficking step. TBC proteins are thought to act as inhibitors of Rab GTPases, which regulate membrane trafficking events. Up to 11 TBC proteins inhibit autophagy when overexpressed and one of these, TBC1D14, acts at an early stage during autophagosome formation and is involved in regulating recycling endosomal traffic. We found that the early acting autophagy proteins ATG9 and ULK1 localize to transferrin receptor (TFR)-positive recycling endosomes (RE), which are tubulated by excess TBC1D14 leading to an inhibition of autophagosome formation. Finally, transferrin (TF)-containing recycling endosomal membranes can be incorporated into newly forming autophagosomes, although it is likely that most of the autophagosome membrane is subsequently acquired from other sources.
Keywords: ATG13, ATG9, RAB11, Rab GTPases, RabGAPs, TBC1D14, ULK1, autophagosome formation, autophagy, recycling endosomes, transferrin receptor
A RabGAP screen reveals 11 RabGAPs that inhibit autophagy
Many autophagy-related (ATG) proteins have been identified that are necessary for induction of autophagosome formation. These are sequentially recruited to the autophagosome precursor membrane termed the phagophore, or isolation membrane, starting with ATG9 and the ULK1/2 kinase complex containing ATG13, followed by recruitment of the class III phosphatidylinositol 3-kinase complex. To address the membrane trafficking steps necessary for double-membrane formation and expansion we set out to analyze all putative and known TBC domain-containing RabGAP proteins for their ability to inhibit starvation-induced autophagy when overexpressed. RabGAPs act as inhibitors of their corresponding Rab GTPase and therefore abolish a specific membrane trafficking step controlled by that Rab GTPase. We found 11 RabGAPs that were able to inhibit autophagosome formation by 40% or more when overexpressed. This number is perhaps unsurprising considering how many different membrane sources for autophagosomes—including the ER, Golgi, mitochondria and plasma membrane—have been identified, and it supports a model where many different membrane trafficking pathways are involved in autophagosome formation. Further evidence for the complexity and diversity involved in autophagosome formation has come from recent findings from the Klionsky and Rubinsztein labs that multiple endocytic and exocytic SNARE proteins, which mediate membrane fusion events, are necessary for autophagosome formation in yeast and mammalian cells. Additionally, the Wade and Dikic laboratories have identified a somewhat overlapping set of TBC proteins that interact through an autophagy network or directly with the autophagosome marker LC3.
TBC1D14 is an effector of RAB11, whose function in recycling endosomal traffic is necessary for autophagosome formation
Our focus turned to the TBC protein TBC1D14, in particular, as it shows extensive colocalization with ULK1 as well as binding it directly. The ULK1 kinase complex together with ATG9 are the earliest acting ATG proteins, and we therefore hypothesized that TBC1D14 is also involved in early steps of autophagy. Although we could not detect GAP activity toward any of the 55 Rab GTPases tested, TBC1D14 binds to activated RAB11 exclusively and tubulates TFR-RAB11-positive recycling endosomes when overexpressed. TBC1D14 most likely acts as an effector of active RAB11. Indeed, overexpressing a constitutively inactive form of RAB11 (RAB11N124I) also leads to inhibition of autophagy and tubulation of TFR-positive REs. Additionally, this tubulation leads to impaired TF recycling to the plasma membrane and sequestration of ULK1 and ATG13 on tubulated REs. Ablation of the RE compartment by DAB/H2O2 precipitation also leads to impaired autophagosome formation, supporting the notion that the main mechanism of autophagy inhibition by TBC1D14 is through impairment of RE function, which is necessary for autophagosome formation (Fig. 1).
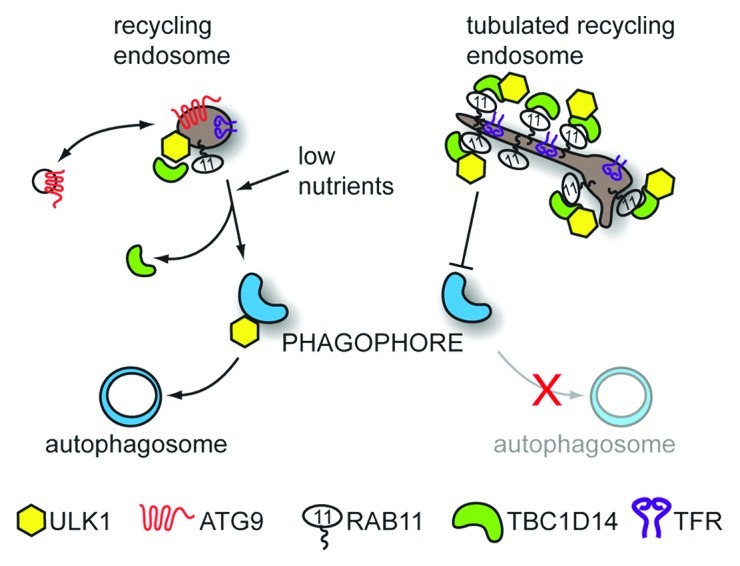
Figure 1. Recycling endosomes contribute to autophagosome formation. REs shuttle internalized cargo such as TF (not shown) with its receptor, TFR, back to the plasma membrane. Additionally, a pool of REs containing the transmembrane protein ATG9 and ULK1 are important in autophagosome formation. Blocking RE traffic by overexpressing the RAB11 effector protein TBC1D14 or inactive RAB11N124I leads to tubulation of REs and inhibition of starvation-induced autophagy. While ULK1 remains associated with REs, ATG9 relocalizes upon starvation, possibly to recruit membrane from other sources. Meanwhile, ULK1-positive REs partially relocalize with the autophagosome marker LC3 (not shown) and the RE membrane can be integrated into forming autophagosomes leading to deposition of TF in the autophagosomal lumen.
ULK1 and ATG9-Positive REs contribute to autophagosome formation
TBC1D14-tubulated REs sequester most of the cellular ULK1, but we have shown ULK1 can normally also be found on a pool of undisturbed TFR-positive REs, which are also ATG9 positive. These REs partially colocalize with the autophagosome marker LC3 upon induction of autophagy, and this colocalization is blocked by overexpression of TBC1D14. Intriguingly, the RE membrane seems to be incorporated into the forming autophagosome although this is most likely followed by further membrane recruitment from other sources. Interestingly, ULK1 remains associated with TFR-positive REs after induction of autophagy, whereas ATG9 seems to be retrieved. This is consistent with the findings of Orsi et al. from our laboratory that ATG9 is not incorporated into the autophagosome, but may traffic elsewhere to recruit more membrane and/or autophagy proteins. In light of our findings and data regarding SNARE proteins involved in autophagosome formation, it seems REs may be placed ideally at the nexus between endo- and exocytosis to coordinate various vesicular trafficking events involved in autophagosome formation. It needs to be noted that this may apply mostly to starvation-induced autophagy where REs may be linked to nutrient sensing, whereas basal autophagy, which does not rely on ATG9, or RAB11-TBC1D14 may use different or additional mechanisms.
Acknowledgment
We thank Cancer Research UK for funding the work to which this article refers and for funding the work in the laboratory of S.A.T.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/21486